
Synthetic Small Molecule Modulators of Hsp70 and Hsp40 Chaperones as Promising Anticancer Agents

1
Institute for Physiology, Charité—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt Universität zu Berlin, Charitéplatz 1, 10117 Berlin, Germany
2
Organische Chemie 1, Universität Bayreuth, Universitätsstrasse 30, 95440 Bayreuth, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(4), 4083; https://doi.org/10.3390/ijms24044083
Submission received: 28 January 2023
/
Revised: 9 February 2023
/
Accepted: 11 February 2023
/
Published: 17 February 2023
(This article belongs to the Special Issue Recent Advances of Novel Pharmaceutical Designs for Anti-cancer Therapies)
Abstract
:A class of chaperones dubbed heat shock protein 70 (Hsp70) possesses high relevance in cancer diseases due to its cooperative activity with the well-established anticancer target Hsp90. However, Hsp70 is closely connected with a smaller heat shock protein, Hsp40, forming a formidable Hsp70-Hsp40 axis in various cancers, which serves as a suitable target for anticancer drug design. This review summarizes the current state and the recent developments in the field of (semi-)synthetic small molecule inhibitors directed against Hsp70 and Hsp40. The medicinal chemistry and anticancer potential of pertinent inhibitors are discussed. Since Hsp90 inhibitors have entered clinical trials but have exhibited severe adverse effects and drug resistance formation, potent Hsp70 and Hsp40 inhibitors may play a significant role in overcoming the drawbacks of Hsp90 inhibitors and other approved anticancer drugs.
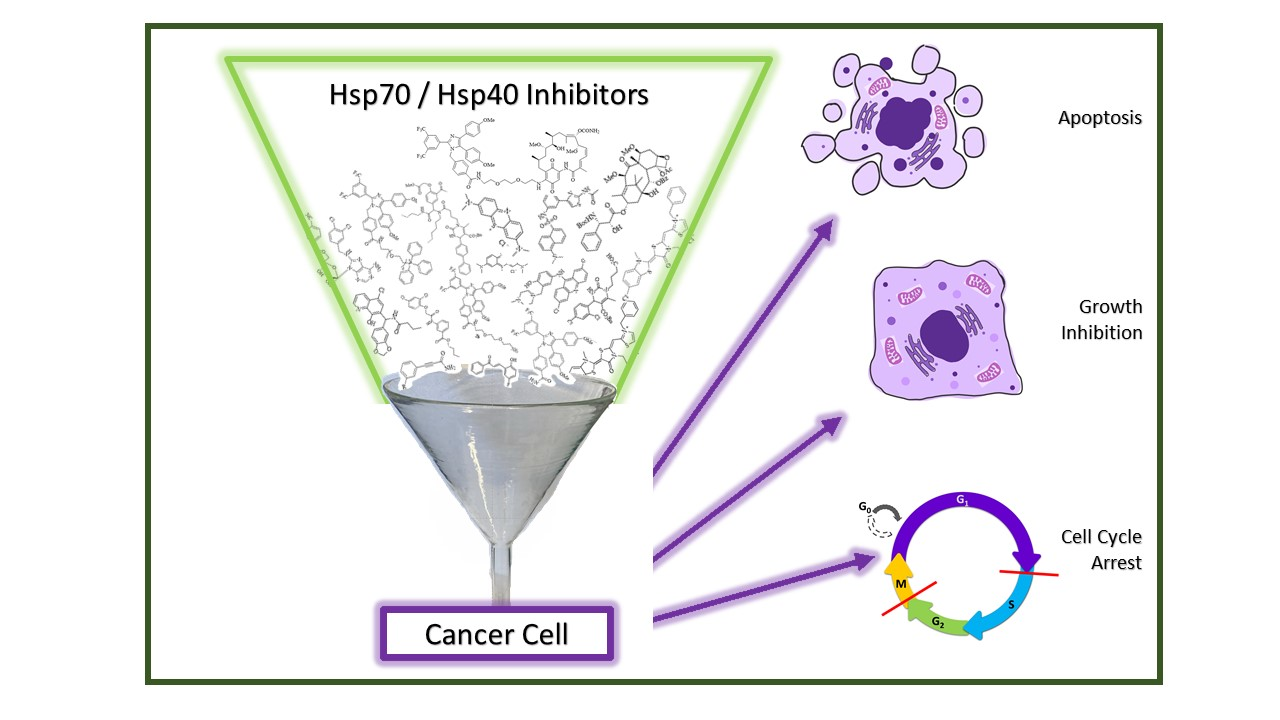
1. Introduction
Stress factors such as heat lead to defensive and protective cellular responses, enabling the cell to cope with the stress [1]. The emergence of chromosomal puffs as a defined heat shock response in Drosophila flies was observed for the first time in 1962 by Ferruccio Ritossa [2]. In 1974, the increased expression of certain heat shock proteins in response to heat and other stress factors was discovered [3]. These heat shock proteins (Hsps) are classified by their molecular weights in kDa (e.g., Hsp90, Hsp70, etc.) and function as chaperones in order to protect important proteins from degradation, to control the quality of protein folding, and to deliver misfolded or damaged proteins to the proteasome for disposal (protein triage), thus vouchsafing cell viability under these conditions [4]. This multi-chaperone system (“epichaperome”) plays an important role in various cancer diseases [5]. Canonical functions of chaperones are linked with the ubiquitin-proteasome system (UPS) and chaperone-mediated autophagy (CMA), while non-canonical functions affect the immune system, including inflammatory and autoimmune mechanisms [6]. Thus, targeting heat shock proteins is a promising strategy to combat cancer.
The Hsp family is subdivided into Hsp110 (HSPH), Hsp90 (HSPC), Hsp70 (HSPA), Hsp40 (DNAJ), small Hsps (HSPB), and the chaperonin family proteins Hsp60/Hsp10 (HSPD/E) and TRiC (CCT) [7]. Hsp90 has already become a valuable chaperone drug target. Prominent examples of anticancer active Hsp90 inhibitors are the natural products (geldanamycin and radicicol) as well as their (semi-)synthetic derivatives, 17-AAG/tanespimycin and ganetespib, the latter have reached clinical trials [8]. Hsp90 regulates the activity and stability of crucial transcription factors such as the tumor suppressor p53 and the androgen receptor (AR), while Hsp90 activity itself is regulated by posttranslational modifications (e.g., lysine acetylation under control by HDAC6) and by other heat shock proteins such as Hsp70 [9]. The protein folding by Hsp90 and Hsp70 is ATP-dependent, while Hsp70 has a crucial function in the protection of cells against various stress factors, including enhanced cell survival. In addition, Hsp70 forms complexes with Hsp90 with the help of HOP (Hsp70–Hsp90 organizing protein) in order to exert its housekeeping activities [10,11,12]. There is growing evidence that Hsp70 inhibitors have the potential to overcome Hsp90 inhibitor resistance [13]. The organelle-specific members of the Hsp70 protein family, such as mitochondrial mortalin (mtHsp70, Grp75) and endoplasmic reticulum/ER-based Grp78, were also identified as possible anticancer drug targets due to their crucial roles in cell proliferation and survival in various cancers [14,15]. In addition, extracellular Hsps attracted increased attention as cancer targets and biomarkers [16].
Client polypeptides are transferred to Hsp70 by the smaller co-chaperone, Hsp40. Hsp40 prevents the aggregation of unfolded polypeptides, but there are also folded proteins among the clients of Hsp40. There are numerous Hsp40 isoforms, also dubbed J-domain proteins (DNAJs), that are structurally different from Hsp70 and Hsp90 proteins [17]. Nevertheless, DNAJ/Hsp40 and Hsp70 proteins form a tight Hsp70/Hsp40 complex in order to fulfill their protein-folding functions [17,18]. Hsp40 binds to the nucleotide-binding domain (NBD) of Hsp70 and accelerates the ATPase activity of Hsp70 enormously [18]. Thus, small-molecule inhibitors of the Hsp70-Hsp40 axis have considerable potential as anticancer drug candidates.
In this review, the current state and recent developments in the field of synthetic Hsp70 and Hsp40 inhibitors are discussed.
2. Hsp70 Inhibitors
The mechanisms of action of proteins of the Hsp70 family and their roles in various cancer diseases were thoroughly reviewed recently [10,11,12]. In line with the mounting knowledge of these proteins, Hsp70 inhibitors are a continuously growing class of compounds, which can be subdivided into Hsp70 and Hsc70 inhibitors on the one hand and inhibitors targeting organelle-specific Hsp70 proteins such as Grp78 and mortalin on the other hand.
2.1. Hsp70i and Hsc70 Inhibitors
Inducible Hsp70 (also abbreviated as Hsp70i) and consecutive Hsc70 inhibitors can be classified according to their binding mode into N-terminal nucleotide binding domain/NBD-targeting inhibitors, C-terminal substrate binding domain/SBD-targeting inhibitors, and allosteric inhibitors (Table 1).
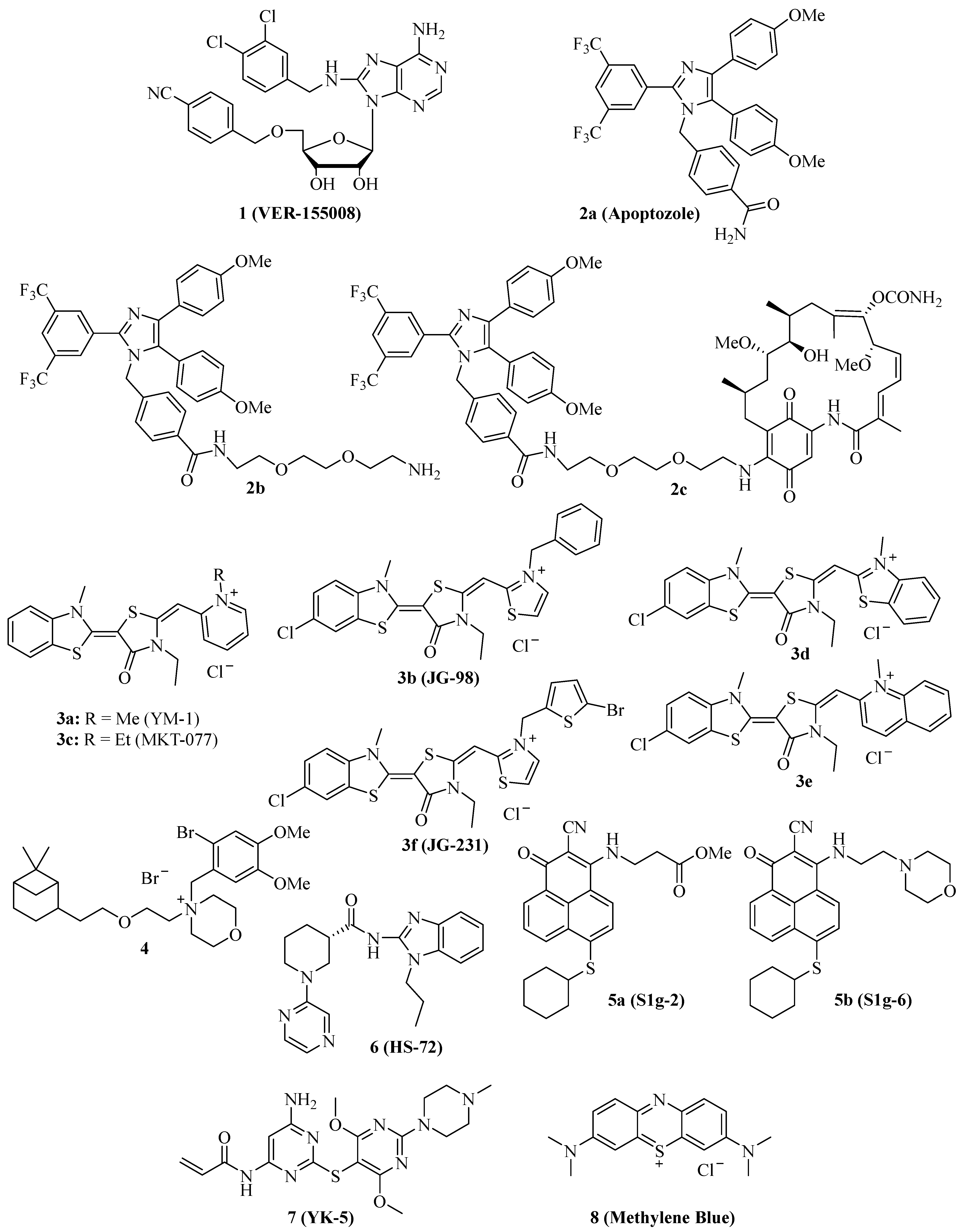
In 2009, the adenosine-derivative 1 (Figure 1, VER-155008) was identified as a selective NBD-targeting Hsc70/Hsp70 inhibitor (IC50 = 0.5 µM), which showed antiproliferative effects on HCT-116 colon carcinoma cells (GI50 = 5.0 µM), reduced Raf-1 and Her2 protein levels, and enhanced the apoptosis induction by the Hsp90 inhibitors 17-AAG and VER-82160 in HCT-116 cells [19,20]. Compound 1 also revealed promising effects on non-small cell lung cancer (NSCLC), such as inhibition of NSCLC proliferation and cell cycle arrest (increased G0/G1 cell percentage) [21]. In addition, compound 1 inhibited pleural mesothelioma cell proliferation and colony formation, which are associated with G1 cell cycle arrest, suppressed phospho-Akt, and induction of macroautophagy [22]. In LNCaP95 prostate cancer cells, the Hsp70 inhibitor 1 induced apoptosis and suppressed the expression of the full-length androgen receptor (AR-FL) and of the androgen receptor splice variant 7 (AR-V7), which are associated with castration-resistant prostate cancer (CRPC) [23]. These AR-suppressing effects were correlated with an inhibition of YB-1 phosphorylation by compound 1, followed by reduced nuclear translocation of YB-1. Another study using the AR-positive LNCaP and the AR-negative PC-3 prostate cancer cell lines showed that compound 1 was antiproliferative and pro-apoptotic in both cell lines, albeit the pro-apoptotic effects were higher in the AR-positive cells [24]. Compound 1 downregulated AR expression and induced G1 cell cycle arrest in the LNCaP cells. Moreover, a distinct suppression was observed for Hsp27 in PC-3 cells and HOP and Hsp90β in both cell lines treated with compound 1. In MCF-7 breast cancer cells, compound 1 induced apoptosis associated with mitochondrial damage, and the anticancer activity of compound 1 was reduced by heat shock [25]. Anaplastic thyroid carcinoma (APC) is the most lethal thyroid cancer with high drug resistance, but compound 1 was able to induce paraptosis in APC cells dependent on de novo protein synthesis [26]. In a panel of glioma cells (1321N1, GOS-3, and U87-MG), compound 1 showed higher antiproliferative activities (IC50 = 12–13 µM) than the approved drug temozolomide (IC50 = 135–180 µM) associated with downregulation of Akt kinase activity and the modulation of certain miRNAs, e.g., the upregulation of miR-215 and miR-194-5p [27]. Compound 1 was evaluated in muscle-invasive bladder cancer (MIBC) models and induced apoptosis along with inhibition of MIBC cell proliferation and migration [28]. The activity of compound 1 against MIBC was associated with the suppression of protein members of p53/Rb, PI3K, and SWI/SFW signaling. Especially strong degrading effects of compound 1 were observed on the demethylase KDMA6 and the histone acetyltransferase EP300, both members of the histone modification pathway.
Due to these promising anticancer effects of compound 1, its combination with various other anticancer drugs was investigated. In combination with the Hsp90 inhibitor radicicol, compound 1 was able to enhance the APC cell killing activity of radicicol, accompanied by suppressed heat shock cognate 70/Hsc70, Akt, and survival [29]. The combination of compound 1 with the Hsp90 inhibitor STA9090 was evaluated in MIBC models and was more efficient than single compound therapy [28]. However, the combination of compound 1 with doxorubicin in canine osteosarcoma (OSA) cells showed no improvements. Already, compound 1 alone displayed strong apoptosis induction, inhibition of colony formation, and antiproliferative activities against OSA cells based on Hsp70 inhibition as well as Akt suppression and BAG1 degradation [30]. In contrast to that, compound 1 showed synergy effects in combination with the Hsp90 inhibitor 17-AAD and sensitized A549 NSCLC cells to radiation therapy [21]. The combination of compound 1 with manumycin A, an anticancer active antibiotic that upregulates Hsp70 in cancer cells, sensitized lung tumor cells to manumycin A treatment [31]. Micelles of compound 1 together with gold nanorods were successfully tested as mild-temperature photothermal therapy in colon cancer, leading to strong colon tumor growth in vivo at a temperature of 45 °C [32]. Compound 1 exhibited considerable activity against multiple myeloma (MM) cells (IC50 = 1.7 µM for OPM2, 3.0 µM for RPMI 8226, and 6.5 µM for MM.1S cells), and the combination of compound 1 with the proteasome inhibitor bortezomib displayed synergy effects in terms of apoptosis induction in MM cells, which was associated with suppression of anti-apoptotic Bcl-2, Bcl-xL, and Mcl-1 and upregulation of pro-apoptotic NOXA and Bim [33]. In addition, the ER stress marker CHOP (CCAAT-enhancer binding protein homologous protein) was induced by this combination treatment. The natural product shikonin was described as a proteasome inhibitor and necroptosis inducer in MM cells while it also upregulated Hsp70, and, thus, the combination with compound 1 enhanced shikonin-induced MM cell death [34]. Compound 1 also exhibited promising effects on acute myeloid leukemia (AML) cells alone (induction of apoptosis, inhibition of cell proliferation, and colony formation) and in combination with the Hsp90 inhibitor 17-DMAG (additive antiproliferative and pro-apoptotic activity) [35]. The release of AML cell growth factors and regulators such as TNF-α, VEGF, IL-3, IL-1β, and IL-1 receptor antagonist was strongly suppressed upon treatment with compound 1. A new structurally related 6,8,9-trisubstituted purine derivative was recently disclosed that induced apoptosis and senescence in luminal A subtype MCF-7 breast carcinoma cells [36].
The substituted imidazole derivative 2a (Figure 1, apoptozole) was discovered as a pro-apoptotic inhibitor of Hsc70 (KD = 210 nM) and Hsp72 (KD = 140 nM) in 2008, which showed high tumor cell growth inhibitory activities with GI50 values in the nanomolar concentration range (GI50 = 220 nM for SK-OV-3, 250 nM for HCT-15, and 130 nM for A549 cells) [37]. Compound 2a was identified as an inhibitor of the Hsp70 ATPase by affinity chromatography upon conjugation of the amino-ethyloxy modified apoptozole derivative 2b (Figure 1) with a resin, leading to a reduced interaction of Hsp70 with APAF-1, while no affinity to Hsp40, Hsp60, or Hsp90 was observed [38]. Time-dependent antiproliferative activity was checked, and amenable IC50 values (0.8 µM for A549, 0.8 µM for HeLa, 0.7 µM for MDA-MB-231, and 0.7 µM for HepG2 cells) were obtained after 72 h of incubation, while the combination with doxorubicin led to a sensitization of A549 and HeLa cells to doxorubicin treatment. At doses of 4 mg/kg/day (i.p.) given for two weeks, compound 2b reduced the tumor growth of A549, RKO, and HeLa tumor xenografts by 61%, 65%, and 68%, respectively; in the latter case, the combination with doxorubicin led to more pronounced tumor growth inhibition (81% tumor growth reduction). The anti-leukemia properties of compound 2b and its hybrid molecules, such as compound 2c (Figure 1), with the Hsp90 inhibitor geldanamycin (2b and geldanamycin connected by ethylene glycol-based linker systems) were studied [39]. Compounds 2b, hybrid 2c, and other related hybrids induced apoptosis in a caspase-dependent way, however, the hybrids were more active against leukemia cells than their parent compounds 2b and geldanamycin. Compound 2c inhibited autophagy, but it also induced apoptosis in HeLa cancer cells based on its selective inhibition of the lysosomal Hsp70 and the degradation of lysosomal membranes associated with cathepsin release followed by caspase activation [40].
Several synthetic allosteric inhibitors of the Hsp70 ATPase domain were described. The rhodacyanine class of the Hsp70-inhibitory dye compounds was established by the discovery of compound 3a (Figure 1, YM-1), in particular, by the blocking of the Hsp70 interaction with the nucleotide exchange factor (NEF) Bag3 by compound 3a (IC50 = 4.8 µM) [41]. 3a binds to an allosteric binding site, stabilizing ADP-bound Hsp70 with a weak Bag3 affinity. Consequently, compound 3a suppressed FoxM1 and HIF1α pathways in MCF-7 and HeLa cells, which is unique for Hsp70-Bag3 inhibition since other Hsp70 inhibitors (e.g., the natural flavonoid myricetin) did not show such mechanistic effects. In MCF-7 breast carcinoma xenografts, compound 3a (25 mg/kg every second day for 6 days, i.p.) inhibited tumor growth associated with induction of p21 and suppression of FoxM1 and survivin. The activity of compound 3a was also investigated in glioma models, and it sensitized U251 and U343 glioma cells to treatment with the Bcl-2 inhibitors (-)-gossypol (AT-101) and ABT-737 [42]. Analogously, 3a sensitized apoptosis-resistant and chemo-resistant breast cancer cells to drug treatment [43]. In doxorubicin-resistant BT-549rDOX cells, compound 3a suppressed Mcl-1 and showed synergistic antiproliferative effects in combination with doxorubicin. In addition to compound 3a, further close Hsp70-inhibitory analogs such as compound 3b (Figure 1, JG-98) with thiazolium moieties were described, which were discovered during the search for optimized Hsp70 inhibitors derived from compound 3a and the mortalin (mitochondrial Hsp70) inhibitor MKT-077 (Figure 1, compound 3c, see below). Compound 3b exhibited prolonged microsomal half-lives and at least 3-fold higher antiproliferative activities against MDA-MB-231 triple-negative breast cancer (TNBC; EC50 = 0.4 µM) and hormone-sensitive MCF-7 breast cancer cells (EC50 = 0.7 µM) than compounds 3a (EC50 = 2.0 for MDA-MB-231 and 5.2 for MCF-7 cells) and 3c (EC50 = 1.4 for MDA-MB-231 and 2.2 for MCF-7 cells) [44]. Compound 3b induced apoptosis in MDA-MB-231 cells by caspase activation and enhanced p62 oligomerization as a hint at forming autophagosomes. Compound 3b also inhibited the Hsp70-Bag3 interaction, which was accompanied by FoxM1 suppression and upregulation of p21 and p27 in treated MCF-7 cells [45]. Doses of 3 mg/kg (every second day for six days) of compound 3b inhibited the growth of MCF-7 breast carcinoma xenografts in vivo. In addition to breast cancer cell lines MCF-7 and MDA-MB-231, further cancer cell lines sensitive to compound 3b were identified (with EC50 < 1 µM), such as HeLa, A375, HT-29, SKOV-3, Jurkat, MM1.R, INA6, RPMI-8226, JJN-3, and U266. Mechanistically, compound 3b exerted its antiproliferative activities against breast cancers both in Bag3-dependent (via ERK activation) and Bag3-independent ways (via suppressed Akt and c-Myc) [46]. Combinations with the proteasome inhibitors MG132 and bortezomib enhanced the antitumor activity of compound 3b in breast cancer models both in vitro and in vivo (5 mg/kg of compound 3b plus 1 mg/kg of bortezomib in MDA-MB-231 TNBC xenografts). Synergy effects in breast cancer cells were also observed in combination with α-amanitin (RNA-polymerase II inhibitor), LY294002 (Akt inhibitor), and sunitinib (RTK inhibitor). In addition, the infiltration of tumor bodies by tumor-associated macrophages (TAMs) was inhibited by compound 3b based on the inactivation of tumor stromal cell Hsp70 proteins [47]. Similar to compound 1, compound 3b also sensitized lung cancer cells to treatment with manumycin A [31]. In a recent effort, new rhodacyanine analogs of compound 3b with benzo-fused N-heterocycle moieties were described, with benzothiazolium compound 3d and quinolinium compound 3e (Figure 1) as the most promising compounds exhibiting high antiproliferative activity against TNBC cells (IC50 = 0.24 µM for 3d and 0.37 µM for 3e in MDA-MB-231 cells), accompanied by a considerable selectivity since non-malignant MCF-10A cells with low levels of Hsp70 were much less sensitive to treatment with 3d [48]. Both compounds are very stable, with half-lives of more than 2 h in microsomes. Apoptosis upon caspase-activation was induced by compounds 3d and 3e in MDA-MB-468 breast cancer cells, while both compounds led to autophagy both in MDA-MB-231 and MDA-MB-468 cells. In addition to FoxM1, survivin, HuR, and Akt suppression, compounds 3d and 3e also degraded KRAS in MDA-MB-231 and MDA-MB-468 cells. Further 3b/JG-98-derived benzothiazole rhodacyanines were described, culminating in the discovery of the bromothienyl analog 3f (JG-231), which showed high activity against MCF-7 (IC50 = 0.12 µM) and MDA-MB-231 breast cancer cells (IC50 = 0.25 µM), disruption of Bag3 interaction, amenable microsomal stability (half-life of more than 60 min), degradation of Akt and HuR in MCF-7 xenografts, amenable in vivo pharmacokinetics parameters and in vivo tumor growth inhibition of MDA-MB-231 xenografts at doses of 4 mg/kg (i.p.) [49].
The cationic spasmolytic drug pinaverium bromide (Figure 1, 4) was repurposed as an inhibitor of constitutively activated Hsc70. Compound 4 inhibited cell proliferation (IC50 of ca. 10 µM) of A2058 melanoma cells and induced apoptosis in these cells [50]. Its binding site was located at the NBD and linker domains of Hsc70.
S1g-2 (Figure 1, 5a) was identified as an inhibitor of Hsp70-Bim interaction (IC50 = 0.4 µM) in CML cells by screening a Bcl-2 inhibitor library [51]. Its allosteric Hsp70-binding site is near the binding site of the rhodacyanines 3. Compound 5a selectively induces apoptosis in CML cells by suppressing oncoprotein clients such as Akt, Raf-1, eIF4E, and RPS16. Hsp70-Bim interaction protected BCR-ABL-independent TKI-resistant CML cells from apoptosis, and, thus, treatment with compound 5a can overcome the resistance of a highly problematic CML type. The close analog compound 5b (Figure 1, S1g-6), which has a morpholino side chain replacing the unstable ester side chain of 5a, was recently described as a new sub-micromolar Hsp70-Bim interaction inhibitor [52]. Compound 5b also induced apoptosis in cancer cells and suppressed Akt and Raf-1.
The benzimidazole derivative 6 (Figure 1, HS-72) selectively inhibits inducible Hsp70 (Hsp70i), which is in stark contrast to its low affinity for the closely related constitutively activated Hsc70 [53]. At doses of 25 and 50 µM, compound 6 inhibited the proliferation of BT474, MCF-7, and SkBr3 breast cancer cells, and the proteins Her2 and Akt were degraded by compound 6 in BT474 and MCF-7 breast cancer cells. In the Her2-overexpressing MMTV-neu spontaneous breast tumor mouse model, compound 6 (20 mg/kg biweekly for 21 days, i.p.) was well tolerated and inhibited breast tumor growth, leading to prolonged survival of treated mice.
The 2,5′-thiodipyrimidine 7 (Figure 1, YK-5) was identified as an irreversible inhibitor of a new allosteric site of the Hsp70 NBD domain and binds covalently to a cysteine residue of the binding site via its reactive acrylamide moiety [54]. It is highly active against Kasumi-1 AML cells (IC50 = 0.9 µM) and SkBr3 breast cancer cells (IC50 = 0.8 µM) and a potent apoptosis inducer by caspase-3/7 activation (IC50 = 1.2 µM) in MOLM13 AML cells. 7 degraded Her2 and Raf-1 in SkBr3 breast cancer cells as a consequence of Hsp70 inhibition.
The dye methylene blue (Figure 1, 8) showed manifold biological activities, and, thus, it was also identified as an inhibitor of the Hsp70 ATPase, leading to a rapid suppression of Tau protein in neurodegenerative cell models [55]. In HeLa cervix carcinoma cells expressing poly-glutaminylated AR (AR112Q), compound 8 inhibited the Hsp70-mediated degradation of AR112Q [56]. In A375 and G361 metastatic melanoma cells, compound 8 suppressed the heat shock response (downregulation of Hsp27, Hsp70, and Hsc70), induced ROS formation, and caused glutathione depletion at a concentration of 10 µM [57]. Geldanamycin is an Hsp90 inhibitor, which increases Hsp70 expression, but compound 8 (10 µM) was able to suppress geldanamycin-induced Hsp70 expression in A375 melanoma cells. Hence, 10 µM of compound 8 also sensitized A375 melanoma cells to geldanamycin treatment, but it also sensitized these cells to treatment with etoposide and doxorubicin. Compound 8 possessed antiproliferative properties against A549 NSCLC cells and induced early apoptosis, yet it enhanced the degradation of N-terminal AR fragments as well as autophagy [58]. In mice, compound 8 inhibited benzo[a]pyrene induced lung carcinogenesis and suppressed Hsp70 as well as the tumor biomarkers ADA and LDH.
The structures of competitive and allosteric NBD binders are shown in Figure 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Effects of NBF-targeting Hsp70 inhibitors on cancers.
| Compound | Cancer Model(s) | Effects |
|---|---|---|
| 1 (VER-155008) | HCT-116 colon carcinoma, A549 NSCLC, pleural mesothelioma, LNCaP95 prostate carcinoma, anaplastic thyroid carcinoma, glioma (1321N1, GOS-3, U87-MG), muscle-invasive bladder cancer, osteosarcoma, multiple myeloma, acute myeloid leukemia | Selective Hsp70/Hsc70 inhibition, antiproliferative, suppression of Her2 and Raf-1, G1 cell cycle arrest, sensitization to 17-AAG and radiation, suppression of Akt and phospho-Akt, macroautophagy induction, suppression of AR-FL and ARV7, apoptosis and paraptosis induction, upregulation of miR-215 and miR-194-5p, degradation of KDMA6 and EP300, degradation of BAG1, upregulation of CHOP, suppression of VEGF release by leukemia cells, synergy effects with drugs (manumycin A, bortezomib, shikonin, 17-DMAG) and PDT [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35] |
| 2a (Apoptozole) | SK-OV-3 ovarian carcinoma, HCT-15 colon carcinoma, A549 NSCLC | Suppression of Hsp70-APAF-1, antiproliferative, pro-apoptotic [37] |
| 2b, 2c | HeLa cervix carcinoma, MDA-MB-231 breast carcinoma, HepG2 hepatoma, A549 NSCLC, RKO colon carcinoma, leukemia | Suppression of Hsp70-APAF-1, antiproliferative, sensitization to doxorubicin, in vivo tumor growth inhibition of A549, RKO, and HeLa xenografts, apoptosis induction, autophagy inhibition, cathepsin release [38,39,40] |
| 3a (YM-1) | MCF-7 breast carcinoma, HeLa cervix carcinoma, U251 and U343 glioma, doxorubicin-resistant BT-549rDOX breast carcinoma | Inhibition of Hsp70-Bag3, suppression of FoxM1 and HIF1α pathways, in vivo inhibition of MCF-7 tumor growth, induction of p21, suppression of FoxM1 and surviving, sensitization of glioma to (−)-gossypol (AT-101) and ABT-737, suppression of Mcl-1, synergistic antiproliferative effects with doxorubicin [41,42,43] |
| 3b (JG-98) | Triple-negative MDA-MB-231 and hormone sensitive MCF-7 breast cancer, lung cancer, miscellaneous | Inhibition of Hsp70-Bag3, FoxM1 suppression, upregulation of p21 and p27, sensitization of breast cancer to bortezomib in vivo, inhibition of TAM infiltration, sensitization of lung cancer to manumycin A [31,44,45,46,47] |
| 3d, 3e | Triple-negative breast cancer (e.g., MDA-MB-231, MDA-MB-468) | Stable, antiproliferative, tumor-selective, induction of apoptosis and autophagy, degradation of KRAS, suppression of FoxM1, survivin, HuR, and Akt [48] |
| 3f (JG-231) | MCF-7 and MDA-MB-231 breast cancer | Stable, antiproliferative, inhibition of Hsp70-Bag3, degradation of Akt and HuR, tumor growth inhibition in vivo [49] |
| 4 (Pinaverium bromide) | A2058 melanoma | Apoptosis induction [50] |
| 5a (S1g-2) | CML | Inhibition of Hsp70-Bim, apoptosis induction, suppression of Akt, Raf-1, eIF4E and RPS16 [51] |
| 5b (S1g-6) | Miscellaneous | Inhibition of Hsp70-Bim, apoptosis induction, degradation of Akt and Raf-1 [52] |
| 6 (HS-72) | BT474, MCF-7 and SkBr3 breast carcinoma, Her2-overexpressing MMTV-neu spontaneous breast tumor mouse model | Selective Hsp70i inhibition, antiproliferative, Her2 and Akt degradation, tumor growth inhibition and prolonged survival in vivo [53] |
| 7 (YK-5) | Kasumi-1 AML, SkBr3 breast carcinoma, MOLM13 AML | Antiproliferative, apoptosis induction, Her2 and Raf-1 degradation [54] |
| 8 (Methylene Blue) | AR112Q-expressing HeLa cervix carcinoma, A375 and G361 melanoma, A549 NSCLC | Heat shock response suppression, ROS formation, glutathione depletion, suppressed geldanamycin-induced Hsp70, sensitization of cancer cells to geldanamycin, etoposide and doxorubicin, apoptosis induction, inhibition of lung carcinogenesis in vivo [56,57,58] |
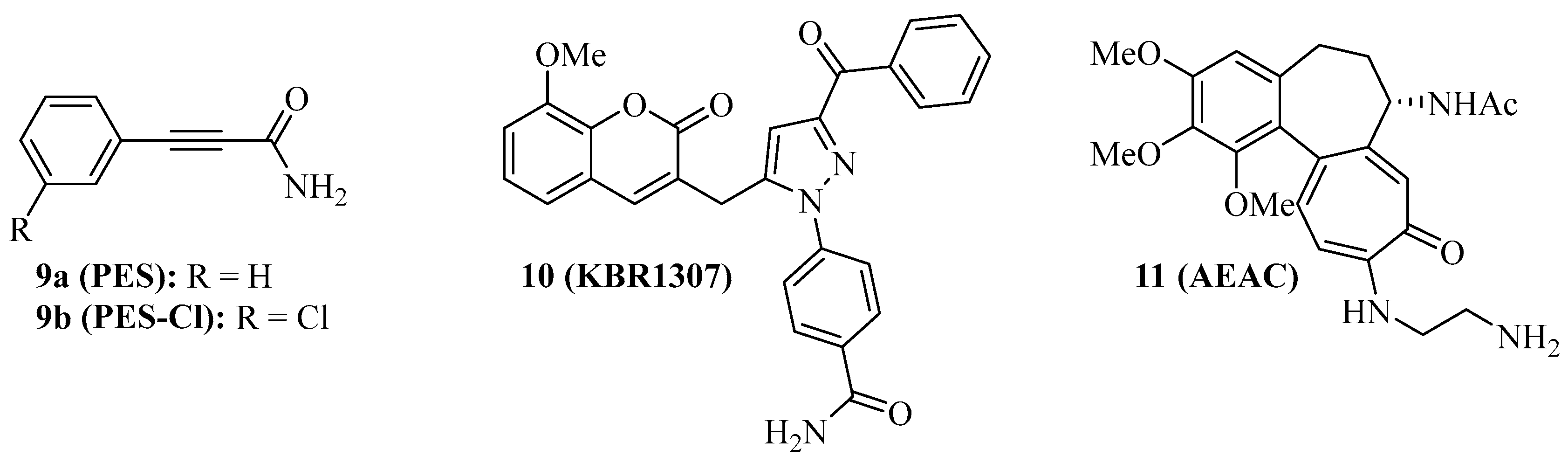
Some Hsp70 inhibitors with SBD-targeting properties were described (Figure 2). Compound 9a (Figure 2, 2-phenylethynesulfonamide, PES, pifithrin-µ) is a prominent example, which was thoroughly studied for its Hsp70-related effects in various cancers. Compound 9a showed antiproliferative activities against various osteosarcoma, breast, and pancreatic carcinoma cell lines at IC50 values of 5–10 µM independent of the p53-state of the tumor cells, induced cell death independent of caspase activation, and led to dysfunctional autophagy by the formation of p62-oligomers/aggregates [59]. It decreased the interaction of Hsp70 with APAF1, p53, and the co-chaperones Hsp40, CHIP, and BAG-1M and suppressed NF-κB signaling and activity. In vivo, compound 9a (40 mg/kg, i.p., every five days for 30 days) blocked Myc-based lymphopathogenesis and led to prolonged survival in Eµ-Myc transgenic mice. Compound 9a exhibited considerable antiproliferative activity against acute leukemia (AML and ALL) cells (IC50 = 2.5–12.7 µM), induced apoptosis in these cells by caspase activation, and led to a degradation of Akt and ERK1/2 [60]. In addition, compound 9a sensitized acute leukemia cells to treatment with cytarabine (an antimetabolite), 17-AAG (a Hsp90 inhibitor), vorinostat (a HDAC inhibitor), and sorafenib (a RTK inhibitor). In primary effusion lymphoma (PEL), compound 9a exerted cytotoxic effects on BC3 and BCBL1 cells by apoptosis and another cell death mechanism, which was associated with immunogenic activity such as activation of dendritic cells [61]. Compound 9a increased lysosome permeabilization and cathepsin D release in PEL cells, accompanied by Bid cleavage, outer mitochondrial depolarization, and AIF translocation to the nucleus. Moreover, compound 9a sensitized PEL cells to bortezomib treatment. The combination of compound 9a with DNA-targeting platinum complexes such as cisplatin and oxaliplatin also revealed synergy effects in HT-29 colon carcinoma and PC-3 prostate carcinoma cells [62]. In prostate cancer cells, compound 9a also increased the antitumor effects of hyperthermia (HT, 43 °C), best when given immediately before HT started, which was accompanied by upregulation of p21 and suppression of c-Myc and cyclin D1 [63]. The combination of compound 9a (100 µg in 50 µL) and HT (43 °C for 1 h, twice on days 0 and 4), led to significant PC-3 prostate carcinoma xenograft growth inhibition. Its modified analog compound 9b (Figure 2, PES-Cl) was antiproliferative against a panel of BRAF-V600E mutant melanoma (IC50 values between 2–5 µM, while inactive against melanocytes) and showed higher antiproliferative activity than compound 9a against SkBr3 breast carcinoma, FaDu head and neck squamous cell carcinoma, and H1299 lung adenocarcinoma cells [64]. The cytotoxic activity of compound 9b is based on apoptosis induction (caspase activation) and inhibition of autophagy (p62 accumulation), while the HeLa cell cycle was arrested in the G2-M phase by compounds 9a and 9b associated with cyclin B1 degradation. At doses of 20 mg/kg (i.p., once per week), compound 9b led to a much higher survival rate of Eµ-Myc mice (71.4% survival) than compound 9a (35% survival) after 210 days. Compound 9b also induced apoptosis in A375 melanoma cells, accompanied by Her2 degradation in these cells [65]. In contrast to compounds 1 and 3c, only compound 9b led to G2-M arrest in H1299 and A375 cells based on cyclin B1 degradation.
Compound 9a does not interact with Hsp70 when no nucleotide is bound to the protein. The coumarin-pyrazole hybrid, compound 10 (Figure 2, KBR1307) was designed, which binds Hsp70 in the presence and absence of nucleotides [66]. Both compounds 9a and 10 reduce the activity of Hsp70 ATPase significantly, but compound 10 was more active against MCF-7 cells than compound 9a. Similar coumarin-thiazole hybrids were described by the same group before as binders to the C-terminus of Hsp70 with activity against DLD-1 colon carcinoma and HepG2 hepatoma cells [67].
The screening of the InterBioScreen compound library for Hsp70 inhibitors revealed that the semi-synthetic colchicine derivative, compound 11 (Figure 2, N-aminoethylaminocolchicine, AEAC), interferes with substrate binding and refolding functions of Hsp70, based on a nanomolar affinity for Hsp70 (KD = 149 nM) [68]. Although the antiproliferative and cytotoxic activities of compound 11 are low, it sensitized C6 rat glioblastoma and B16 mouse melanoma cells to doxorubicin treatment. The combination of compound 11 (2 mg/kg) and doxorubicin (1 mg/kg) inhibited in vivo B16 tumor growth by 71% and increased the lifespan of treated mice by ca. 15 days when compared with untreated mice.
2.2. Grp78 Inhibitors
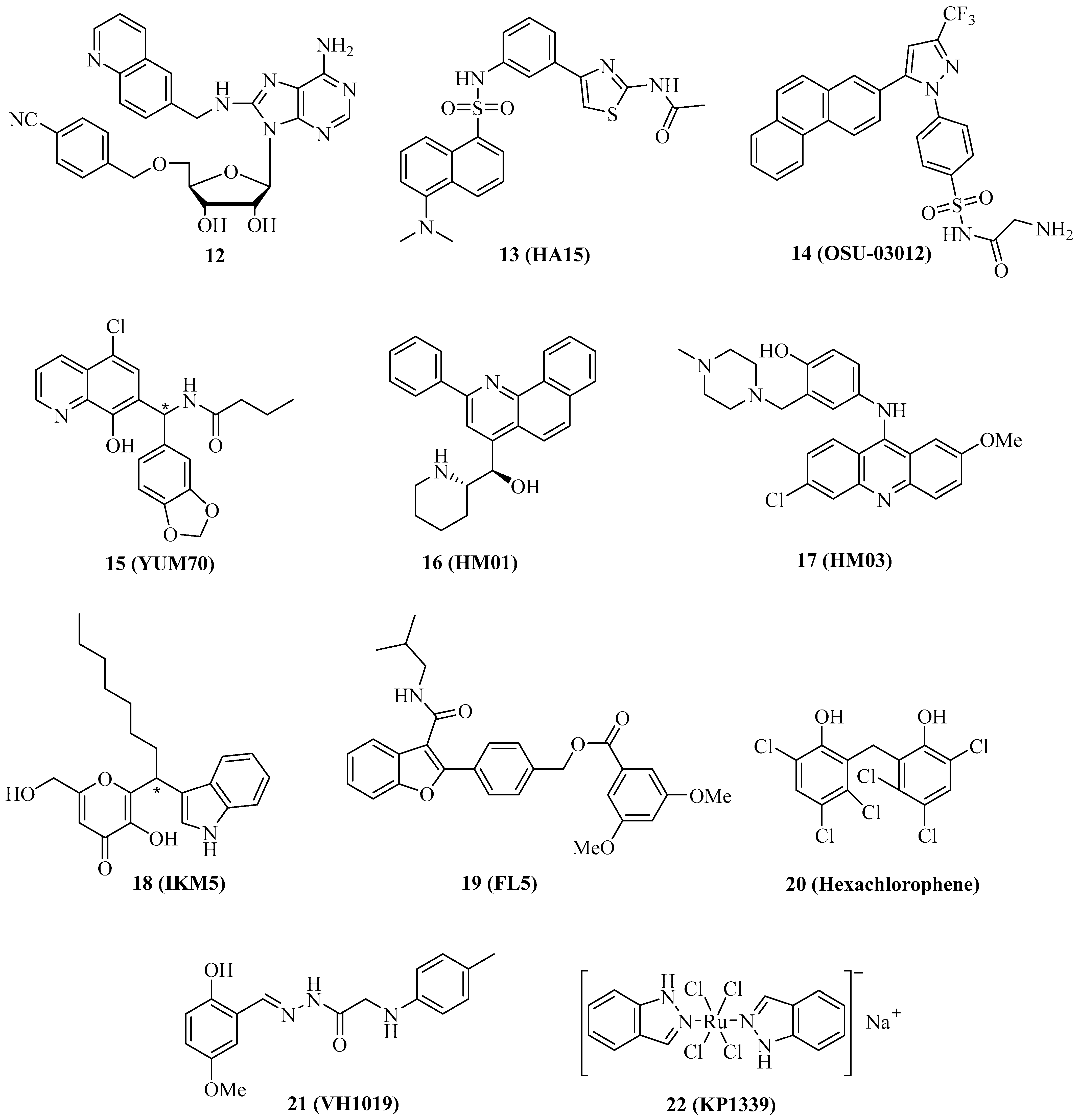
The specific targeting of organelle-specific Hsp70 isoform proteins such as Grp78 (ER) and mortalin (mitochondria) has become a valuable strategy to combat cancer. Grp78 is the “master protein” of UPR (unfolded protein response) and redirects misfolded polypeptides for degradation or refolding [14]. Some of the already described Hsp70 inhibitors also inhibit Grp78. For instance, compound 1 is likewise a Grp78 inhibitor. In OSA cells, compound 1 inhibited Grp78 in addition to Hsp70, followed by antiproliferative and proapoptotic effects [29]. In MCF-7 and MDA-MB-231 breast cancer cells, Grp78 was upregulated upon tamoxifen treatment, leading to resistance; however, Grp78-inhibitory 1 enhanced apoptosis induction by tamoxifen in these cells, accompanied by suppression of tamoxifen-induced phosphor-GSK-3β, which is a downstream factor of Akt signaling [69]. The quinoline analog 12 (Figure 3) of compound 1 was slightly more active against and selective for Grp78 (KD = 0.6 µM for Grp78, 0.3 µM for Hsp70) than compound 1 (KD = 0.8 µM for Grp78, 0.1 µM for Hsp70), however, compound 12 showed no antiproliferative activity against HCT-116 colon cancer cells in contrast to compound 1 (GI50 = 5.0 µM) [70]. The absence of antiproliferative activity in compound 12 was explained by the detrimental physicochemical properties of this compound, which need to be improved in order to obtain a valuable adenosine-based Grp78-selective inhibitor in the future.
The thiazole benzenesulfonamide 13 (Figure 3, HA15) was identified as a Grp78 inhibitor that enhanced ER stress associated with autophagy and apoptosis, leading to cell death in melanoma cells, in particular in BRAF-mutant cells, at a concentration of 10 µM [71]. In vivo, compound 13 (0.7 mg/mouse/day for 2 weeks, i.p.) inhibited A375 melanoma growth without causing side effects such as mouse weight loss and liver damage. Grp78 was found to be upregulated in lung cancers, and treatment with compound 13 led to antiproliferative effects on A549 lung cancer cells as well as to apoptosis induction, autophagy, and increased ER stress [72]. The GRp78 inhibitor 13 suppressed KRAS expression and revealed antiproliferative and pro-apoptotic activities in various KRAS-mutant cancer cell lines (A427 lung adenocarcinoma, H460 non-small cell lung carcinoma, HCT-116 and LS180 colon carcinomas, PANC-1 and CFPAC-1 pancreatic ductal adenocarcinomas), and induced apoptosis in A427, HCT-116, and PANC-1 cells by caspase activation [73]. Compound 13 also suppressed cell proliferation and steroidogenesis in adrenocortical carcinoma (ACC) cells, and showed synergy effects in combination with the approved drug mitotane, which is also an activator of ER stress [74].
In carboplatin-resistant canine osteosarcoma cells (HMPOS-2.5R and HMPOS-10R), compound 13 and the atypical Grp78 inhibitor 14 (Figure 3, the celecoxib derivative OSU-03012, which binds directly to the Grp78 ATPase domain) still showed considerable activity (EC50 = 1.9–3.5 µM for 13 and 5.3–8.5 µM for 14), while the Hsp70 inhibitor 1 was distinctly less active against these resistant cells (EC50 = 25–30 µM) than against the parent HMPOS cells (EC50 = 1.8 µM) [75,76]. Compound 14 reduced the expression of Bag2, and treatment with compound 14, especially in combination with the PDE5 inhibitor sildenafil, formed toxic autophagosomes that inducing cell death in GBM5 and GBM12 glioblastoma cells [77]. Similar effects were observed for the multi-kinase inhibitor sorafenib, which is an approved anticancer drug and also able to bind to the N-terminal domain of Grp78.
Analogously to compound 13, the hydroxyquinoline derivative 15 (Figure 3, YUM70) was able to downregulate KRAS, leading to antiproliferative and pro-apoptotic effects on KRAS-mutant cancer cells [73]. Initially, compound 15 was identified as an antiproliferative and caspase-dependent pro-apoptotic compound in pancreas cancer cells (IC50 = 2.8 µM for MiaPaCa-2, 4.5 µM for PANC-1, and 9.6 µM for BxPC-3 cells) based on Grp78 inhibition (by binding to the SBD) followed by ER stress via eIF2α phosphorylation as well as AT4 and CHOP activation [78]. Synergy effects of compound 15 on MiaPaCa-2 cells were observed in combination with vorinostat or topotecan. In the MiaPaCa-2 xenograft model, compound 15 (30 mg/kg, 5 days a week for 7 weeks, i.p.) inhibited tumor growth and caused weight loss in treated mice.
The N-heteroaromatic compounds 16 (Figure 3, HM01) and 17 (Figure 3, HM03) were identified as Grp78 inhibitor hits from the virtual screening of an NCI diversity set, and they showed considerable affinity to the substrate-binding channel of Grp78 [79]. Both compounds exerted moderate antiproliferative activity against HCT-116 colon cancer cells (IC50 between 10 and 25 µM) but may serve as lead compounds for the design of more potent Grp78 inhibitors.
The indolylkojyl derivative 18 (Figure 3, IKM5) was prepared by a simple three-component reaction and identified as a potent Grp78 inhibitor (Ki = 1.4 µM), which showed high antiproliferative activities against a panel of breast cancer cell lines (IC50 = 0.15 µM for MCF-7, 0.21 µM for MDA-MB-231, 0.54 µM for MDA-MB-468, and 3.5 µM for BT474 cells) [80]. Compound 18 induced TIMP-1 by blocking its interaction with Grp78, suppressed EMT markers such as MMP-2, Twist1, and vimentin, and upregulated the Par-4 tumor suppressor, which controls NF-κB signaling. Compound 18 increased the activity of doxorubicin against invasive breast cancer cells. In vivo, compound 18 (30 mg/kg, b.w.) inhibited 4T1 breast tumor growth by 79.2% and lung metastasis formation by 84.5%.
The benzofuran derivative 19 (Figure 3, FL5) is a strong binder and stabilizer of Grp78 (Tm increase > 2 °C), which was associated with its anticancer and antiangiogenic activities against renal cell carcinoma (RCC) cells (10 µM of 19 led to 50% cell death) and HUVECs (EC50 = 1.5 µM), while it was inactive against mouse fibroblasts [81]. Docking studies showed that compound 19 does not interfere with the ATPase activity of Grp78.
A high-throughput substrate binding assay led to the identification of the disinfectant hexachlorophene (Figure 3, 20) as a Grp78 inhibitor binding to the SBD [82]. Compound 20 was cytotoxic against HCT-116 colon carcinoma cells (CC50 = 3.4 µM) and induced apoptosis and autophagy, as well as an unfolded protein response associated with upregulated ATF4, XBP1s, and CHOP.
In silico methods for the design of new Grp78 inhibitors led to the identification of 21 (Figure 3, VH1019) with antiproliferative activity against MCF-7 breast cancer cells (IC50 = 12.7 µM) [83]. It is noteworthy that compound 21 mimicked ATP in its binding to Grp78.
In addition to organic compounds, metal complexes can be a valuable source of Grp78 inhibitors. The anionic ruthenium complex 22 (Figure 3, KP1339, BOLD-100) is an efficient Grp78 suppressor that has already demonstrated promising anticancer activity in phase 1 clinical trials, both as compound 22 and in its benzindazolium salt form, KP1019 [84]. A phase 1b/2 clinical trial with the metallodrug compound 22 for patients suffering from gastrointestinal cancer is ongoing (NCT04421820). Mechanistic studies revealed apoptosis induction in sensitive cancer cell lines by activation of caspase-8, which was associated with disruption of the ER upon suppression of key chaperones, leading to the degradation of vital proteins [85]. In contrast, cancer cells with a low response to complex compound 22-induced G2 cell cycle arrest may serve as a general hint at cancers resistant to Grp78 inhibitors. Interactions with the ribosomal proteins RPL10 and RPL24 and with the transcription factor GTF2I were identified in HCT-116 colon carcinoma cells and associated with ribosomal disturbance and ER stress induction [86]. Grp78 is upregulated in asbestos-associated pleural mesothelioma, and, thus, the activity of 22 against this cancer was evaluated. Complex compound 22 was cytotoxic against mesothelioma cells (EC50 = 71 µM for REN and 90 µM for MM98 cells), inhibited REN cell colony formation at a concentration of 100 µM, induced apoptosis in REN cells by activation of caspase-3/7 and caspase-8, increased ROS formation and cytosolic Ca2+ levels, and suppressed Grp78 expression in REN cells [87]. CHOP and XPB1 expression were upregulated by compound 22 in REN cells. Sensitive HCT-116 colon carcinoma (IC50 = 76 µM) and Capan1 pancreatic carcinoma cells (IC50 = 40 µM) were used to study cell-based resistance to treatment with compound 22 [88]. Increased glucose uptake and upregulated glycolysis were observed in sensitive cancer cells upon treatment, but especially in the resistant cell lines HCTR and CapanR obtained from HCT-116 and Capan1 cells, respectively, upon exposure to compound 22. However, this specific mechanism made the resistant tumor cells highly vulnerable to the treatment with the glycolysis inhibitor and ER stress inducer 2-dexyglucose, which led to synergy effects of combinations of compound 22 with 2-deoxyglucose in resistant HCTR cells.
2.3. Mortalin Inhibitors
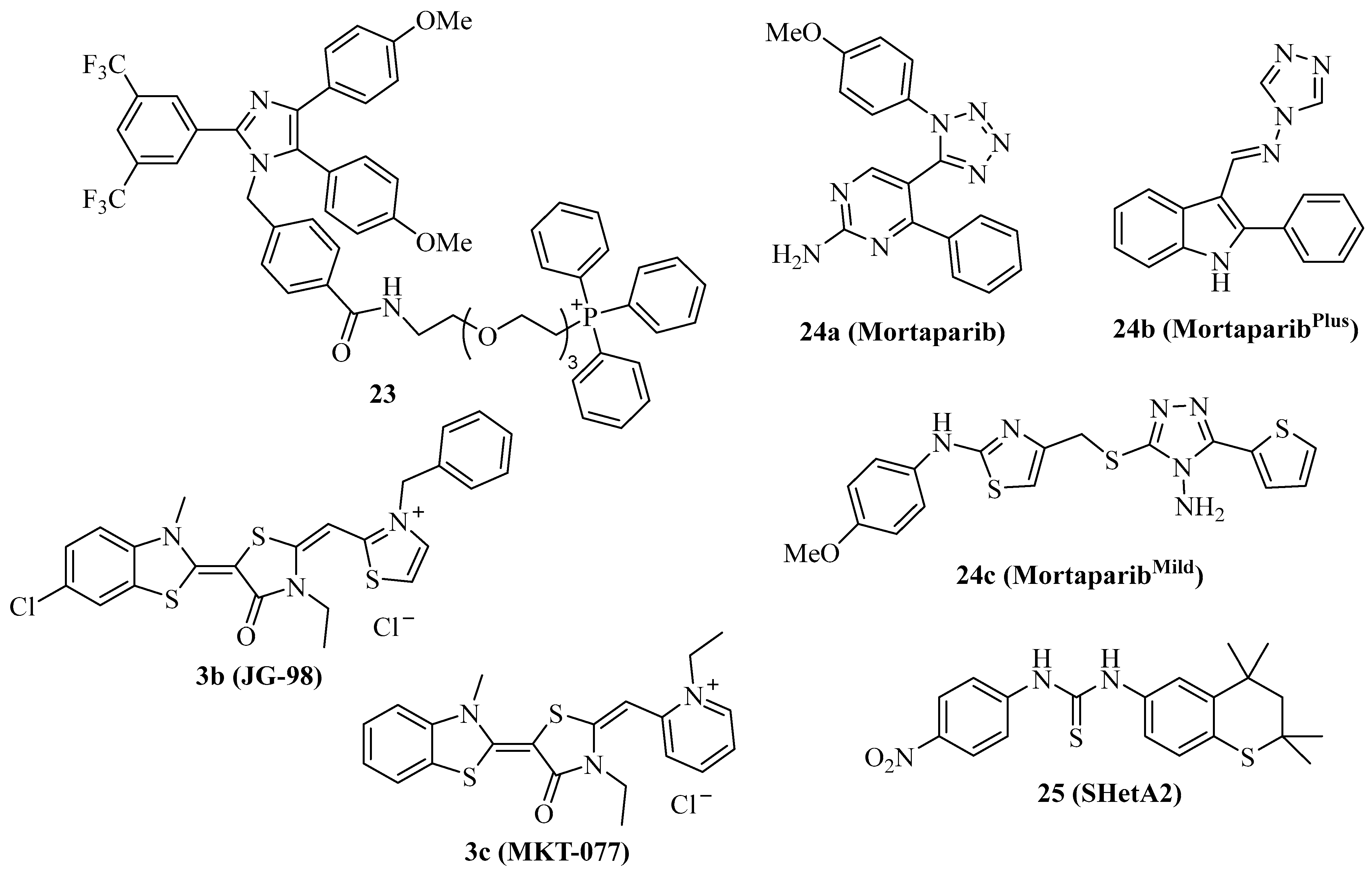
Mortalin is the mitochondrial Hsp70 isoform with an N-terminal mitochondrial localization motif and has become an important target for cancer therapy [15]. Due to its preferred localization in mitochondria, mortalin is involved in the regulation of mitochondrial metabolism and of key tumor factors such as p53, PI3K/AKT, Raf/MEK/ERK, and JAK/STAT pathways [15,89]. The triphenylphosphonium (TPP) moiety was described as a mitochondria-targeting device, and, thus, compound 2b was conjugated with TPP in order to obtain the mortalin-targeting apoptozole conjugate compound 23 (Figure 4) [40]. Compound 23 showed higher antiproliferative activity against a panel of 20 cancer cell lines (IC50 = 0.5–1.5 µM) than compound 2b and induced caspase-dependent apoptosis in HeLa cells by blocking the interaction of p53 with mortalin in the mitochondria, followed by Bak-mediated mitochondrial outer membrane permeabilization.
Cationic Hsp70 inhibitors such as compound 3b (Figure 4, JG-98) were identified as mortalin inhibitors with pronounced activity against proteasome inhibitor-resistant MM cells associated with 55S mitoribosome degradation [90]. Compound 3c (Figure 4, MKT-077) overlaps with the p53-binding region of mortalin, releasing active p53 followed by upregulation of p21 in treated cancer cells [91,92]. In addition, compound 3c sensitized K562 leukemia cells to complement-mediated lysis by inhibition of mortalin and its interaction with the C9 complement protein [93]. Compound 3c was identified already in the 1990s as a mitochondria-targeting anticancer compound with preference for ras-associated cancers, and entered clinical trials after promising in vivo results [94,95,96,97]. However, complications such as renal toxicity prevented the approval of compound 3c as an anticancer drug [98].
The tetrazole derivative 24a (Figure 4, mortaparib) is a relatively new dual mortalin and PARP1 inhibitor that was developed in 2019 [99]. Compound 24a led to the activation and nuclear accumulation of p53 by inhibiting mortalin. In addition, PARP1 was downregulated by compound 24a, followed by increased double-strand breaks and apoptosis induction in HeLa cervix carcinoma and SKOV-3 ovarian cancer cells. In vivo, compound 24a (20 mg/kg i.p.) was well tolerated, inhibited the tumor growth of SKOV-3 xenografts, and suppressed the formation of metastases in the lungs and kidneys. Based on these promising anticancer activities of compound 24a, the same group developed further mortaparib derivatives. The 1,2,4-triazole 24b (Figure 4, mortaparibPlus) blocked the interaction of p53 with mortalin, leading to p53 activation and suppression of PARP1 and CARF in HCT-116 cells [100]. Compound 24b induced apoptosis and activated p21 in HCT-116 (p53 wildtype) and DLD-1 (p53S241F) colon carcinoma cells in p53-dependent and –independent ways. In contrast to colon cancer cells, in breast cancer cells, compound 24b upregulated only p21 in the p53-wildtype MCF-7 cells, while T47D cells (p53L194F) treated with compound 24b showed no p21 changes but activation of PARP1, albeit compound 24b inhibited the mortalin-p53 interaction in both cell lines [101]. More recently, compound 24c (Figure 4, mortaparibmild) was identified as an inhibitor of PARP1 and mortalin-p53 interaction in HCT-116 cells, however, at higher concentrations than compounds 24a and 24c, thus leading to the attribute “mild” for compound 24c [102].
The thiochromane derivative 25 (Figure 4, SHetA2) is an orally bioavailable mortalin inhibitor that blocks the interaction of mortalin with p53 in ovarian cancer cells [103]. Due to its promising chemo-preventive and selective anticancer activities, including apoptosis induction, compound 25 has entered clinical phase 1 studies for patients with advanced/recurrent cervical, endometrial, or ovarian cancer [15]. Since mutant p53 led to drug resistance upon treatment with compound 25, strategies to overcome this resistance are sought. A promising strategy is the combination of compound 25 with the p53 reactivator PRIMA-1Met, which was studied in a panel of ovarian cancer cell lines [104]. PRIMA-1Met reduced resistance to compound 25 and exhibited synergy effects in combination with compound 25 in p53-mutant and p53-wildtype ovarian cancer cells, accompanied by caspase activation, increased ROS formation, and reduced ATP. The combination of compound 25 (60 mg/kg by gavage every day for 2 weeks, then every second day for 3 weeks) and PRIMA-1Met (10 mg/kg, i.p. every other day) inhibited MESOV tumor growth (tumor free rate of 67%) in an additive way without toxicity to the liver and kidneys.
3. Modulators of Co-Chaperone Hsp40
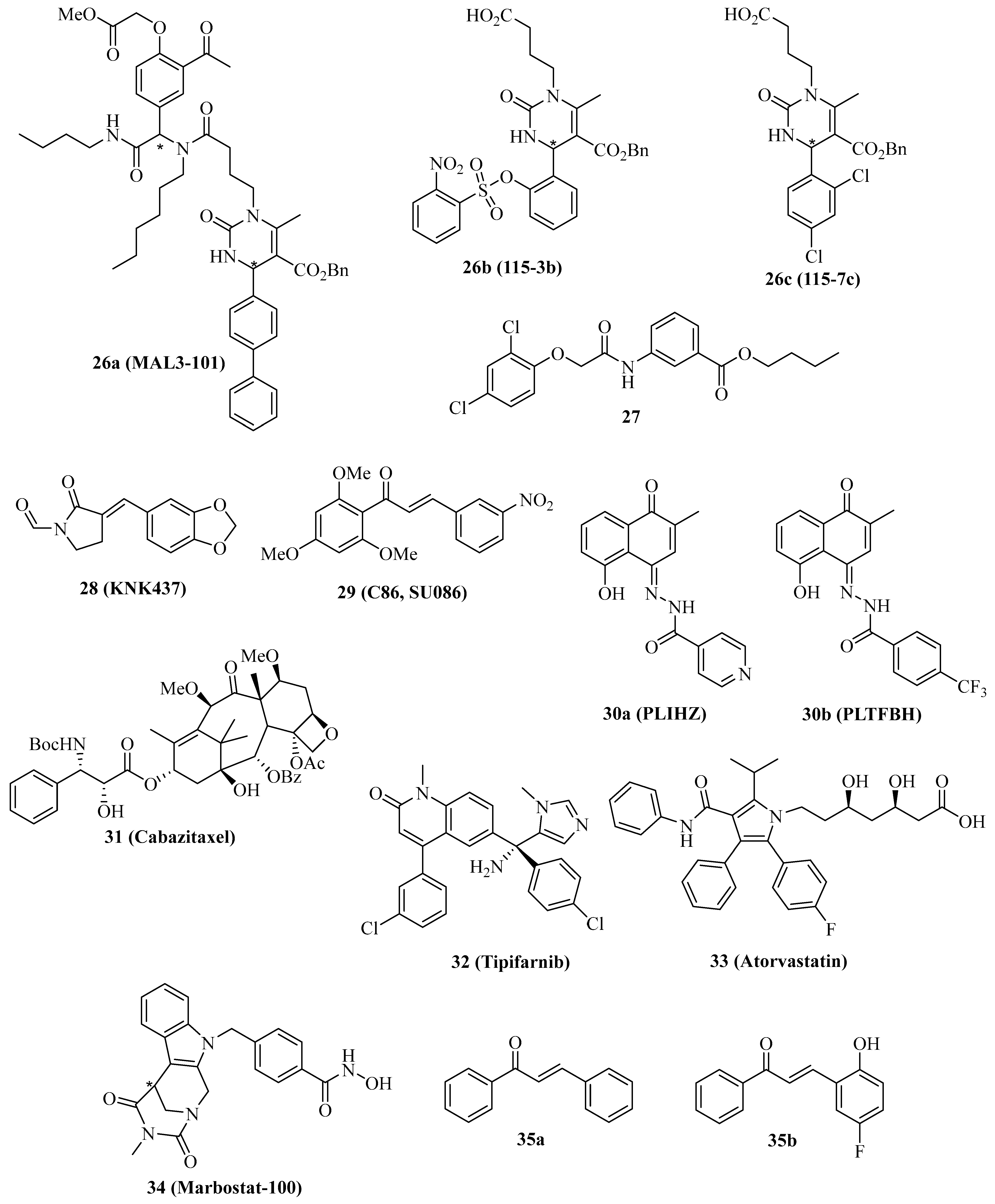
The co-chaperones of the Hsp40 family are vital for the ATPase activity of Hsp70 proteins, and modulation of Hsp40 activity has tremendous effects on the Hsp70 network of protein interaction and integrity. The natural Hsp70 ATPase stimulator 15-deoxyspergualin served as a lead compound for the design of the uracil derivative NSC-630668-R/1, which inhibits endogenous and Hsp40-induced ATPase activity of Hsp70 [105,106]. Further synthetic derivatives were prepared using the straightforward Biginelli multicomponent reaction, leading to the identification of 26a (Figure 5, MAL3-101), which specifically blocks Hsp70 ATPase in an allosteric way by inhibition of the Hsp40 co-chaperone protein TAg [107]. In this way, the strategy to target the Hsp70/Hsp40 axis as a possible treatment for cancer diseases was established. In contrast, the synthetic small-molecule dihydropyrimidine compounds 26b (Figure 5, 115-3b) and 26c (Figure 5, 115-7c), which lack the amide side chain of 26a, stimulated protein folding by the bacterial DnaK (Hsp70) chaperone in the presence of the DnaJ (Hsp40) and GrpE co-chaperones, while compound 26a inhibited protein folding in this assay as expected with an EC50 value of 3.2 µM [108,109].
Compound 26a showed various anticancer properties, e.g., antiproliferative activity against Merkel cell carcinoma (MCC) accompanied by apoptosis induction [110]. In addition, compound 26a (40 mg/kg i.p. every other day) inhibited WaGa MCC growth in mice, again associated with apoptosis induction in vivo. Moreover, compound 26a (10 µM) showed antiproliferative activity against T24 and SW780 MIBC cells, and the combination of compound 26a with the Hsp70 inhibitor 1 or with the Hsp90 inhibitor STA-9090 led to synergistic antiproliferative activity against MIBC cells, which were resistant to compound 26a alone [111]. RMS13 rhabdomyosarcoma cells were also sensitive to treatment with 26a based on the induction of UPR and apoptosis; however, dose-escalation led to the isolation of a resistant RMS13-R cell line with upregulated autophagy and an ER-associated degradation pathway [112]. Inhibition of autophagy restored the sensitivity to 26a in the RMS13-R cells.
The phenoxy-N-arylacetamide 27 (Figure 5) was found to inhibit Hsp70 by direct binding to the co-chaperone DnaJ (IC50 = 0.13 µM) [113].
In 2000, the synthetic lactam derivative 28 (Figure 5, KNK437) was identified as a suppressor of heat-induced Hsp70 and Hsp40 in COLO 320DM human colon cancer cells, whose effect was more pronounced than for the known natural Hsp inhibitor quercetin [114]. In vivo, compound 28 (200 mg/kg i.p.) showed synergy effects in combination with heat treatment (44 °C) in mice bearing SCC VII squamous cell carcinomas [115]. At a non-toxic concentration of 300 µM, compound 28 inhibited colony formation of p53-mutant SAS/mp53 human squamous cell carcinoma cells at a temperature of 42 °C [116]. This inhibitory effect of compound 28 in combination with heat was stronger than in the p53-wildtype SAS/neo cells and accompanied by the induction of apoptosis at a reduced concentration (100 µM). Compound 28 (100 µM) also inhibited heat-induced (44 °C) transcription-activating histone H3-Lys4 methylation both in thermoresistant HSC4 and in thermosensitive KB oral squamous cell carcinoma cells, which was associated with Hsp70 suppression by compound 28 [117]. In immortalized Cos-1 cells, 100 µM of compound 28 inhibited mTOR and S6K phosphorylation and suppressed mTORC1 activity, leading to apoptosis induction [118]. Compound 28 (50 µM) sensitized MDA-MB-231 breast cancer cells to ionizing radiation by mechanisms independent from Hsp suppression [119]. Instead, hypoxia-related AKT and HIF-1α survival pathways were inhibited by compound 28, which were interesting off-targets of this compound. The Hsp40 protein DNAJA1 was upregulated in colorectal cancer cells, but treatment with compound 28 strongly inhibited the level of the Hsp40 protein DNAJA1 in SW480 (IC50 = 24.7 µM), RKO (IC50 = 25.5 µM), LOVO (IC50 = 56.0 µM) and SW620 cells (IC50 = 48.3 µM), while no effects on Hsp70 and Hsp90 were found [120]. DNAJA1-overexpressing SW480 and SW620 cells exhibited much faster and stronger tumor growth in vivo, which was inhibited by treatment with compound 28 (20 mg/kg i.p. for 20 days). The combination of 5-FU/L-OHP with compound 28 (20 mg/kg i.p.) efficiently suppressed liver metastasis formation in DNAJA1-overexpressing SW480 colorectal tumors. Mechanistically, compound 28 suppressed CDC45 and upregulated ubiquitin in DNAJA1-overexpressing SW480 cells as a consequence of its strong DNAJA1 downregulating activity.
The chalcone compound 29 (Figure 5, C86/SU086) was initially identified as a synthetic xanthohumol analog with antiproliferative activity against HeLa cervix carcinoma cells (IC50 = 1.4 µM), pro-apoptotic activity by caspase-3 activation, and thioredoxin reductase inhibitory activity (IC50 = 3.5 µM) followed by induction of ROS formation in treated HeLa cells [121]. The effects of compound 29 on castration-resistant prostate cancer (CRPC) were studied in more detail, revealing strong antiproliferative activity against 22Rv1 CRPC cells (IC50 between 1 and 2.5 µM) and a suppression of FL-AR/ARv7 signaling based on enhanced degradation of FL-AR and ARv7 in 22Rv1 CRPC cells treated with compound 29 (10 µM) [122]. Experiments using biotinylated 29 confirmed a direct binding as a pan-Hsp40/DNAJ inhibitor, which interacts with DNAJA, DNAJB, and DNAJC proteins likely via the J domains of the Hsp40 proteins. In addition, compound 29 is bound to Hsp40 in complexes with AR and Arv7 and with Hsp70/Bag3/CHIP. Similar effects were observed for the Hsp70 inhibitor 3b (JG98, IC50 = 0.4–0.5 µM for 22Rv1 cells, destabilization of FL-AR/ARv7 proteins). Both compounds 29 (15 mg/kg, i.v. 3 x per week) and 3f (8 mg/kg, i.p. every other day) inhibited the growth of 22Rv1 CRPC xenografts in mice; however, the combination of both drug candidates surpassed the antitumor activity of the single compounds. More recently, compound 29 was also identified as a Hsp90 inhibitor in prostate cancer cells, which interferes with tumor cell glycolysis [123]. In C4-2 prostate cancer xenografts, compound 29 (50 mg/kg/day i.p.) inhibited tumor growth similar to the approved prostate cancer drugs enzalutamide (10 mg/kg/day p.o.) and abiraterone (200 mg/kg/day p.o.). Yet, the combination of compound 29 with either enzalutamide or abiraterone led to synergistic and additive tumor growth inhibitory effects, respectively.
The semi-synthetic hydrazones 30a (Figure 5, PLIHZ) and 30b (Figure 5, PLTFBH), which were made of the natural naphthoquinone plumbagin, were identified as Hsp40 inhibitors by molecular docking [124]. Compounds 30a and 30b revealed antiproliferative activities against HN31 pharyngeal squamous cell carcinoma cells (IC50 = 1.2 µM for 30a and 0.6 µM for 30b) and suppressed DNAJA1 and conformational mutant p53 levels in HN31 cells, accompanied by suppression of tumor cell migration based on downregulation of active Cdc42 and Rac. No effects were observed for wild-type p53 or DNA-contact mutant p53 proteins, indicating a selective action against conformational mutant p53. In addition to DNAJA1, compound 30b showed distinct inhibitory activities against DNAJA2, DNAJA3, DNAJB1, DNAJB12, and DNAJC3.
The semi-synthetic 3rd generation taxane derivative cabazitaxel (Figure 5, 31) was able to inhibit LNCaP and PC-3 prostate cancer cell proliferation and suppress Hsp40, HOP, and AR in prostate cancer cells at very low doses (0.3 nM) [125].
The inhibition of farnesylation of HDJ-2 Hsp40 proteins has become a reasonable marker for the clinical outcome of anticancer active farnesyl transferase inhibitors [126,127,128]. Farnesyl transferase inhibitors such as tipifarnib (Figure 5, 32) entered clinical trials for various cancer diseases because they inhibit the farnesylation of Ras proteins, leading to their inactivation [129,130]. The radio-sensitizing effect on SF763 glioblastoma cells treated with compound 32 was associated with a suppression of the radio-induced translocation of HDJ-2 by compound 32 based on reduced levels of farnesylated HDJ-2 [131]. In addition, compound 32 inhibited U87 and SF763 glioblastoma cell proliferation (IC50 = 3.1 µM for U87 and 1.9 µM for SF763) and led to G2/M arrest in these cells based on p21 induction. The HMG-CoA reductase inhibitory statin drug 33 (Figure 5, atorvastatin), which is applied for the treatment of cardiovascular disease based on its cholesterol depletion activity, was also studied for its inhibition of DNAJA1 farnesylation in pancreatic cancer cells expressing wild-type or mutant p53 proteins [132]. Compound 33 induced apoptosis, upregulated p21, and degraded mutant p53 (R172H) and cyclin D1 in PO3 cells based on the suppression of farnesylated DNAJA1. In addition, compound 33 suppressed mutant p53 in SU 86.86 (G245S), BXPC-3 (Y220C), and Pan 10.05 (I255N) pancreatic cancer cells, while mutant p53 levels in MIA-PaCa-2 (R248W) and PANC-1 (R273H) were not affected by compound 33. Nuclear translocation of mutant p53 was inhibited in PO-3 cells by compound 33, which also suppressed PO-3 cell migration and invasion.
Selective inhibition of cytoplasmic HDAC6 by the HDAC6 inhibitor 34 (Figure 5, marbostat) led to MYC degradation and apoptosis in MYC-overexpressing B-cell lymphoma cells. It was shown that HDAC6 inhibition by compound 34 led to hyperacetylation of tubulin followed by enhanced binding of DNAJA3 to hyperacetylated tubulin in the cytoplasm of B-cell lymphoma cells, which led to enhanced Myc degradation [133].
4. Discussion
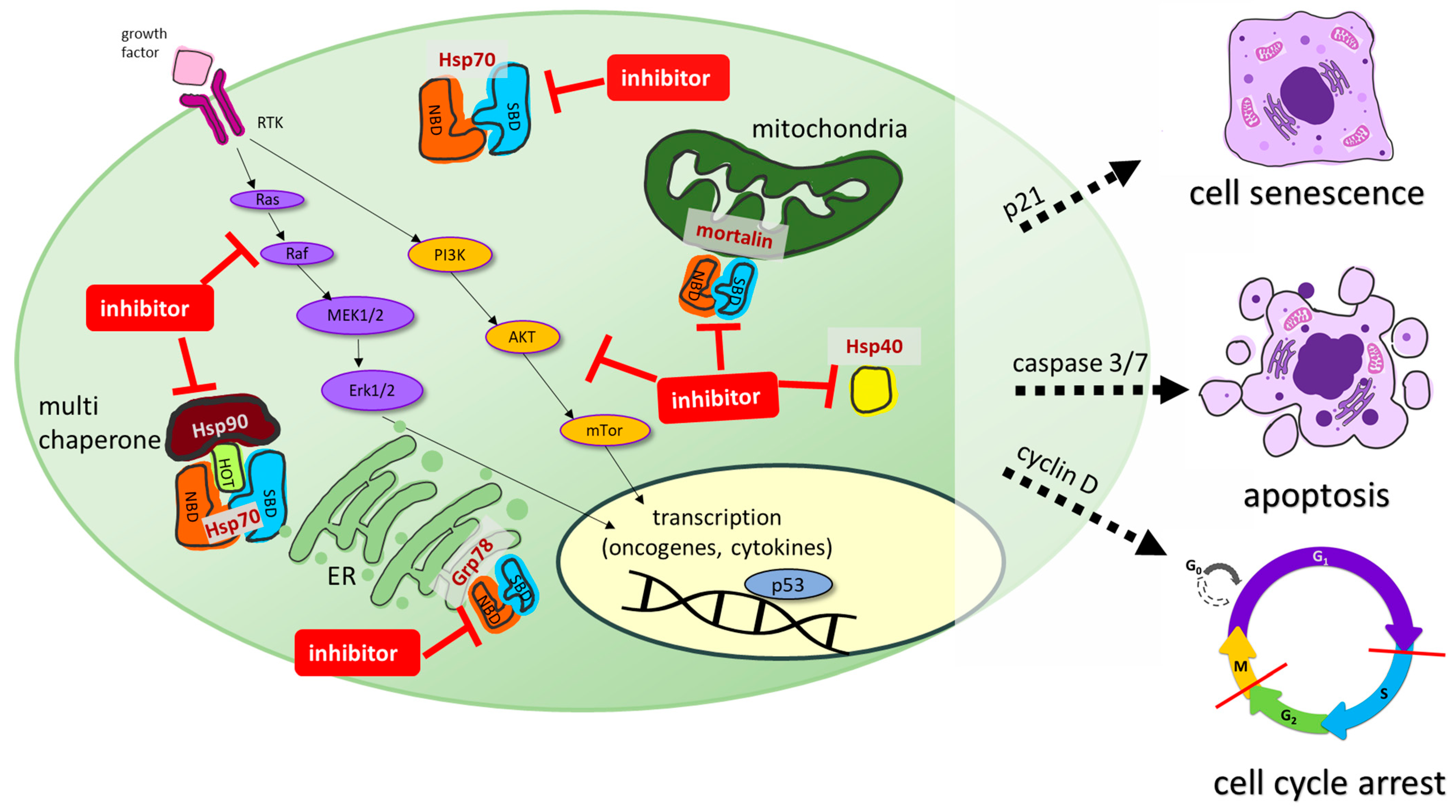
The design and development of inhibitors of the Hsp70-Hsp40 axis is a prospering field of research. Numerous Hsp70 inhibitors were described, and the number of Hsp40 inhibitors and modulators is also growing. Hsp70 is essential for the survival of proliferating cancer cells, where it is often overexpressed, while it is more or less dispensable for non-transformed cells, making it an excellent anticancer drug target for tumor-selective drug candidates. Hsp70 is composed of two distinct domains, a 40 kDa N-terminal nucleotide-binding domain (NBD) that regulates client association and a 25 kDa C-terminal substrate-binding domain (SBD). Hsp70 is found in the cytosol and possesses multiple cellular functions. Together with Hsp90, it acts as a multi chaperone complex to modulate cellularly-protective heat stress responses. Organelle-specific Hsp70 isoforms such as Grp78 in the endoplasmic reticulum or mortalin in the mitochondria were also identified as promising anticancer drug targets for Hsp70 inhibitors [14,15]. Hsp40 is another member of the Hsp family and functions as a client protein of Hsp70. It is a small co-chaperone of Hsp70 that prevents the aggregation of unfolded polypeptides and proteins before transferring them to Hsp70 [17]. Hsp40 also binds to the nucleotide-binding domain of Hsp70, thereby increasing the ATPase activity of Hsp70. Moreover, Hsp70 is also involved in modulating receptor tyrosine kinase (RTK) signaling pathway activity, such as the Ras/Raf-MAPK or the AKT pathway. Synthetic Hsp70 inhibitors can interact with the N-terminal NBD or the C-terminal SBD of Hsp70. Some inhibitors also interfere with Hsp40 or act on organelle-specific Hsp70 isoforms such as Grp78 (ER) or mortalin (mitochondria), leading to pronounced anticancer effects such as apoptosis, cell cycle arrest, or cellular senescence (Figure 6) [14,15].
These effects are often synergistic in combination with well-established Hsp90 inhibitors, and have the potential to overcome the drawbacks of single Hsp90 inhibition in cancer therapy. A conjugate of 17-AAG with apoptozole (2c) designed as a dual Hsp70/Hsp90 inhibitor was superior to Hsp70 inhibitor 2b in terms of anticancer activity [39,40]. Synthetic efforts led to promising compounds with intrinsic dual or multimodal activities, such as dual mortalin/PARP1 inhibition, which are pointing the way to more efficient anticancer drug candidates [99,100,101].
Synthetic conjugate strategies also led to subcellular isoform targeting by attachment of a mitochondria-specific triphenylphosphinium moiety to the apoptozole scaffold in the selective mortalin inhibitor 23 [40]. It is noteworthy that mitochondria-targeting cationic dyes such as rhodacyanines (3a–f) and methylene blue (8) were identified as Hsp70 inhibitors, which were initially developed for other applications than cancer therapy. Methylene blue was the first synthetic drug applied for the treatment of malaria and the lead compound for the development of tricyclic anti-depressant drugs [135]. Meanwhile, several rhodacyanine derivatives have also been investigated as antiprotozoal agents [136]. Thus, the repurposing of drugs appears promising and is not a one-way road. As far as drug repurposing is concerned, the disinfectant hexachlorophene (20) and the spasmolytic drug pinaverium bromide (4) are further interesting examples of Hsp70 inhibitory activity [50,82]. In contrast to the cationic mitochondria-targeting compounds with preference for mortalin binding, the anionic ruthenium complex 22 (KP1339) is an efficient Grp78 suppressor [85,86,87,88]. However, the exact anticancer mechanisms of action of such pleiotropic ruthenium complexes are only partially understood. In terms of Grp78 isoform selectivity, the modification of compounds 1 through 12 already showed a simple way to improve selectivity for Grp78 [70].
From a chemical point of view, the (semi-)synthetic Hsp70 and Hsp40 inhibitors described in this review are structurally heterogenic and comprise various compound classes. While natural Hsp70 and Hsp40 inhibitors were largely excluded from this review for clarity’s sake, some promising chemically modified natural products (11, 30a, 30b) and synthetic surrogates (26a, 29) were mentioned. In terms of Hsp40 protein targeting, structurally related synthetic compounds can have opposite effects on their target. While 26a inhibited Hsp40, its analogs compounds, 26b and 26c, activated Hsp40 [107,108,109,110,111,112]. In addition, chalcone 29 inhibited Hsp40, while chalcones 35a and 35b activated Hsp40 [121,122,123,134]. Interestingly, in the Chalcone case, both effects led to anticancer activity. Posttranslational modifications of Hsp40 (farnesylation) and its binding partners (acetylated tubulin) were also described as targetable Hsp40-modulatory mechanisms [125,131,132,133]. In this way, potential combination partners such as farnesyltransferase inhibitors and HDAC6 inhibitors emerged as modulators of the Hsp70-Hsp40 axis, which might be considered for future in vivo experiments.
As mentioned above, the combination of Hsp70 and/or Hsp40 inhibitors with various approved anticancer drugs revealed promising antitumor effects both in vitro and in vivo, suggesting suitable therapy regimens for new clinical trials. The Hsp70 inhibitors 3c and 22 (in its benzindazolium form, KP1019) already underwent early-stage clinical trials decades ago, even before their Hsp70 inhibitory activity was discovered [84,94]. Meanwhile, compound 22 experienced a revival and is currently in phase 1b/2 clinical trials (NCT04421820) in combination with FOLFOX (folinic acid, 5-fluorouracil, and oxaliplatin) for the treatment of advanced gastrointestinal tumors. However, with various new inhibitors and optimized analogs being available now, more valuable outcomes can be expected for Hsp70 and Hsp40 inhibitors from future clinical studies when designed and conducted properly.
Hsp40 is another member of the Hsp protein family and functions as a client protein of Hsp70. It is a small co-chaperone of Hsp70 that prevents the aggregation of unfolded polypeptides and proteins before transferring them to Hsp70. Hsp40 also binds to the nucleotide-binding domain (NBD) of Hsp70, thereby increasing the ATPase activity of Hsp70. Moreover, Hsp70 is also involved in modulating receptor tyrosine kinase (RTK) signaling pathway activity, such as the Ras/Raf-MAPK or the AKT pathway.
Synthetic Hsp70 inhibitors can act at the N-terminal (NBD) or substrate-binding C-terminal (SBD) binding domains of Hsp70. Some inhibitors also interfere with Hsp40 or act on organelle specific Hsp isoforms such as Grp78 (ER) or mortalin (mitochondria), leading to pronounced anticancer effects such as apoptosis, cell cycle arrest, or cellular senescence.
Author Contributions
Writing—original draft preparation, B.B.; writing—review and editing, B.N. and M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Santoro, M.G. Heat shock factors and the control of stress response. Biochem. Pharmacol. 2000, 59, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Tissiéres, A.; Mitchell, H.K.; Tracy, U.M. Protein synthesis in salivary glands of Drosophila melanogaster: Relation to chromosome puffs. J. Mol. Biol. 1974, 84, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Hasday, J.D.; Singh, I.S. Fever and the heat shock response: Distinct, partially overlapping processes. Cell Stress Chaperones 2000, 5, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.S.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Macario, A.J.L.; Conway de Macario, E. Chaperone Proteins and Chaperonopathies. In Stress: Physiology, Biochemistry, and Pathology Handbook of Stress Series; Fink, G., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 3, pp. 135–152. [Google Scholar]
- Kampinga, H.H.; Hageman, J.; Vos, V.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat shock protein 90 inhibitors: An update on achievements, challenges, and future directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of Hsp90 in cancer. Int. J. Mol. Sci. 2021, 22, 10317. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Albakova, Z.; Armeev, G.A.; Kanevsky, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. Hsp70 multi-functionality in cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Liu, J.; DiSanto, M.E.; Zhang, X. Heat shock protein 70 and 90 family in prostate cancer. Life 2022, 12, 1489. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, H.; Cao, L. Recent advance in heat shock proteins in cancer diagnosis, prognosis, metabolism and treatment. Biomed. Pharmacother. 2021, 142, 112074. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A.; Baghdady, A.M.; Ali, S.A.; Ahmed, M.I. GRP78 targeting: Hitting two birds with a stone. Life Sci. 2020, 260, 118317. [Google Scholar] [CrossRef] [PubMed]
- Elwakeel, A. Abrogating the interaction between p53 and mortalin (Grp75/HSPA9/mtHsp70) for cancer therapy: The story so far. Front. Cell Dev. Biol. 2022, 10, 879632. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Siam, M.K.S.; Sacitharan, P.K.; Ziganshin, R.H.; Ryazantsev, D.Y.; Sapozhnikov, A.M. Extracellular heat shock proteins and cancer: New perspectives. Transl. Oncol. 2021, 14, 100995. [Google Scholar] [CrossRef]
- Li, J.; Qian, X.; Sha, B. Heat shock protein 40: Structural studies and their functional implications. Protein Pept. Lett. 2009, 16, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liang, C.; Zhou, L. Structural and functional analysis of the Hsp70/Hsp40 chaperone system. Protein Sci. 2020, 29, 378–390. [Google Scholar] [CrossRef]
- Williamson, D.S.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Foloppe, N.; Francis, G.L.; Graham, C.J.; Howes, R.; et al. Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J. Med. Chem. 2009, 52, 1510–1513. [Google Scholar] [CrossRef]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; GEoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.; Liu, W.; Shao, Y.; Chen, L. VER-155008, a small molecule inhibitor of HSP70 with potent antio-cancer activity on lung cancer cell lines. Exp. Biol. Med. 2014, 239, 638–645. [Google Scholar] [CrossRef]
- Sakai, K.; Inoue, M.; Mikami, S.; Nishimura, H.; Kuwabara, Y.; Kojima, A.; Toda, M.; Ogawa-Kobayashi, Y.; Kikuchi, S.; Hirata, Y.; et al. Functional inhibition of heat shock protein 70 by VER-155008 suppresses pleural mesothelioma cell proliferation via an autophagy mechanism. Thoracic Cancer 2021, 12, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Shiota, M.; Tanaka, M.; Otsuka, A.; Matsumoto, M.; Kato, M.; Tamada, S.; Iwao, H.; Miura, K.; Nakatani, T.; et al. Heat shock protein 70 inhibitors suppress androgen receptor expression in LNCAP95 prostate cancer cells. Cancer Sci. 2017, 108, 1820–1827. [Google Scholar] [CrossRef] [Green Version]
- Brünnert, D.; Langer, C.; Zimmermann, L.; Bargou, R.C.; Burchardt, M.; Chatterjee, M.; Stope, M.B. The heat shock protein 70 inhibitor VER155008 suppresses the expression of HSP27, HOP and HSP90β and the androgen receptor, induces apoptosis, and attenuates prostate cancer cell growth. J. Cell. Biochem. 2020, 121, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, H.; Zhang, X.; Li, H. Visualizing and quantifying the effect of the inhibition of HSP70 on breast cancer cells based on laser scanning microscopy. Technol. Cancer Res. Treat. 2018, 17, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.-H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. The hsp70 inhibitor VER155008 induces paraptosis requiring de novo protein synthesis in anaplastic thyroid carcinoma cells. Biochem. Biophys. Res. Commun. 2014, 454, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Shervington, L.; Patil, H.; Shervington, A. Could the anti-chaperone VER155008 replace temozolomide for glioma treatment. J. Cancer 2015, 6, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Cavanaugh, A.; Juengst, B.; Sheridan, K.; Danella, J.F.; Williams, H. Combined inhibition of heat shock proteins 90 and 70 leads to simultaneous degradation of the oncogenic signaling proteins involved in muscle invasive bladder cancer. Oncotarget 2015, 6, 39821–39838. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.-H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. Hsp70 inhibition potentiates radicicol-induced cell death in anaplastic thyroid carcinoma cells. Anticancer Res. 2014, 34, 4829–4838. [Google Scholar]
- Asling, J.; Morrison, J.; Mutsaers, A.J. Targeting HSP70 and GRP78 in canine osteosarcoma cells in combination with doxorubicin chemotherapy. Cell Stress Chaperones 2016, 21, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Sojka, D.R.; Hastorek, S.; Vydra, N.; Toma-Jonik, A.; Wieczorek, A.; Gogler-Piglowska, A.; Scieglinska, D. Inhibition of the heat shock protein A (HSPA) family potentiates the anticancer effects of manumycin A. Cells 2021, 10, 1418. [Google Scholar] [CrossRef]
- Tang, X.; Tan, L.; Shi, K.; Peng, J.; Xiao, Y.; Li, W.; Chen, L.; Yang, Q.; Qian, Z. Gold nanorods together with HSP inhibitor-VER-155008 micelles for colon cancer mild-temperature photothermal therapy. Acta Pharmaceut. Sin. 2018, 8, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 synergistically enhances bortezomib-induced cytotoxicity in multiple myeloma. Cell Stress Chaperones 2020, 25, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Kawano, Y.; Fujiwara, S.; Kikukawa, Y.; Okuno, Y.; Tasaki, M.; Ueda, M.; Ando, Y.; Yoshinaga, K.; Ri, M.; et al. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int. J. Oncol. 2015, 46, 963–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reikyam, H.; Nepstad, I.; Sulen, A.; Gjertsen, B.T.; Hatfield, K.J.; Bruserud, O. Increased antileukemic effects in human acute myeloid leukemia by combining HSP70 and HSP90 inhibitors. Exp. Opin. Investig. Drugs 2013, 22, 551–563. [Google Scholar] [CrossRef]
- Kul, P.; Tuncbilek, M.; Ergul, M.; Tunoglu, E.N.Y.; Tutar, Y. A novel 6,8,9.trisubstituted purine analogue drives breast cancer luminal A subtype MCF-7 to apoptosis and senescence through Hsp70 inhibition. Anticancer Agents Med. Chem. 2023, 23, 585–598. [Google Scholar] [CrossRef]
- Williams, D.R.; Ko, S.-K.; Park, S.; Lee, M.-R.; Shin, I. An apoptosis-inducing small molecule that binds to heat shock protein 70. Angew. Chem. Int. Ed. 2008, 47, 7466–7469. [Google Scholar] [CrossRef]
- Ko, S.-K.; Kim, J.; Na, D.C.; Park, S.; Park, S.-H.; Hyun, J.Y.; Baek, K.-H.; Kim, N.D.; Kim, N.-K.; Park, Y.N.; et al. A small molecule inhibitor of ATPase activity of HSP70 induces apoptosis and has antitumor activities. Chem. Biol. 2015, 22, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-H.; Kim, W.-J.; Li, H.; Seo, W.; Park, S.-H.; Kim, H.; Shin, S.C.; Zuiderweg, E.R.P.; Kim, E.E.; Sim, T.; et al. Anti-leukemia activity of a Hsp70 inhibitor and its hybrid molecules. Sci. Rep. 2017, 7, 3537. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-H.; Baek, K.-H.; Shin, I.; Shin, I. Subcellular Hsp70 inhibitors promote cancer cell death via different mechanisms. Cell Chem. Biol. 2018, 25, 1242–1254. [Google Scholar] [CrossRef] [Green Version]
- Colvin, T.A.; Gabai, V.L.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; Garcia-Marcos, M.; et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef] [Green Version]
- Antonietti, P.; Linder, B.; Hehlgans, S.; Mildenberger, I.C.; Burger, M.C.; Fulda, S.; Steinbach, J.P.; Gessler, F.; Rödel, F.; Mittelbronn, M.; et al. Interference with the HSF1/HSP70/BAG3 pathway primes glioma cells to matrix detachment and BH3 mimetic-induced apoptosis. Mol. Cancer Ther. 2016, 16, 156–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, C.K.; Linder, B.; Bonn, F.; Rothweiler, F.; Dikic, I.; Michaelis, M.; Cinatl, J.; Mandal, M.; Kögel, D. BAG3 overexpression and cytoprotective autophagy mediate apoptosis resistance in chemoresistant breast cancer cells. Neoplasia 2018, 20, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Srinivasan, S.R.; Connarn, J.; Ahmad, A.; Young, Z.T.; Kabza, A.M.; Zuiderweg, E.R.P.; Sun, D.; Gestwicki, J.E. Analogues of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077, as anti-cancer agents. ACS Med. Chem. Lett. 2013, 4, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Colvin, T.; Rauch, J.N.; Acosta-Alvear, D.; Kampmann, M.; Dunyak, B.; Hann, B.; Aftab, B.T.; Murnane, M.; Cho, M.; et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol. Cancer Ther. 2015, 14, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Yaglom, J.A.; Wang, Y.; Li, A.; Li, Z.; Monti, S.; Alexandrov, I.; Lu, X.; Sherman, M.Y. Cancer cell responses to Hsp70 inhibitor JG-98: Comparison with Hsp90 inhibitors and finding synergistic drug combinations. Sci. Rep. 2018, 8, 3010. [Google Scholar] [CrossRef] [Green Version]
- Gabai, V.L.; Yaglom, J.A.; Wang, Y.; Meng, L.; Shao, H.; Kim, G.; Colvin, T.; Gestwicki, J.; Sherman, M.Y. Anticancer effects of targeting Hsp70 in tumor stromal cells. Cancer Res. 2016, 76, 5926–5932. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-S.; Kumar, V.; Lee, D.-Y.; Chen, Y.; Wu, Y.-C.; Gao, J.-Y.; Chu, P.-C. Development of novel rhodacyanine-based heat shock 70 inhibitors. Curr. Med. Chem. 2021, 28, 5431–5446. [Google Scholar] [CrossRef]
- Shao, H.; Li, X.; Moses, M.A.; Gilbert, L.A.; Kalyanaraman, C.; Young, Z.T.; Chernova, M.; Journey, S.N.; Weissman, J.S.; Hann, B.; et al. Exploration of benzothiazole rhodacyanines as allosteric inhibitors of protein-protein interactions with heat shock protein 70 (Hsp70). J. Med. Chem. 2018, 61, 6163–6177. [Google Scholar] [CrossRef]
- Dublang, L.; Underhaug, J.; Flydal, M.I.; Velasco-Carneros, L.; Maréchal, J.-D.; Moro, F.; Boyano, M.D.; Martinez, A.; Muga, A. Inhibition of the humN Hac/0 system by small ligands as a potential anticancer approach. Cancers 2021, 13, 2936. [Google Scholar] [CrossRef]
- Song, T.; Guo, Y.; Xue, Z.; Guo, Z.; Wang, Z.; Lin, D.; Zhang, H.; Pan, H.; Zhang, X.; Yin, F.; et al. Small-molecule inhibitor targeting the Hsp70-Bim protein-protein interaction in CML cells overcomes BCR-ABL-independent TKI resistance. Leukemia 2021, 35, 2862–2874. [Google Scholar] [CrossRef]
- Wang, Z.; Song, T.; Guo, Z.; Uwituze, L.B.; Guo, Y.; Zhang, H.; Wang, H.; Zhang, X.; Pan, H.; Ji, T.; et al. A novel Hsp70 inhibitor specifically targeting the cancer-related Hsp70-Bim protein-protein interaction. Eur. J. Med. Chem. 2021, 220, 113452. [Google Scholar] [CrossRef]
- Howe, M.K.; Bodoor, K.; Carlson, D.A.; Hughes, P.F.; Alwarawrah, Y.; Loiselle, D.R.; Jaeger, A.M.; Darr, D.B.; Jordan, J.L.; Hunter, L.M.; et al. Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem. Biol. 2014, 21, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Taldone, T.; Patel, H.J.; Patel, P.D.; Rodina, A.; Gozman, A.; Maharaj, R.; Clement, C.C.; Patel, M.R.; Brodsky, J.L.; et al. Heat shock protein 70 inhibitors. 1. 2,5′-Thiopyrimidine and 5-(phenylthio)pyrimidine acrylamides as irreversible binders to an allosteric site on heat shock protein 70. J. Med. Chem. 2014, 57, 1188–1207. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Miyata, Y.; Koren III, J.; Jones, J.R.; Trotter, J.H.; Chang, L.; O’Leary, J.; Morgan, D.; Lee, D.C.; Shults, C.L.; et al. Chemical manipulation of Hsp70 ATPase activity regulates Tau stability. J. Neurosci. 2009, 29, 12079–12088. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.M.; Morishima, Y.; Clapp, K.M.; Peng, H.-M.; Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Inhibition of Hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J. Biol. Chem. 2010, 285, 15714–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.L.; Cabello, M.C.; Qiao, S.; Azimian, S.; Wondrak, G.T. Phenotypic identification of the redox dye methylene blue as an antagonist of heat shock response gene expression in metastatic melanoma cells. Int. J. Mol. Sci. 2013, 14, 4185–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchala, D.; Bhatt, L.K.; Pethe, P.; Shelat, R.; Kulkarni, Y.A. Anticancer activity of methylene blue via inhibition of heat shock protein 70. Biomed. Pharmacother. 2018, 107, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Kühnl, A.; Reins, J.; Fischer, S.; Ortiz-Tanchez, J.; Schlee, C.; Mochmann, L.H.; Heesch, S.; Benlasfer, O.; Hofmann, W.-K.; et al. Antileukemic activity of the HSP70 inhibitor pifithrin-µ in acute leukemia. Blood Cancer J. 2011, 1, e28. [Google Scholar] [CrossRef] [Green Version]
- Granato, M.; Lacconi, V.; Peddis, M.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Santarelli, R.; Trivedi, P.; Frati, L.; D’Orazi, G.; et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013, 4, e730. [Google Scholar] [CrossRef] [Green Version]
- McKeon, A.M.; Egan, A.; Chandanshive, J.; McMahon, H.; Griffith, D.M. Novel improved synthesis of HSP70 inhibitor, Pifithrin-µ. In vitro synergy quantification of Pifithrin-µ combined with Pt drugs in prostate and colorectal cancer cells. Molecules 2016, 21, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekihara, K.; Harashima, N.; Tongu, M.; Tamaki, Y.; Uchida, N.; Inomata, T.; Harada, M. Pifithrin-µ, and inhibitor of heat-shock protein 70, can increase the antitumor effects of hyperthermia against human prostate cancer cells. PLoS ONE 2013, 8, e78772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaburski, G.M.; Leu, J.I.; Beeharry, N.; Hayik, S.; Andrake, M.D.; Zhang, G.; Herlyn, M.; Villanueva, J.; Dunbrack, R.L., Jr.; Yen, T.; et al. A modified HSP70 inhibitor shows broad activity as an anticancer agent. Mol. Cancer Ther. 2013, 11, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budina-Kolomets, A.; Balaburski, G.M.; Bondar, A.; Beeharry, N.; Yen, T.; Murphy, M.E. Comparison of the activity of three different HSP70 inhibitors on apoptosis, cell cycle arrest, autophagy inhibition, and HSP90 inhibition. Cancer Biol. Ther. 2014, 15, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Coskun, K.A.; Koca, I.; Gümüs, M.; Tutar, Y. Designing specific HSP70 substrate binding domain inhibitor for perturbing protein folding pathways to inhibit cancer mechanism. Anticancer Agents Med. Chem. 2021, 21, 1472–1480. [Google Scholar] [CrossRef]
- Kocam, I.; Gümüs, M.; Özgür, A.; Disli, A.; Tutar, Y. A noverl approach to inhibit heat shock response as anticancer strategy by coumarine compounds containing thiazole skeleton. Anticancer Agents Med. Chem. 2015, 15, 916–930. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Sverchinsky, D.V.; Mikhaylova, E.R.; Semenyuk, P.I.; Komarova, E.Y.; Niskanen, S.A.; Nikotina, A.D.; Burakov, A.V.; Kartsev, V.G.; Guzhova, I.V.; et al. Sensitizing tumor cells to conventional drugs: HSP70 chaperone inhibitors, their selection and application in cancer models. Cell Death Dis. 2018, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Pujari, R.; Jose, J.; Bhavnani, V.; Kumar, N.; Shastry, P.; Pal, J.K. Tamoxifen-induced cytotoxicity in breast cancer cells is mediated by glucose-regulated protein 78 (GRP78) via AKT (Thr308) regulation. Int. J. Biochem. Cell Biol. 2016, 77, 57–67. [Google Scholar] [CrossRef]
- Macias, A.T.; Williamson, D.S.; Allen, N.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Francis, G.L.; Graham, C.J.; et al. Adenosine-derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: Insights into isoform selectivity. J. Med. Chem. 2011, 54, 4034–4041. [Google Scholar] [CrossRef]
- Cerezo, M.; Lehraki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wu, Y.; Lian, X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol. Open 2020, 9, bio053298. [Google Scholar] [CrossRef]
- Ha, D.P.; Huang, B.; Wang, H.; Rangel, D.F.; Van Krieken, R.; Liu, Z.; Samanta, S.; Neamati, N.; Lee, A.S. Targeting GRP78 suppresses oncogenic KRAS protein expression andreduced viability of cancer cells bearing various KRAS mutations. Neoplasia 2022, 33, 100837. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.; Doghman-Bouguerra, M.; Ronco, C.; Benhida, R.; Rocchi, S.; Lalli, E. The GRP78/BiP inhibitor HA15 synergizes with mitotane action against adrenocortical carcinoma cells through convergent activation of ER stress pathways. Mol. Cell. Endocrinol. 2018, 474, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Mattos, D.R.; Weinman, M.A.; Wan, X.; Goodall, C.P.; Serrill, J.D.; McPhail, K.L.; Milovancev, M.; Bracha, S.; Ishmael, J.E. Canine osteosarcoma cells exhibit basal accumulation of multiple chaperone proteins and are sensitive to small molecule inhibitors of GRP78 and heat shock protein function. Cell Stress Chaperones 2022, 27, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Devi, A.; Mishra, S. Molecular docking and molecular dynamics studies reveal structural basis pof inhibition and selectivity of inhibitors ECGC and OSU-03012 towards glucose regulated protein-78 (GRP78) overexpressed in glioblastoma. J. Mol. Model. 2015, 21, 272. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Shuch, B.; Albers, T.; Roberts, J.L.; Tavallai, M.; Proniuk, S.; Zukiwski, A.; Wang, D.; Chen, C.-S.; Bottaro, D.; et al. Multi-kinase inhibitors can associate with heat shock proteins through their NH2-termini by which they suppress chaperone function. Oncotarget 2016, 7, 12975–12996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, S.; Yang, S.; Debnath, B.; Xue, D.; Kuang, Y.; Ramkumar, K.; Lee, A.S.; Ljungman, M.; Neamati, N. The hydroxyquinoline analogue YUM70 inhibits GRP78 to induce ER stress-mediated apoptosis in pancreatic cancer. Cancer Res. 2021, 81, 1883–1895. [Google Scholar] [CrossRef]
- Huang, M.; Li, Z.; Li, D.; Walker, S.; Greenan, C.; Kennedy, R. Structure-based design of HSPA5 inhibitors: From peptide to small molecule inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3044–3050. [Google Scholar] [CrossRef]
- Nayak, D.; Katoch, A.; Sharma, D.; Faheem, M.M.; Chakraborty, S.; Sahu, P.K.; Chikan, N.A.; Amin, H.; Gupta, A.P.; Gandhi, S.G.; et al. Indolylkojyl methane analogue IKM5 potentially inhibits invasion of breast cancer cells via attenuation of GRP78. Breast Cancer Res. Treat. 2019, 177, 307–323. [Google Scholar] [CrossRef]
- Qiao, Y.; Dsouza, C.; Matthews, A.A.; Jin, Y.; He, W.; Bao, J.; Jiang, F.; Chandna, R.; Ge, R.; Fu, L. Discovery of small molecules targeting GRP78 for antiangiogenic and anticancer therapy. Eur. J. Med. Chem. 2020, 193, 112228. [Google Scholar] [CrossRef]
- Ambrose, A.J.; Zerio, C.J.; Sivinski, J.; Schmidlin, C.J.; Shi, T.; Ross, A.B.; Widrick, K.J.; Johnson, S.M.; Zhang, D.D.; Chapman, E. A high throughput substrate binding assay reveals hexachlorophene as an inhibitor of the ER-resident HSP70 chaperone GRP78. Bioorg. Med. Chem. Lett. 2019, 29, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, A.N.I.; Lim, J.W.; Seo, S.H.; Lee, J.Y.; Lim, S.M.; Pae, A.N. GRP78-targeted in-silico virtual screening of novel anticancer agents. Chem. Biol. Drug. Des. 2018, 92, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent—Preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Schoenhacker-Alte, B.; Mohr, T.; Pirker, C.; Kryezu, K.; Kuhn, P.-S.; Buck, A.; Hofmann, T.; Gerner, C.; Hermann, G.; Koellensperger, G.; et al. Sensitivity towards the Grp78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption or ER homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef]
- Neuditschko, B.; Legin, A.A.; Baier, D.; Schintlmeister, A.; Reipert, S.; Wagner, M.; Keppler, B.K.; Berger, W.; Meier-Menches, S.M.; Gerner, C. Interaction with ribosomal proteins accompanies stress induction of the anticancer metallodrug BOLD-100/KP1339 in the endoplasmic reticulum. Angew. Chem. Int. Ed. 2021, 60, 5063–5068. [Google Scholar] [CrossRef]
- Ranzato, E.; Bonsignore, G.; Martinotti, S. ER stress response and induction of apoptosis in malignant pleural mesothelioma: The Achilles heel targeted by the anticancer ruthenium drug BOLD-100. Cancers 2022, 14, 4126. [Google Scholar] [CrossRef]
- Baier, D.; Schoenhacker-Alte, B.; Rusz, M.; Pirker, C.; Mohr, T.; Mendrina, T.; Kirchhofer, D.; Meier-Menches, S.M.; Hohenwallner, K.; Schaier, M.; et al. The anticancer ruthenium compound BOLD-100 targets glycolysis and generates a metabolic vulnerability towards glucose deprivation. Pharmaceutics 2022, 14, 238. [Google Scholar] [CrossRef]
- Yoon, A.-R.; Wadhwa, R.; Kaul, S.C.; Yun, C.-O. Why is mortalin a potential therapeutic target for cancer? Front. Cell Dev. Biol. 2022, 10, 914540. [Google Scholar] [CrossRef]
- Ferguson, I.D.; Tin, Y.-H.T.; Lam, C.; Shao, H.; Tharp, K.M.; Hale, M.; Kasap, C.; Mariano, M.C.; Kishishita, A.; Escobar, B.P.; et al. Allosteric HSP70 inhibitors perturb mitochondrial proteostasis and overcome proteasome inhibitor resistance in multiple myeloma. Cell Chem. Biol. 2022, 29, 1288–1302. [Google Scholar] [CrossRef]
- Kaul, S.C.; Reddel, R.R.; Mitsui, Y.; Wadhwa, R. An N-terminal region of Mot-2 binds to P53 in vitro. Neoplasia 2001, 3, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, R.; Sugihara, T.; Yoshida, A.; Nomura, H.; Reddel, R.R.; Simpson, R.; Maruta, H.; Kaul, S.C. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the Hsp70 family protein Mot-2 and reactivation of p53 function. Cancer Res. 2000, 60, 6818–6821. [Google Scholar]
- Pilzer, D.; Saar, M.; Koya, K.; Fishelson, Z. Mortalin inhibitors sensitize K562 leukemia cells to complement-dependent cytotoxicity. Int. J. Cancer 2010, 126, 1428–1435. [Google Scholar] [CrossRef]
- Koya, K.; Li, Y.; Wang, H.; Ukai, T.; Tatsuda, N.; Kawakami, M.; Shishido, T.; Chen, L.B. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996, 56, 538–543. [Google Scholar]
- Modica-Napolitano, J.S.; Koya, K.; Weisberg, E.; Brunelli, B.T.; Li, Y.; Chen, L.B. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-007. Cancer Res. 1996, 56, 544–550. [Google Scholar]
- Chiba, Y.; Kubota, T.; Watanabe, M.; Matsuzaki, S.W.; Otani, Y.; Teramoto, T.; Matsumoto, Y.; Koya, K.; Kitajima, M. MKT-077, localized lipophilic cation: Antitumor activity against human tumor xenografts serially transplanted into nude mice. Anticancer Res. 1998, 18, 1047–1052. [Google Scholar] [PubMed]
- Tikoo, A.; Shakri, R.; Connolly, L.; Hirokawa, Y.; Shishido, T.; Bowers, B.; Ye, L.H.; Kohama, K.; Simpson, R.J.; Maruta, H. Treatment of ras-induced cancers, by the F-actin-bundling drug MKT-077. Cancer J. 2000, 6, 162–168. [Google Scholar] [PubMed]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumors. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Putri, J.F.; Bhargava, P.; Dhanjal, J.K.; Yaguchi, T.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Mortaparib, a novel dual inhibitor of mortalin and PARP1, is a potential drug candidate for ovarian and cervical cancers. J. Exp. Clin. Cancer Res. 2019, 28, 499. [Google Scholar] [CrossRef] [PubMed]
- Sari, A.N.; Elwakeel, A.; Dhanjal, J.K.; Kumar, V.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Identification and characterization of mortaparibPlus—A novel triazole derivative that targets mortalin-p53 interaction and inhibits cancer-cell proliferation by wild-type p53-dependent and -independent mechanisms. Cancers 2021, 13, 835. [Google Scholar] [CrossRef]
- Elwakeel, A.; Sari, A.N.; Dhanjal, J.K.; Meidinna, H.N.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Mutant p53L194F harboring luminal-A breast cancer cells are refractory to apoptosis and cell cycle arrest in response to mortaparibPlus, a multimodal small molecule inhibitor. Cancers 2021, 13, 3043. [Google Scholar] [CrossRef] [PubMed]
- Meidinna, H.N.; Shefrin, S.; Sari, A.N.; Zhang, H.; Dhanjal, J.K.; Kaul, S.C.; Sundar, D.; Wadhwa, R. Identification of a new member of mortaparib class of inhibitors that target mortalin and PARP1. Front. Cell Dev. Biol. 2022, 10, 918970. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.M.; Nammalwar, B.; Long, A.; Matsumoto, H.; Singh, A.; Bunce, R.A.; Berlin, K.D. SHetA2 interference with mortalin binding to p66shc and p53 identified using drug-conjugated magnetic microspheres. Investig. New Drugs 2013, 32, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramraj, S.K.; Elayapillai, S.P.; Pelikan, R.C.; Zhao, Y.D.; Isingizwe, Z.R.; Kennedy, A.L.; Lightfoot, S.A.; Benbrook, D.M. Novel ovarian cancer maintenance therapy targeted at mortalin and mutant p53. Int. J. Cancer 2020, 147, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L. Selectivity of the molecular chaperone-specific immunosuppressive agent 15-deoxyspergualin: Modulation of Hsc70 ATPase activity without compromising DnaJ chaperone interactions. Biochem. Pharmacol. 1999, 57, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Fewell, S.W.; Day, B.W.; Brodsky, J.L. Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. J. Biol. Chem. 2001, 276, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fewell, S.W.; Smith, C.M.; Lyon, M.A.; Dumitrescu, T.P.; Wipf, P.; Day, B.W.; Brodsky, J.L. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J. Biol. Chem. 2004, 279, 51131–51140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisén, S.; Gestwicki, J.E. Identification of small molecules that modify the protein folding activity of heat shock protein 70. Anal. Biochem. 2008, 374, 371–377. [Google Scholar] [CrossRef]
- Wisén, S.; Bertelsen, E.B.; Thompson, A.D.; Patury, S.; Ung, P.; Chang, L.; Evans, C.G.; Walter, G.M.; Wipf, P.; Carlson, H.A.; et al. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of Hsp70-Hsp40. ACS Chem. Biol. 2010, 5, 611–622. [Google Scholar] [CrossRef]
- Adam, C.; Baeurle, A.; Brodsky, J.L.; Wipf, P.; Schrama, D.; Becker, J.C.; Houben, R. The Hsp70 modulator MAL3-101 inhibits Merkel cell carcinoma. PLoS ONE 2014, 9, e92041. [Google Scholar] [CrossRef] [Green Version]
- Prince, T.; Ackerman, A.; Cavanaugh, A.; Schreiter, B.; Juengst, B.; Andolino, C.; Danella, J.; Chernin, M.; Williams, H. Dual targeting of HSP70 does not induce the heat shock response and synergistically reduces viability in muscle invasive bladder cancer. Oncotarget 2018, 9, 32702–32717. [Google Scholar] [CrossRef] [Green Version]
- Sannino, S.; Guerriero, C.J.; Sabnis, A.J.; Stolz, D.B.; Wallace, C.T.; Wipf, P.; Watkins, S.C.; Bivona, T.G.; Brodsky, J.L. Compensatory increases of select proteostasis networks, after Hsp70 inhibition in cancer cells. J. Cell Sci. 2018, 131, jcs217760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassel, J.A.; Ilyin, S.; McDonnell, M.E.; Reitz, A.B. Novel inhibitors of heat shock protein Hsp70-mediated luciferase refolding that bind to DnaJ. Bioorg. Med. Chem. 2012, 20, 3609–3614. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Kitahara, M.; Nagata, K. Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res. 2000, 60, 2942–2948. [Google Scholar] [PubMed]
- Koishi, M.; Yokota, S.; Mae, T.; Nishimura, S.; Kanamori, S.; Horii, N.; Shibuya, K.; Sasai, K.; Hiraoka, M. The effects of KNK437, a novel inhibitor of heat shock protein synthesis, on the acquisition of thermotolerance in a murine transplantable tumor in vivo. Clin. Cancer Res. 2001, 7, 215–219. [Google Scholar] [PubMed]
- Ohnishi, K.; Takahashi, A.; Yokota, S.; Ohnishi, T. Effects of a heat shock protein inhibitor KNK437 on heat sensitivity and heat tolerance in human squamous cell carcinoma cell lines differing in p53 status. Int. J. Radiat. Biol. 2004, 80, 607–614. [Google Scholar] [CrossRef]
- Matsuda, K.; Nakagawa, S.-Y.; Nakano, T.; Asaumi, J.-I.; Jagetia, G.C.; Kawasaki, S. Effects of KNK437 on heat-induced methylation of histone H3 in human oral squamous cell carcinoma cells. Int. J. Hyperthermia 2006, 22, 729–735. [Google Scholar] [CrossRef]
- Inoue, H.; Uyama, T.; Hayashi, J.; Watanabe, A.; Kobayashi, K.-I.; Tadokoro, T.; Yamamoto, Y. N-Formyl-3,4-methylenedioxy-benzylidene-γ-butyrolactam, KNK437 induces caspase-3 activation through inhibition of mTORC1 activity in Cos-1 cells. Biochem. Biophys. Res. Commun. 2010, 395, 56–60. [Google Scholar] [CrossRef]
- Oommen, D.; Prise, K.M. KNK437, abrogates hypoxia-induced radioresistance by dual targeting of the AKT and HIF-1α survival pathways. Biochem. Biophys. Res. Commun. 2012, 421, 538–543. [Google Scholar] [CrossRef]
- Yang, S.; Ren, X.; Liang, Y.; Yan, Y.; Zhou, Y.; Hu, J.; Wang, Z.; Song, F.; Wang, F.; Liao, W.; et al. KNK437 restricts the growth and metastasis of colorectal cancer via targeting DNAJA1/CDC45 axis. Oncogene 2020, 39, 249–261. [Google Scholar] [CrossRef]
- Zhang, B.; Duan, D.; Ge, C.; Yao, J.; Liu, Y.; Li, X.; Fang, J. Synthesis of xanthohumol analogues and discovery of potent thiredoxin reductase inhibitor as potential anticancer agent. J. Med. Chem. 2015, 58, 1795–1805. [Google Scholar] [CrossRef]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 chaperone axis as a novel strategy to treat castration-resistant prostate cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, M.A.; Kumar, V.; Tailor, D.; Garcia-Marques, F.J.; Hsu, E.-C.; Liu, S.; Bermudez, A.; Kanchustambham, V.; Shankar, V.; Inde, Z.; et al. SU086, an inhibitor of HSP90, impairs glycolysis and represents a treatment strategy for advanced prostate cancer. Cell Rep. Med. 2022, 3, 100502. [Google Scholar] [CrossRef] [PubMed]
- Alalem, M.; Bhosale, M.; Ranjan, A.; Yamamoto, S.; Kaida, A.; Nishikawa, S.; Parrales, A.; Farooki, S.; Anant, S.; Padhye, S.; et al. Mutant p53 depletion by novel inhibitors for HSP40/J-domain proteins derived from the natural compound plumbagin. Cancers 2022, 14, 4187. [Google Scholar] [CrossRef] [PubMed]
- Rottach, A.-M.; Ahrend, H.; Martin, B.; Walther, B.; Zimmermann, U.; Burchardt, M.; Stope, M.B. Cabazitaxel inhibits prostate cancer cell growth by inhibition of androgen receptor and heat shock protein expression. World J. Urol. 2019, 37, 2137–2145. [Google Scholar] [CrossRef]
- Adjei, A.A.; Davis, J.N.; Erlichman, C.; Svingen, P.A.; Kaufmann, S.H. Comparison of potential markers of farnesyltransferase inhibition. Clin. Cancer Res. 2000, 6, 2318–2325. [Google Scholar]
- Karp, J.E.; Lancet, J.E.; Kaufmann, S.H.; End, D.W.; Wright, J.J.; Bol, K.; Horak, I.; Tidwell, M.L.; Liesveld, J.; Kottke, T.J.; et al. Clinical and biologic activity of the farnesyltransferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: A phase 1 clinical-laboratory correlative trial. Blood 2001, 97, 3361–3369. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.J.; Ho, L.; Ranganathan, S.; Abbruzzese, J.L.; Alpaugh, R.K.; Beard, M.; Lewis, N.L.; McLaughlin, S.; Rogatko, A.; Perez-Ruixo, J.J.; et al. Phase II and pharmacodynamics study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2003, 21, 1301–1306. [Google Scholar] [CrossRef]
- Wang, J.; Yao, X.; Huang, J. New tricks for human farnesyltransferase inhibitor: Cancer and beyond. Med. Chem. Commun. 2017, 8, 841–854. [Google Scholar] [CrossRef]
- Marchwicka, A.; Kaminska, D.; Monirialamdari, M.; Blazewska, K.M.; Gendaszewska-Darmach, E. Protein prenyltransferases and their inhibitors: Structural and functional characterization. Int. J. Mol. Sci. 2022, 23, 5424. [Google Scholar] [CrossRef]
- Wang, C.-C.; Liao, Y.-P.; Mischel, P.S.; Iwamoto, K.S.; Cacalano, N.A.; McBride, W.H. HDJ-2 as a target for radiosensitization of glioblastoma multiforme cells by the farnesyltransferase inhibitor R115777 and the role of the p53/p21 pathway. Cancer Res. 2006, 66, 6756–6762. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Tong, X.; Sun, L.; Li, H.; Jones, R.D.; Liao, J.; Yang, G.-Y. Inhibition of mutant Kras and p53-driven pancreatic carcinogenesis by atorvastatin: Mainly via targeting of the farnesylated DNAJA1 in chaperoning mutant p53. Mol. Carcinogen. 2019, 58, 2052–2064. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Mägdefrau, A.-S.; Piskor, E.-M.; Kleemann, M.; Beyer, M.; Linke, K.; Hansen, L.; Schaffer, A.-M.; Hoffmann, M.E.; Poepsel, S.; et al. Targeting the MYC interaction network in B-cell lymphoma via histone deacetylase 6 inhibition. Oncogene 2022, 41, 4560–4572. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Marins, M.; Chaichanasak, N.; Yoon, Y.; Fachin, A.L.; Pinhanelli, V.C.; Regasini, L.O.; dos Santos, M.B.; Ayusso, G.M.; de Carvalho Marques, B.; et al. Trans-chalcone increases p53 activity via DNAJB1/HSP40 induction and CRM1 inhibition. PLoS ONE 2018, 13, e0202263. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, R.H.; Coulibaly, B.; Stich, A.; Scheiwein, M.; Merkle, H.; Eubel, J.; Becker, H.; Müller, O.; Zich, T.; Schiek, W.; et al. Methylene blue as an antimalarial agent. Redox Rep. 2003, 8, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Takasu, K. π-Delocalized lipophilic cations as new candidates for antimalarial, antitrypanosomal and antileishmanial agents: Synthesis, evaluation of antiprotozoal potency, and insight into their action mechanisms. Chem. Pharm. Bull. 2016, 64, 656–667. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Structures of NBD-binding and interfering compounds 1–8.

Figure 2.
Structures of SBD-interacting Hsp70 inhibitors 9–11.

Figure 3.
Structures of Grp78 inhibitors 12–22. *: chiral center or asymmetric carbon atom.

Figure 4.
Structures of mortalin inhibitors 3b, 3c, 23–25.

Figure 5.
Structures of Hsp40 modulators 26–35. *: chiral center or asymmetric carbon atom.

Figure 6.
Cellular effects of Hsp70 isoforms and their inhibition by synthetic small molecules. Hsp70 is composed of two distinct domains, a 40 kDa N-terminal nucleotide-binding domain (NBD) that regulates client association and a 25 kDa C-terminal substrate-binding domain (SBD). Hsp70 is found in the cytosol with multicellular functions. Together with Hsp90 it acts as a multi-chaperone complex to modulate heat stress responses. Organelle-specific Hsp70 isoforms such as Grp78 in the endoplasmic reticulum or mortalin in mitochondria were identified as promising anticancer drug targets. Inhibition of these multicellular processes by (semi-)synthetic inhibitors (red lines) leads to cell senescence, apoptosis and cell cycle arrest in various cancers.
Figure 6.
Cellular effects of Hsp70 isoforms and their inhibition by synthetic small molecules. Hsp70 is composed of two distinct domains, a 40 kDa N-terminal nucleotide-binding domain (NBD) that regulates client association and a 25 kDa C-terminal substrate-binding domain (SBD). Hsp70 is found in the cytosol with multicellular functions. Together with Hsp90 it acts as a multi-chaperone complex to modulate heat stress responses. Organelle-specific Hsp70 isoforms such as Grp78 in the endoplasmic reticulum or mortalin in mitochondria were identified as promising anticancer drug targets. Inhibition of these multicellular processes by (semi-)synthetic inhibitors (red lines) leads to cell senescence, apoptosis and cell cycle arrest in various cancers.

Table 2.
SBD-interacting Hsp70 inhibitors and their effects on cancer model(s).
| Compound | Cancer Model(s) | Effects |
|---|---|---|
| 9a (PES) | Colon, breast, prostate and pancreas carcinoma, osteosarcoma, lymphoma, acute leukemia | Antiprolierative independent from p53-state, caspase activation, dysfunctional autophagy, prolonged survival of Eµ-Myc mice, NF-κB suppression, degradation of Akt and ERK1/2, immunogenic activity, sensitization of cancers to drugs and hyperthermia [59,60,61,62,63] |
| 9b (PES-Cl) | BRAF-V600E mutant melanoma, SkBr3 breast carcinoma, FaDu head and neck squamous cell carcinoma, H1299 lung adenocarcinoma, lymphoma | More antiproliferative than 9a, apoptosis induction, autophagy inhibition, G2-M phase arrest, degradation of cyclin B1 and Her2, prolonged survival of Eµ-Myc mice [64,65] |
| 10 (KBR1307) | MCF-7 breast carcinoma | More antiproliferative than 9a, binds Hsp70 in absence of nucleotide [66] |
| 11 (AEAC) | C6 rat glioblastoma, B16 mouse melanoma | Increased doxorubicin activity in vitro and in vivo, tumor growth inhibition (71%) and prolonged survival in B16 mice [68] |
Table 3.
Grp78 inhibitors and their effects on cancer model(s).
| Grp78 Inhibitor | Cancer Model(s) | Effects |
|---|---|---|
| 1 (VER-155008) | Osteosarcoma, MCF-7 and MDA-MB-231 breast cancer | Antiproliferative, apoptosis induction, suppression of tamoxifen-induced phosphor-GSK-3β [30,69] |
| 12 | HCT-116 colon carcinoma | More selective for Grp78 than 1, no antiproliferative activity [70] |
| 13 (HA15) | Melanoma (BRAF-mutant), A549 NSCLC, KRAS-mutant cancer, adrenocortical carcinoma | ER stress, induction of apoptosis and autophagy, in vivo inhibition of A375 melanoma growth, suppression of KRAS and steroidogenesis, synergy with mitotane [71,72,73,74] |
| 14 (OSU-03012) | Carboplatin-resistant canine osteosarcoma (HMPOS-2.5R and HMPOS-10R), GBM5 and GBM12 glioblastoma | Antiproliferative, Bag2 suppression, formation of toxic autophagosomes [75,76,77] |
| 15 (YUM70) | KRAS-mutant cancer, pancreatic cancer (Mia-PaCa-2, PANC-1, BxPC-3) | Antiproliferative, ER stress, eIF2α phosphorylation, AT4 and CHOP activation, synergy with vorinostat and topotecan, in vivo MiaPaCa-2 tumor growth inhibition [73,78] |
| 16 (HM01), 17 (HM03) | HCT-116 colon carcinoma | Antiproliferative [79] |
| 18 (IKM5) | Breast cancer (MCF-7, MDA-MB-231, MDA-MB-468, BT474, 4T1) | Antiproliferative, suppression of MMP-2, Twist1 and vimentin, induction of TIMP-1 and Par-4, in vivo inhibition of breast tumor growth and lung metastasis formation [80] |
| 19 (FL5) | Renal cell carcinoma, HUVECs | Cell death, anti-angiogenic [81] |
| 20 (Hexachlorophene) | HCT-116 colon carcinoma | Cytotoxic, induction of apoptosis and autophagy, upregulated ATF4, XBP1s, and CHOP [82] |
| 21 (VH1019) | MCF-7 breast carcinoma | ATP-mimic, antiproliferative [83] |
| 22 (KP1339/BOLD-100) | Miscellaneous, HCT-116 colon carcinoma, REN pleural mesothelioma, Capan1 pancreatic carcinoma | Apoptosis induction and ER disruption in 22-sensitive cells, G2 cell cycle arrest in 22-resistant cells, binding to ribosomal proteins, ER stress, cytotoxic, ROS formation, induction of CHOP and XPB1, glycolysis upregulation, synergy with 2-deoxyglucose [85,86,87,88] |
Table 4.
Mortalin inhibitors and their anticancer effects.
| Mortalin Inhibitor | Cancer Model(s) | Effects |
|---|---|---|
| 3b (JG-98) | Multiple myeloma | Antiproliferative, 55S mitoribosome degradation [90] |
| 3c (MKT-077) | Miscellaneous, ras-induced cancer, K562 leukemia | Antiproliferative, mitochondria accumulation, activation of p53 and p21, inhibition of mortalin-C9 [91,92,93,94,95,96,97] |
| 23 | Miscellaneous, HeLa cervix carcinoma | Antiproliferative, p53 and Bak activation [40] |
| 24a (Mortaparib) | HeLa cervix and SKOV-3 ovarian carcinoma | Dual mortalin and PARP1 inhibitor, p53 activation, apoptosis induction, in vivo inhibition of SKOV-3 tumor growth and metastases [99] |
| 24b (MortaparibPlus) | HCT-116 and DLD-1 colon carcinoma | Inhibition of mortalin-p53 and PARP1, CARF-1 suppression, induction of apoptosis and p21 [100] |
| 24c (MortaparibMild) | HCT-116 colon carcinoma | Inhibition of mortalin-p53 and PARP1 [102] |
| 25 (SHetA2) | Ovarian cancer | Inhibition of mortalin-p53, synergy with p53-reactivator PRIMA-1Met, caspase-activation, increased ROS formation, reduced ATP, in vivo inhibition of MESOV tumor growth, clinical phase 1 studies (advanced/recurrent cervical, endometrial and ovarian cancer) [15,103,104] |
Table 5.
Hsp40 modulators and their anticancer effects.
| Hsp40 Modulator | Cancer Model(s) | Effects |
|---|---|---|
| 26a (MAL3-101) | Merkel cell carcinoma, muscle invasive bladder cancer, RMS13 rhabdomyosarcoma | Antiproliferative, apoptosis induction, in vivo MCC growth inhibition, synergy with 1 and STA9090, UPR induction [110,111,112] |
| 28 (KNK437) | Squamous cell carcinoma (KB, SCC VII and SAS/mp53), immortalized Cos-1, MDA-MB-231 breast carcinoma, colon carcinoma (COLO 320DM, SW480, SW620, RKO, LOVO) | Suppression of Hsp70 and Hsp40, in vivo inhibition of SCC VII squamous cell carcinoma at 44 °C, apoptosis induction and colony formation inhibition at 42 °C, inhibition of heat-induced H3-Lys4 methylation, suppression of AKT and HIF-1α pathways, selective inhibition of DNAJA1, in vivo inhibition of DNAJA1-overexpressing SW480 and SW620 tumors, suppressed CDC45, upregulated ubiquitin, in vivo suppressed liver metastasis formation with 5-FU/L-OHP [113,114,115,116,117,118,119,120] |
| 29 (C86/SU086) | HeLa cervix carcinoma, 22Rv1 and C4-2 prostate cancer | Antiproliferative, pan-Hsp40/DNAJ inhibition, apoptosis induction, ROS formation, inhibition of thioredoxin reductase, degradation of FL-AR and ARv7, Hsp90 inhibition, in vivo 22Rv1 and C4-2 prostate tumor growth inhibition [121,122,123] |
| 30a (PLIHZ), 30b (PLTFBH) | HN31 pharyngeal squamous cell carcinoma | Antiproliferative, suppression of DNAJA1, mutant p53, Cdc42 and Rac [124] |
| 31 (Cabazitaxel) | LNCaP and PC-3 prostate cancer | Antiproliferative, suppression of Hsp40, HOP and AR [125] |
| 32 (Tipifarnib) | SF763 and U87 glioblastoma | Farnesyltransferase inhibition, reduction of farnesylated HDJ-2, radio-sensitizing, antiproliferative, p21 induction, G2/M arrest [131] |
| 33 (Atrovastatin) | Pancreatic carcinoma (PO3, SU 86.86, BXPC-3, Pan 10.05) | HMG-CoA reductase inhibition, suppression of DNAJA1 farnesylation, induction of apoptosis and p21, degradation of mutant p53 (blocked nuclear transport), inhibition of migration [132] |
| 34 (Marbostat-100) | MYC-overexpressing B-cell lymphoma | HDAC6 inhibition, apoptosis induction, MYC- degradation, tubulin hyperacetylation, relocation of DNAJA3 to acetyltubulin [133] |
| 35a, 35b | U2OS osteosarcoma | Activation of Hsp40 and p53, suppression of CRM1 [134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nitzsche, B.; Höpfner, M.; Biersack, B. Synthetic Small Molecule Modulators of Hsp70 and Hsp40 Chaperones as Promising Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 4083. https://doi.org/10.3390/ijms24044083
AMA Style
Nitzsche B, Höpfner M, Biersack B. Synthetic Small Molecule Modulators of Hsp70 and Hsp40 Chaperones as Promising Anticancer Agents. International Journal of Molecular Sciences. 2023; 24(4):4083. https://doi.org/10.3390/ijms24044083
Chicago/Turabian StyleNitzsche, Bianca, Michael Höpfner, and Bernhard Biersack. 2023. "Synthetic Small Molecule Modulators of Hsp70 and Hsp40 Chaperones as Promising Anticancer Agents" International Journal of Molecular Sciences 24, no. 4: 4083. https://doi.org/10.3390/ijms24044083
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.