
Targeted Delivery of Butyrate Improves Glucose Homeostasis, Reduces Hepatic Lipid Accumulation and Inflammation in db/db Mice
 , , and
, , and
Abstract
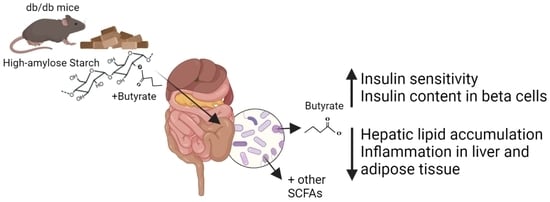
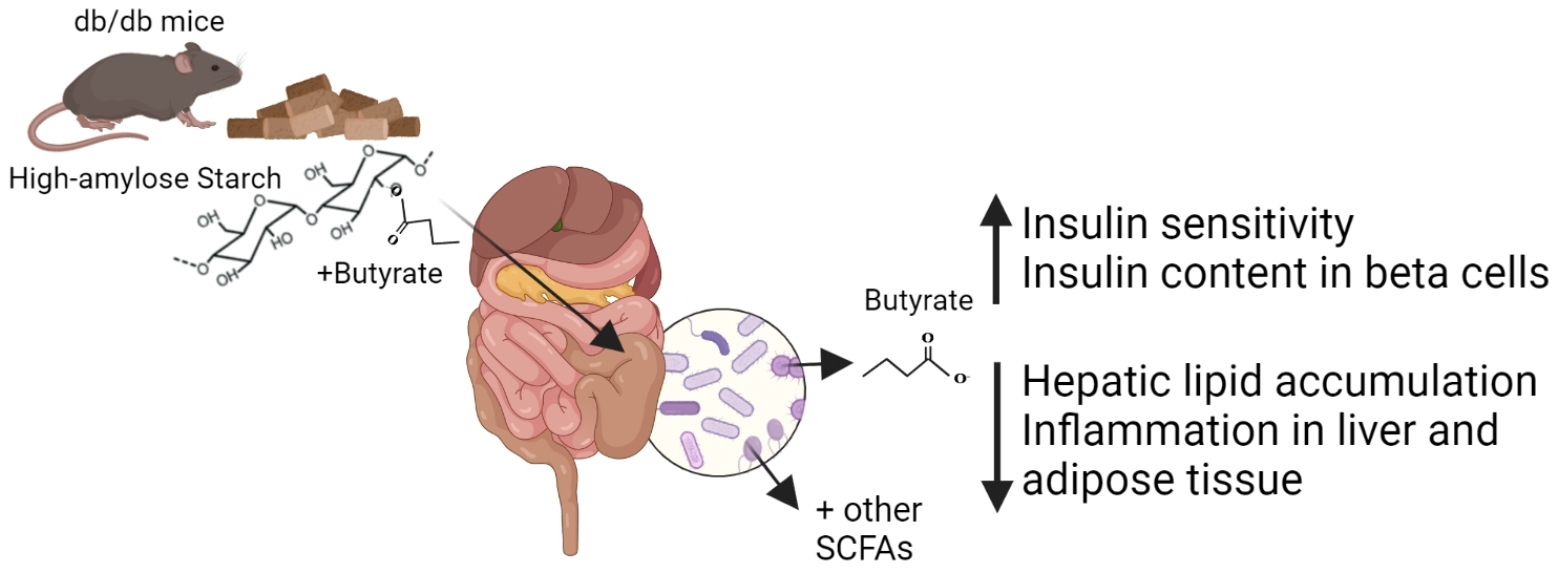
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
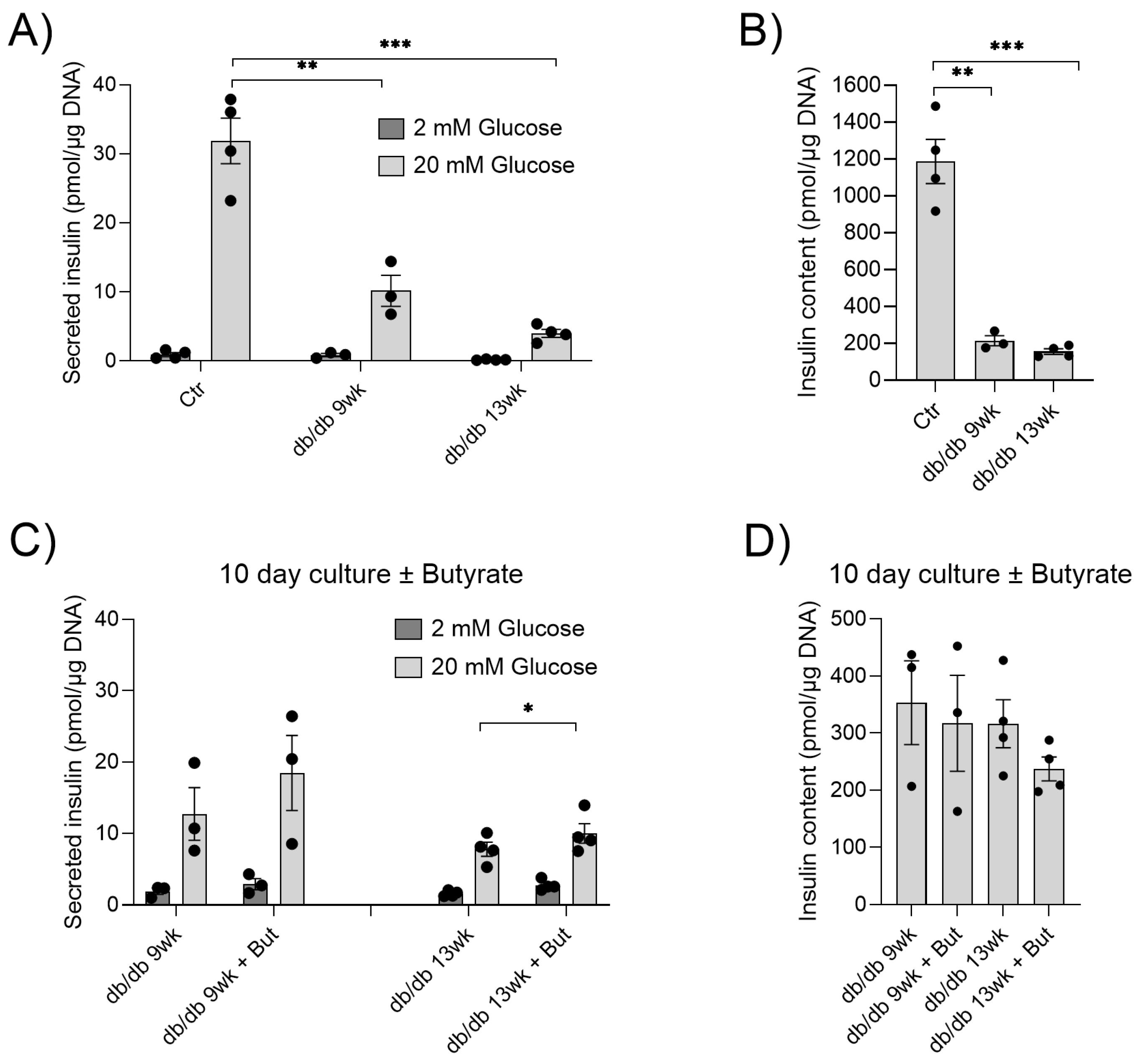
2.1. Butyrate Improves the Function of Islets in db/db Mice In Vitro
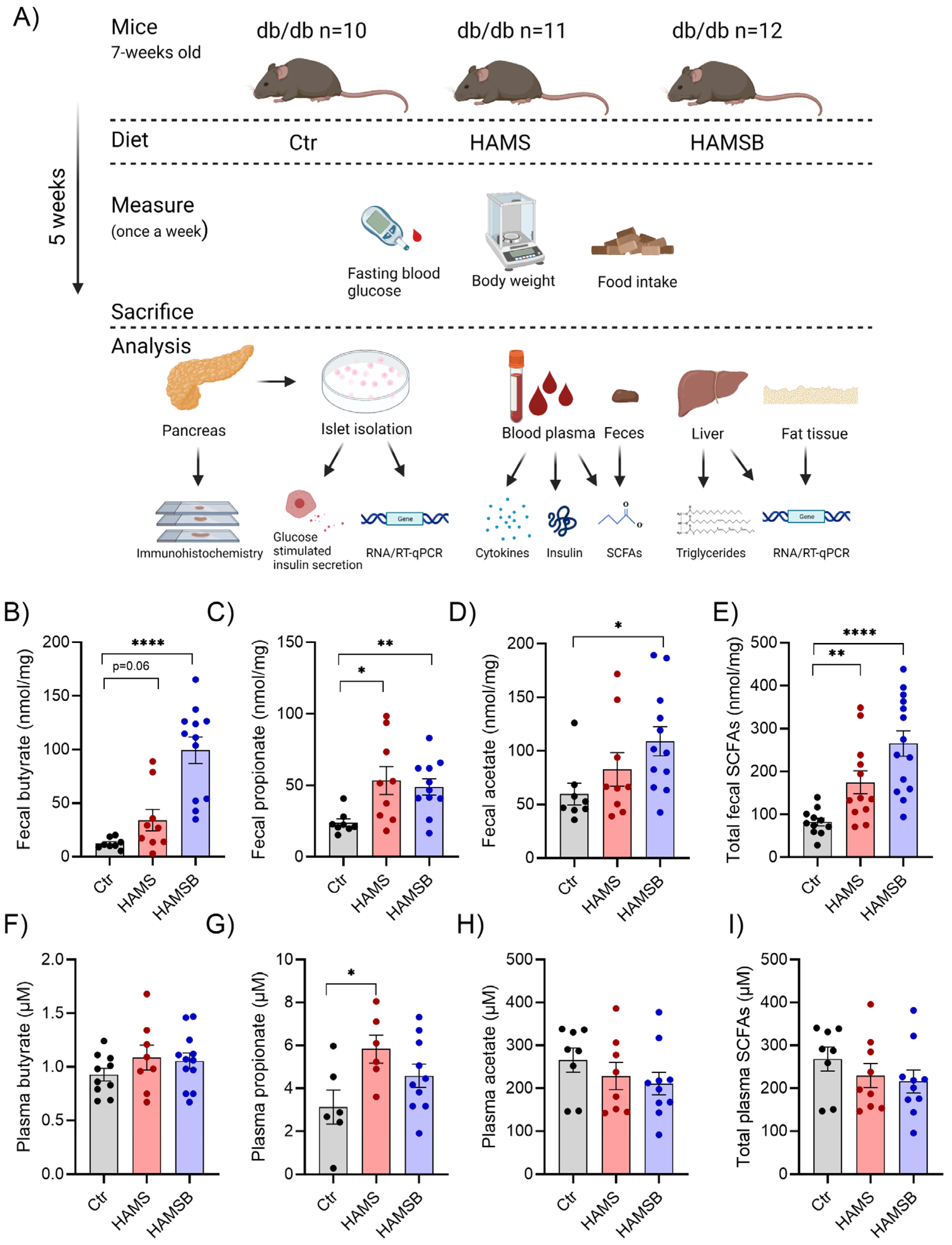
2.2. Effect of HAMSB on SCFA Production
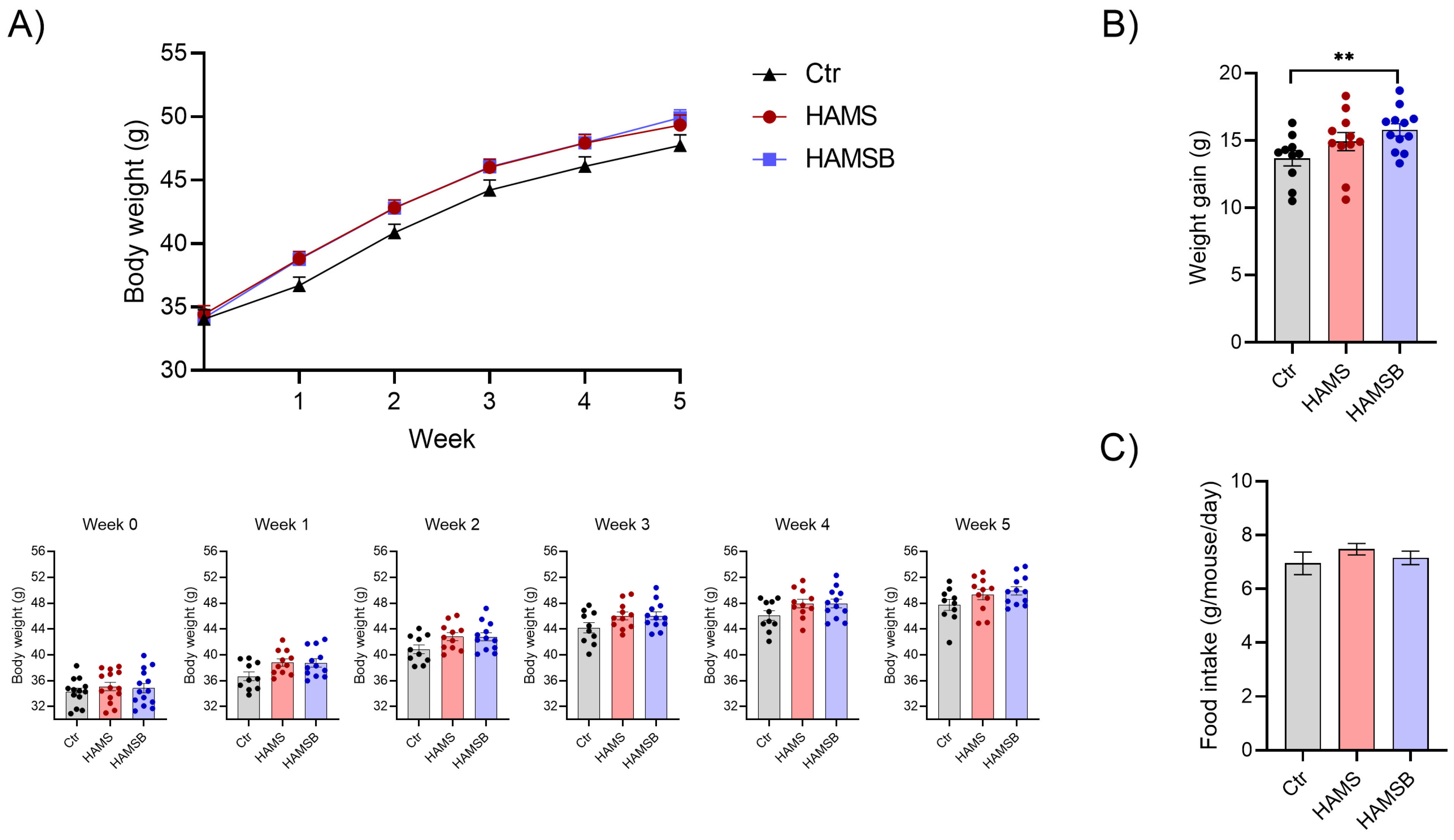
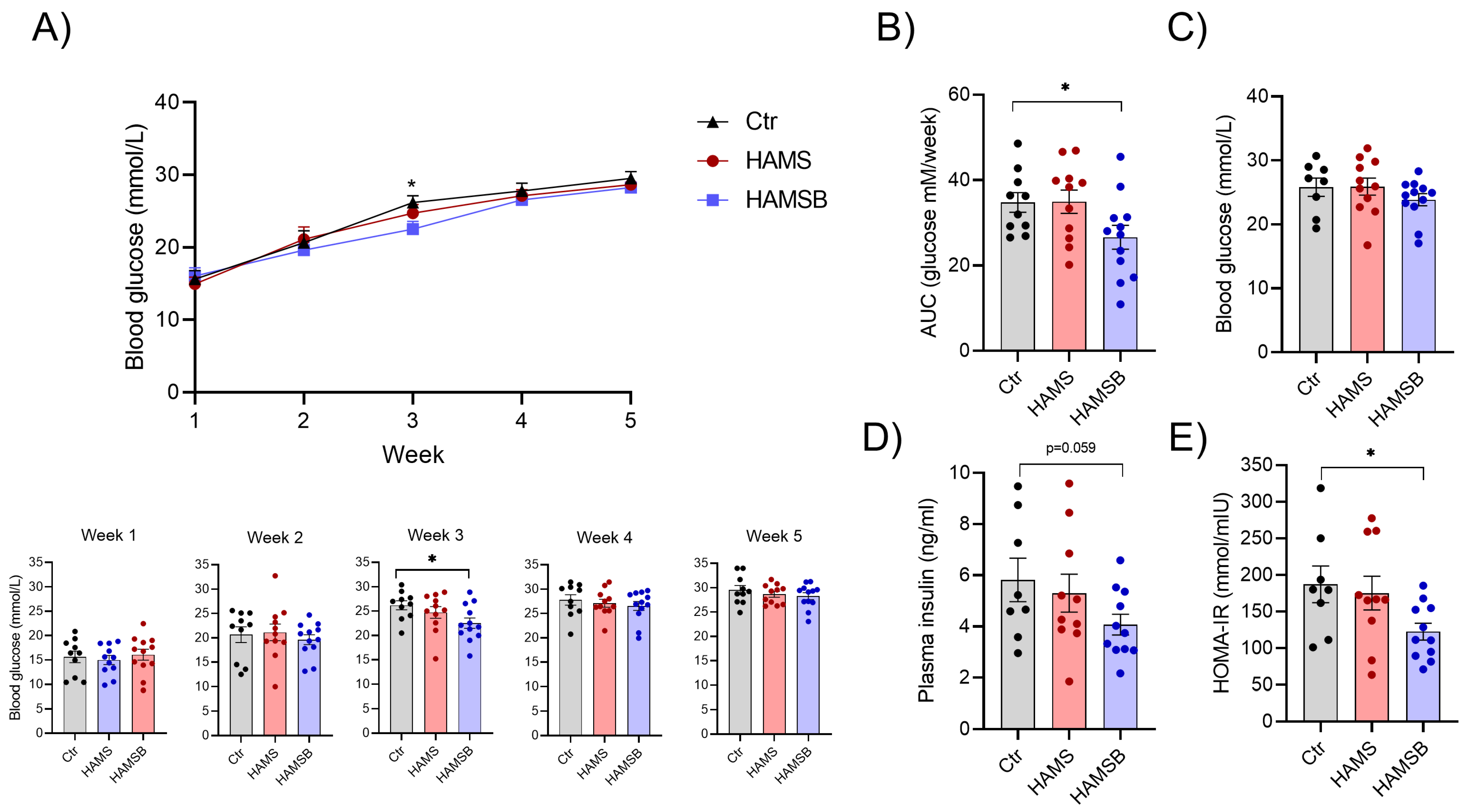
2.3. Effect of HAMSB on Body Weight and Glycemic Control
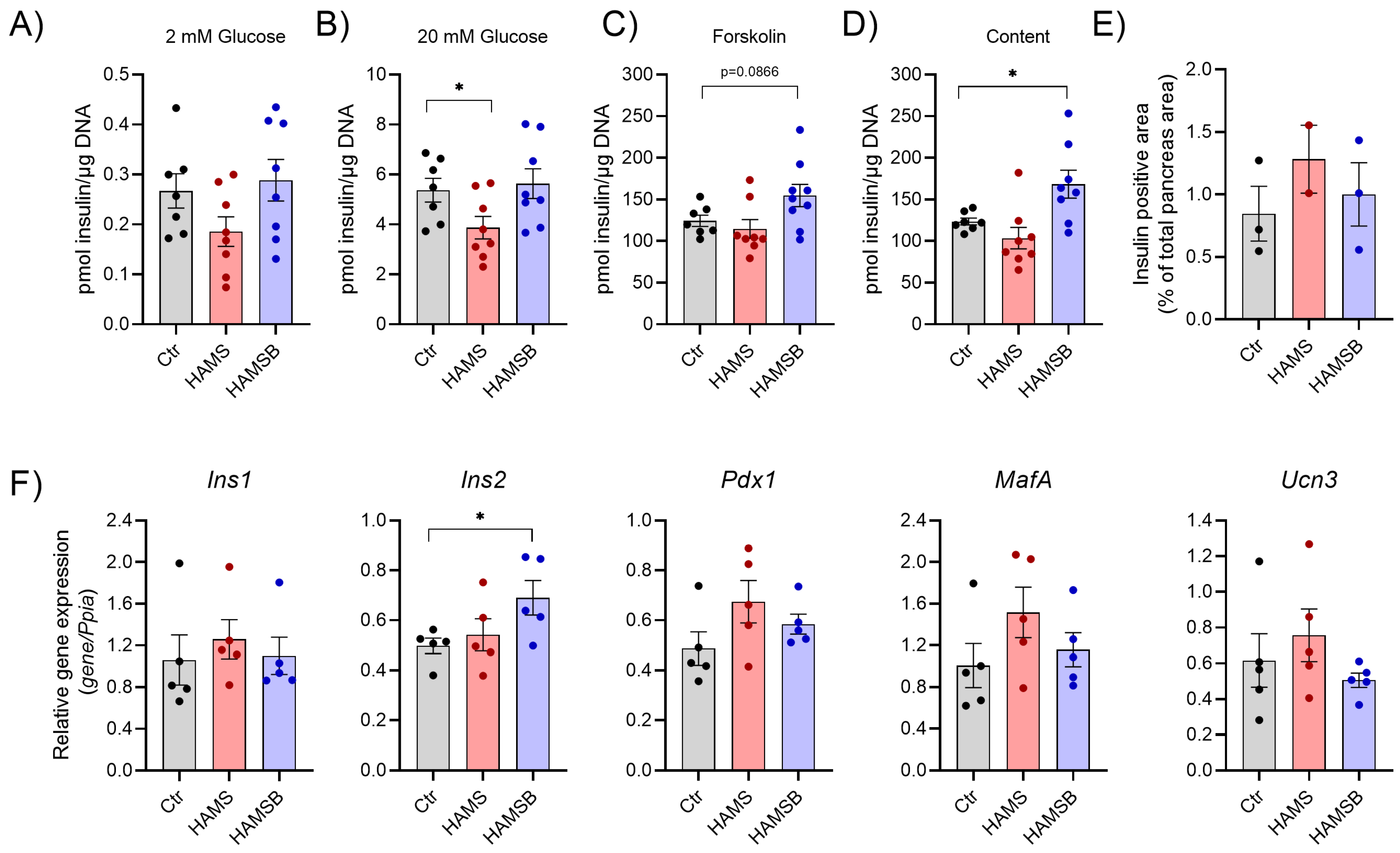
2.4. Effect of HAMSB on Islet Function and Identity
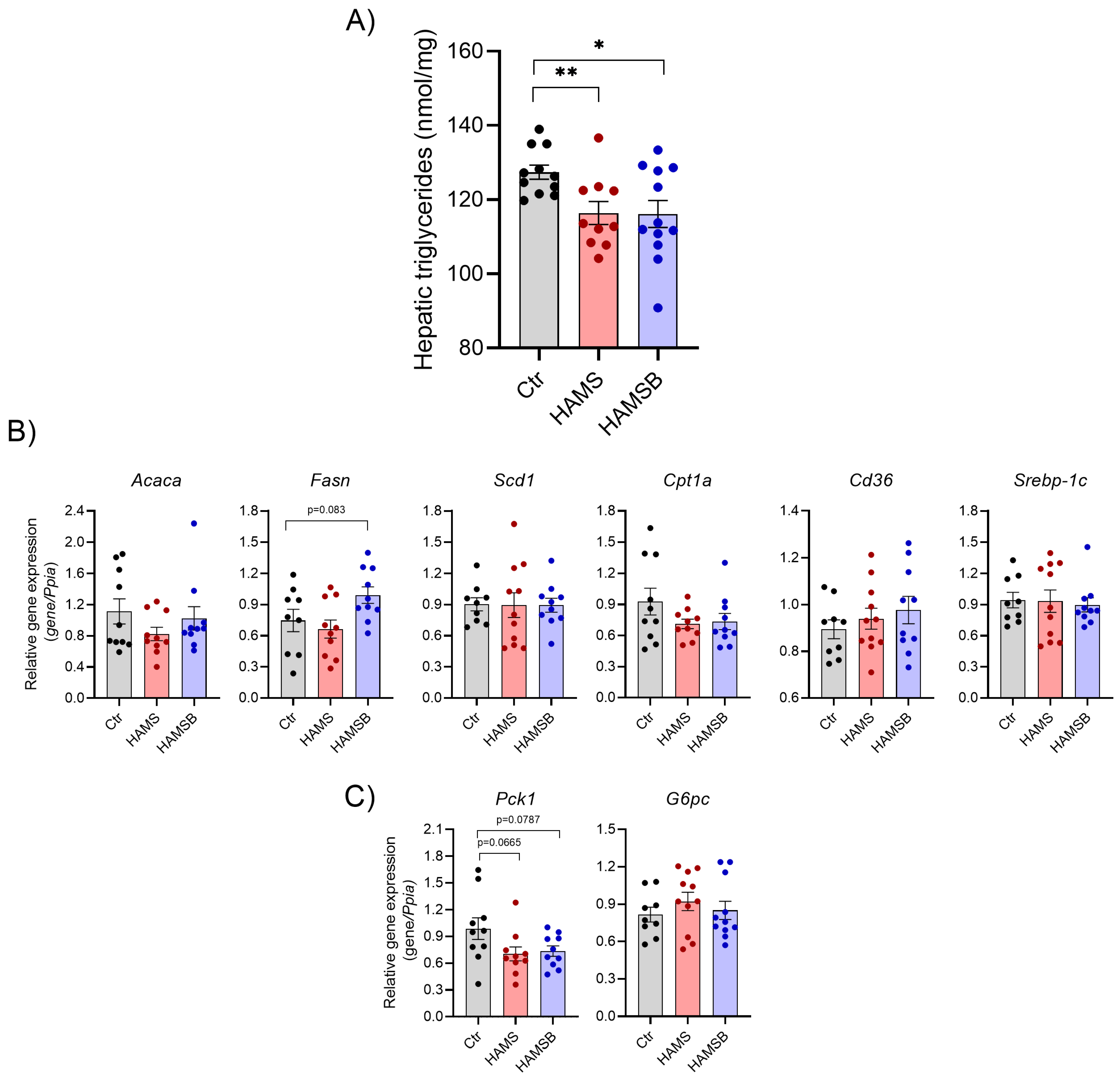
2.5. Effect of HAMSB on Lipid and Glucose Metabolism in the Liver
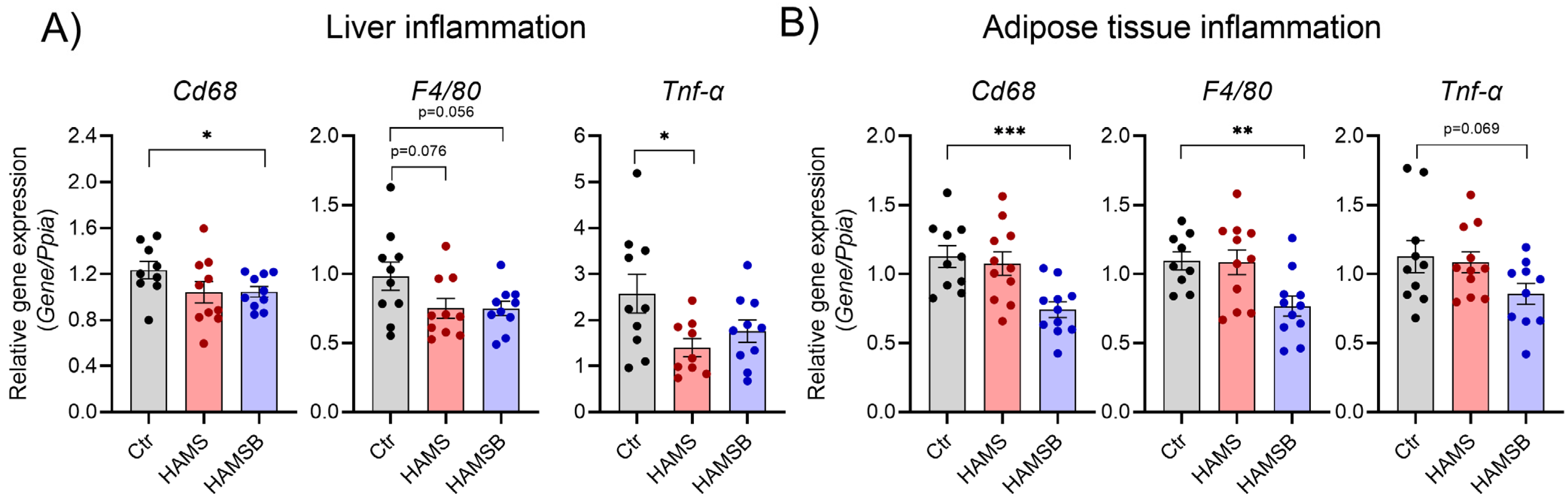
2.6. Effect of HAMSB on Inflammation
3. Discussion
4. Materials and Methods
4.1. Ex Vivo Stimulation of Islets of db/db Mice
4.2. Experimental Design of Mouse Study
4.3. Short-Chain Fatty Acid Analyses
4.4. Blood Samples
4.5. Glucose-Stimulated Insulin Secretion
4.6. RNA Isolation and Gene Expression
4.7. Immunohistochemistry and Beta-Cell Area
4.8. Hepatic Triglycerides
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. [Google Scholar]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global Aetiology and Epidemiology of Type 2 Diabetes Mellitus and Its Complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.A.; Gannon, M. The Beta-cell in Type 2 Diabetes. Curr. Diabetes Rep. 2019, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- McCarville, J.L.; Chen, G.Y.; Cuevas, V.D.; Troha, K.; Ayres, J.S. Microbiota Metabolites in Health and Disease. Annu. Rev. Immunol. 2020, 38, 147–170. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut Microbiota in Human Metabolic Health and Disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Mariño, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect against Type 1 Diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef]
- Bell, K.J.; Saad, S.; Tillett, B.J.; McGuire, H.M.; Bordbar, S.; Yap, Y.A.; Nguyen, L.T.; Wilkins, M.R.; Corley, S.; Brodie, S.; et al. Metabolite-Based Dietary Supplementation in Human Type 1 Diabetes Is Associated with Microbiota and Immune Modulation. Microbiome 2022, 10, 9. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The Short-Chain Fatty Acid Acetate Reduces Appetite via a Central Homeostatic Mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Tremaroli, V.; Schmidt, C.; Lundqvist, A.; Olsson, L.M.; Krämer, M.; Gummesson, A.; Perkins, R.; Bergström, G.; Bäckhed, F. The Gut Microbiota in Prediabetes and Diabetes: A Population-Based Cross-Sectional Study. Cell Metab. 2020, 32, 379–390.e3. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut Metagenome in European Women with Normal, Impaired and Diabetic Glucose Control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Sanna, S.; van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vich Vila, A.; Võsa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.M.A.E.; Oosting, M.; et al. Causal Relationships among the Gut Microbiome, Short-Chain Fatty Acids and Metabolic Diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.A.H.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; Dantoft, T.M.; et al. Aberrant Intestinal Microbiota in Individuals with Prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; et al. A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheus, V.A.; Monteiro, L.C.S.; Oliveira, R.B.; Maschio, D.A.; Collares-buzato, C.B. Butyrate Reduces High-Fat Diet-Induced Metabolic Alterations, Hepatic Steatosis and Pancreatic Beta-cell and Intestinal Barrier Dysfunctions in Prediabetic Mice. Exp. Biol. Med. 2017, 242, 1214–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.H.; Gao, C.L.; Guo, H.L.; Zhang, W.Q.; Huang, W.; Tang, S.S.; Gan, W.J.; Xu, Y.; Zhou, H.; Zhu, Q. Sodium Butyrate Supplementation Ameliorates Diabetic Inflammation in Db/Db Mice. J. Endocrinol. 2018, 238, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.Q.; Zhao, T.T.; Gui, D.K.; Gao, C.L.; Gu, J.L.; Gan, W.J.; Huang, W.; Xu, Y.; Zhou, H.; Chen, W.N.; et al. Sodium Butyrate Improves Liver Glycogen Metabolism in Type 2 Diabetes Mellitus. J. Agric. Food Chem. 2019, 67, 7694–7705. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Raso, G.M.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of Sodium Butyrate and Its Synthetic Amide Derivative on Liver Inflammation and Glucose Tolerance in an Animal Model of Steatosis Induced by High Fat Diet. PLoS ONE 2013, 8, e68626. [Google Scholar] [CrossRef] [Green Version]
- Henagan, T.M.; Stefanska, B.; Fang, Z.; Navard, A.M.; Ye, J.; Lenard, N.R.; Devarshi, P.P. Sodium Butyrate Epigenetically Modulates High-Fat Diet-Induced Skeletal Muscle Mitochondrial Adaptation, Obesity and Insulin Resistance through Nucleosome Positioning. Br. J. Pharmacol. 2015, 172, 2782–2798. [Google Scholar] [CrossRef] [Green Version]
- Sato, F.T.; Yap, Y.A.; Crisma, A.R.; Portovedo, M.; Murata, G.M.; Hirabara, S.M.; Ribeiro, W.R.; Marcantonio Ferreira, C.; Cruz, M.M.; Pereira, J.N.B.; et al. Tributyrin Attenuates Metabolic and Inflammatory Changes Associated with Obesity through a GPR109A-Dependent Mechanism. Cells 2020, 9, 2007. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.A.; van Zelm, M.C.; Muir, J.G.; Gibson, P.R. Review Article: Short Chain Fatty Acids as Potential Therapeutic Agents in Human Gastrointestinal and Inflammatory Disorders. Aliment. Pharmacol. Ther. 2018, 48, 15–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorin, M.J.; Yuan, Z.M.; Sentz, D.L.; Plaisance, K.; Eiseman, J.L. Plasma Pharmacokinetics of Butyrate after Intravenous Administration of Sodium Butyrate or Oral Administration of Tributyrin or Sodium Butyrate to Mice and Rats. Cancer Chemother. Pharmacol. 1999, 43, 445–453. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, H.W.J.; Naylor, C.P.E.; MacFarlane, G.T. Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canfora, E.E.; Van Der Beek, C.M.; Jocken, J.W.E.; Goossens, G.H.; Holst, J.J.; Olde Damink, S.W.M.; Lenaerts, K.; Dejong, C.H.C.; Blaak, E.E. Colonic Infusions of Short-Chain Fatty Acid Mixtures Promote Energy Metabolism in Overweight/Obese Men: A Randomized Crossover Trial. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, Z.; Hadi, A.; Tutunchi, H.; Asghari-Jafarabadi, M.; Naeinie, F.; Roshanravan, N.; Ostadrahimi, A.; Fadel, A. The Effects of Butyrate Supplementation on Glycemic Control, Lipid Profile, Blood Pressure, Nitric Oxide Level and Glutathione Peroxidase Activity in Type 2 Diabetic Patients: A Randomized Triple -Blind, Placebo-Controlled Trial. Clin. Nutr. ESPEN 2022, 49, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Bouter, K.E.C.; Bakker, G.J.; Levin, E.; Hartstra, A.V.; Kootte, R.S.; Udayappan, S.D.; Katiraei, S.; Bahler, L.; Gilijamse, P.W.; Tremaroli, V.; et al. Differential Metabolic Effects of Oral Butyrate Treatment in Lean versus Metabolic Syndrome Subjects Article. Clin. Transl. Gastroenterol. 2018, 9, e155. [Google Scholar] [CrossRef] [PubMed]
- Tougaard, N.H.; Frimodt-Møller, M.; Salmenkari, H.; Stougaard, E.B.; Zawadzki, A.D.; Mattila, I.M.; Hansen, T.W.; Legido-Quigley, C.; Hörkkö, S.; Forsblom, C.; et al. Effects of Butyrate Supplementation on Inflammation and Kidney Parameters in Type 1 Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Clin. Med. 2022, 11, 3573. [Google Scholar] [CrossRef]
- de Groot, P.F.; Nikolic, T.; Imangaliyev, S.; Bekkering, S.; Duinkerken, G.; Keij, F.M.; Herrema, H.; Winkelmeijer, M.; Kroon, J.; Levin, E.; et al. Oral Butyrate Does Not Affect Innate Immunity and Islet Autoimmunity in Individuals with Longstanding Type 1 Diabetes: A Randomised Controlled Trial. Diabetologia 2020, 63, 597–610. [Google Scholar] [CrossRef]
- Felizardo, R.J.F.; de Almeida, D.C.; Pereira, R.L.; Watanabe, I.K.M.; Doimo, N.T.S.; Ribeiro, W.R.; Cenedeze, M.A.; Hiyane, M.I.; Amano, M.T.; Braga, T.T.; et al. Gut Microbial Metabolite Butyrate Protects against Proteinuric Kidney Disease through Epigenetic- and GPR109a-Mediated Mechanisms. FASEB J. 2019, 33, 11894–11908. [Google Scholar] [CrossRef] [Green Version]
- Bajka, B.H.; Topping, D.L.; Cobiac, L.; Clarke, J.M. Butyrylated Starch Is Less Susceptible to Enzymic Hydrolysis and Increases Large-Bowel Butyrate More than High-Amylose Maize Starch in the Rat. Br. J. Nutr. 2006, 96, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Annison, G.; Illman, R.J.; Topping, D.L. Acetylated, Propionylated or Butyrylated Starches Raise Large Bowel Short-Chain Fatty Acids Preferentially When Fed to Rats. J. Nutr. 2003, 133, 3523–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, F.; Wang, J.; Wang, A.; Yao, X.; Strappe, P.; Zhou, Z.; Wu, Q.; Guo, T. Starch Acylation of Different Short-Chain Fatty Acids and Its Corresponding Influence on Gut Microbiome and Diabetic Indexes. Food Chem. 2022, 389, 133089. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.M.; Topping, D.L.; Christophersen, C.T.; Bird, A.R.; Lange, K.; Saunders, I.; Cobiac, L. Butyrate Esterified to Starch Is Released in the Human Gastrointestinal Tract. Am. J. Clin. Nutr. 2011, 94, 1276–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, J.M.; Bird, A.R.; Topping, D.L.; Cobiac, L. Excretion of Starch and Esterified Short-Chain Fatty Acids by Ileostomy Subjects after the Ingestion of Acylated Starches. Am. J. Clin. Nutr. 2007, 86, 1146–1151. [Google Scholar] [CrossRef] [Green Version]
- Jama, H.A.; Rhys-jones, D.; Nakai, M.; Yao, C.K.; Climie, R.E.; Sata, Y.; Anderson, D.; Creek, D.J.; Head, G.A.; Kaye, D.M.; et al. Prebiotic Intervention with HAMSAB in Untreated Essential Hypertensive Patients Assessed in a Phase II Randomized Trial. Nat. Cardiovasc. Res. 2023, 2, 35–43. [Google Scholar] [CrossRef]
- Prause, M.; Pedersen, S.S.; Tsonkova, V.; Qiao, M.; Billestrup, N. Butyrate Protects Pancreatic Beta-cells from Cytokine-Induced Dysfunction. Int. J. Mol. Sci. 2021, 22, 10427. [Google Scholar] [CrossRef]
- Pedersen, S.S.; Prause, M.; Williams, K.; Barrès, R.; Billestrup, N. Butyrate Inhibits IL-1β-Induced Inflammatory Gene Expression by Suppression of NF-ΚB Activity in Pancreatic Beta-cells. J. Biol. Chem. 2022, 298, 102312. [Google Scholar] [CrossRef]
- Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Noland, R.C.; Eder, A.E.; Boulos, M.S.; Karlstad, M.D.; Jason Collier, J. Db/Db Mice Exhibit Features of Human Type 2 Diabetes That Are Not Present in Weight-Matched C57BL/6J Mice Fed a Western Diet. J. Diabetes Res. 2017, 2017, 8503754. [Google Scholar] [CrossRef] [Green Version]
- Geisler, C.E.; Renquist, B.J. Hepatic Lipid Accumulation: Cause and Consequence of Dysregulated Glucoregulatory Hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Püschel, G.P.; Klauder, J.; Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. J. Clin. Med. 2022, 11, 4358. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Duncan, S.H.; Carol McWilliam Leitch, E.; Child, M.W.; Flint, H.J. PH and Peptide Supply Can Radically Alter Bacterial Populations and Short-Chain Fatty Acid Ratios within Microbial Communities from the Human Colon. Appl. Environ. Microbiol. 2005, 71, 3692–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firrman, J.; Liu, L.S.; Mahalak, K.; Tanes, C.; Bittinger, K.; Tu, V.; Bobokalonov, J.; Mattei, L.; Zhang, H.; Van Den Abbeele, P. The Impact of Environmental PH on the Gut Microbiota Community Structure and Short Chain Fatty Acid Production. FEMS Microbiol. Ecol. 2022, 98, fiac038. [Google Scholar] [CrossRef]
- Boets, E.; Gomand, S.V.; Deroover, L.; Preston, T.; Vermeulen, K.; De Preter, V.; Hamer, H.M.; Van den Mooter, G.; De Vuyst, L.; Courtin, C.M.; et al. Systemic Availability and Metabolism of Colonic-Derived Short-Chain Fatty Acids in Healthy Subjects: A Stable Isotope Study. J. Physiol. 2017, 595, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruppin, H.; Bar-Meir, S.; Soergel, K.H.; Wood, C.M.; Schmitt, M.G. Absorption of Short-Chain Fatty Acids by the Colon. Gastroenterology 1980, 78, 1500–1507. [Google Scholar] [CrossRef]
- Bloemen, J.G.; Venema, K.; van de Poll, M.C.; Olde Damink, S.W.; Buurman, W.A.; Dejong, C.H. Short Chain Fatty Acids Exchange across the Gut and Liver in Humans Measured at Surgery. Clin. Nutr. 2009, 28, 657–661. [Google Scholar] [CrossRef]
- Boland, B.B.; Rhodes, C.J.; Grimsby, J.S. The Dynamic Plasticity of Insulin Production in β-Cells. Mol. Metab. 2017, 6, 958–973. [Google Scholar] [CrossRef]
- Weickert, M.O.; Pfeiffer, A.F.H. Signalling Mechanisms Linking Hepatic Glucose and Lipid Metabolism. Diabetologia 2006, 49, 1732–1741. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; Bleeker, A.; Gerding, A.; Van Eunen, K.; Havinga, R.; Van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect against High-Fat Diet-Induced Obesity via a Pparg-Dependent Switch from Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gart, E.; van Duyvenvoorde, W.; Toet, K.; Caspers, M.P.M.; Verschuren, L.; Nielsen, M.J.; Leeming, D.J.; Lima, E.S.; Menke, A.; Hanemaaijer, R.; et al. Butyrate Protects against Diet-Induced NASH and Liver Fibrosis and Suppresses Specific Non-Canonical TGF-β Signaling Pathways in Human Hepatic Stellate Cells. Biomedicines 2021, 9, 1954. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosteli, A.; Sugaru, E.; Haemmerle, G.; Martin, J.F.; Lei, J.; Zechner, R.; Ferrante, A.W. Weight Loss and Lipolysis Promote a Dynamic Immune Response in Murine Adipose Tissue. J. Clin. Investig. 2010, 120, 3466–3479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, S.; Mande, S.S. Host-Microbiome Interactions: Gut-Liver Axis and Its Connection with Other Organs. Npj Biofilms Microbiomes 2022, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Jiang, Z.; Tang, X.; Wang, P.; Li, Y.; Sun, Y.; Le, G.; Zou, S. Sodium Butyrate Protects against Oxidative Stress in HepG2 Cells through Modulating Nrf2 Pathway and Mitochondrial Function. J. Physiol. Biochem. 2016, 73, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Gu, J.; Zhang, H.; Wang, Z.; Zhang, W.; Zhao, Y.; Zheng, Y.; Zhang, W.; Zhou, H.; Zhang, G.; et al. Sodium Butyrate-Modulated Mitochondrial Function in High-Insulin Induced HepG2 Cell Dysfunction. Oxid. Med. Cell. Longev. 2020, 2020, 1904609. [Google Scholar] [CrossRef]
- Yang, T.; Yang, H.; Heng, C.; Wang, H.; Chen, S.; Hu, Y.; Jiang, Z.; Yu, Q.; Wang, Z.; Qian, S.; et al. Amelioration of Non-Alcoholic Fatty Liver Disease by Sodium Butyrate Is Linked to the Modulation of Intestinal Tight Junctions in Db/Db Mice. Food Funct. 2020, 11, 10675–10689. [Google Scholar] [CrossRef]
- Yadav, H.; Lee, J.H.; Lloyd, J.; Walter, P.; Rane, S.G. Beneficial Metabolic Effects of a Probiotic via Butyrate-Induced GLP-1 Hormone Secretion. J. Biol. Chem. 2013, 288, 25088–25097. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The Impact of Short-Chain Fatty Acids on Glp-1 and Pyy Secretion from the Isolated Perfused Rat Colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–G65. [Google Scholar] [CrossRef] [Green Version]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-Chain Fatty Acids Stimulate Glucagon-like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Liu, L.; Zhou, W.; Yang, C.; Mai, G.; Li, H.; Chen, Y. Gut Microbiota-Derived Butyrate Regulates Gut Mucus Barrier Repair by Activating the Macrophage/WNT/ERK Signaling Pathway. Clin. Sci. 2022, 136, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia Drives Intestinal Barrier Dysfunction and Risk for Enteric Infection. Science 2018, 1383, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. [Google Scholar] [CrossRef]
- Park, J.W.; Kim, H.Y.; Kim, M.G.; Jeong, S.; Yun, C.H.; Han, S.H. Short-Chain Fatty Acids Inhibit Staphylococcal Lipoprotein-Induced Nitric Oxide Production in Murine Macrophages. Immune Netw. 2019, 19, e9. [Google Scholar] [CrossRef]
- Yin, L.; Laevsky, G.; Giardina, C. Butyrate Suppression of Colonocyte NF-ΚB Activation and Cellular Proteasome Activity. J. Biol. Chem. 2001, 276, 44641–44646. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Li, J.; Liu, Y.; Xiao, N.; Suo, H.; Xie, K.; Yang, C.; Wu, C. Short-Chain Fatty Acids Suppress Lipopolysaccharide-Induced Production of Nitric Oxide and Proinflammatory Cytokines through Inhibition of NF-KB Pathway in RAW264.7 Cells. Inflammation 2012, 35, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Kato, A.; Kato, C.; Fujii, E.; Adachi, K.; Miyoshi, A.; Suzuki, M. Segmentation of the Pathophysiological Stages of Diabetic Changes in the Db/Db Mouse. J. Toxicol. Pathol. 2009, 22, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skovsø, S. Modeling Type 2 Diabetes in Rats Using High Fat Diet and Streptozotocin. J. Diabetes Investig. 2014, 5, 349–358. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, S.S.; Prause, M.; Sørensen, C.; Størling, J.; Moritz, T.; Mariño, E.; Billestrup, N. Targeted Delivery of Butyrate Improves Glucose Homeostasis, Reduces Hepatic Lipid Accumulation and Inflammation in db/db Mice. Int. J. Mol. Sci. 2023, 24, 4533. https://doi.org/10.3390/ijms24054533
Pedersen SS, Prause M, Sørensen C, Størling J, Moritz T, Mariño E, Billestrup N. Targeted Delivery of Butyrate Improves Glucose Homeostasis, Reduces Hepatic Lipid Accumulation and Inflammation in db/db Mice. International Journal of Molecular Sciences. 2023; 24(5):4533. https://doi.org/10.3390/ijms24054533
Chicago/Turabian StylePedersen, Signe Schultz, Michala Prause, Christina Sørensen, Joachim Størling, Thomas Moritz, Eliana Mariño, and Nils Billestrup. 2023. "Targeted Delivery of Butyrate Improves Glucose Homeostasis, Reduces Hepatic Lipid Accumulation and Inflammation in db/db Mice" International Journal of Molecular Sciences 24, no. 5: 4533. https://doi.org/10.3390/ijms24054533