
The Oxygen Cascade from Atmosphere to Mitochondria as a Tool to Understand the (Mal)adaptation to Hypoxia


1
MAGI GROUP, San Felice del Benaco, 25010 Brescia, Italy
2
School of Medicine and Surgery, University of Milano Bicocca, 20126 Milan, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(4), 3670; https://doi.org/10.3390/ijms24043670
Submission received: 20 January 2023
/
Revised: 5 February 2023
/
Accepted: 9 February 2023
/
Published: 12 February 2023
(This article belongs to the Special Issue Adaptation to Hypoxia: Beyond the Chimera)
Abstract
:Hypoxia is a life-threatening challenge for about 1% of the world population, as well as a contributor to high morbidity and mortality scores in patients affected by various cardiopulmonary, hematological, and circulatory diseases. However, the adaptation to hypoxia represents a failure for a relevant portion of the cases as the pathways of potential adaptation often conflict with well-being and generate diseases that in certain areas of the world still afflict up to one-third of the populations living at altitude. To help understand the mechanisms of adaptation and maladaptation, this review examines the various steps of the oxygen cascade from the atmosphere to the mitochondria distinguishing the patterns related to physiological (i.e., due to altitude) and pathological (i.e., due to a pre-existing disease) hypoxia. The aim is to assess the ability of humans to adapt to hypoxia in a multidisciplinary approach that correlates the function of genes, molecules, and cells with the physiologic and pathological outcomes. We conclude that, in most cases, it is not hypoxia by itself that generates diseases, but rather the attempts to adapt to the hypoxia condition. This underlies the paradigm shift that when adaptation to hypoxia becomes excessive, it translates into maladaptation.
1. Introduction
About 1% of the world human population live permanently at >2500 m altitudes, mainly in non-Western countries [1]. The major health problem these people constantly face stem from breathing an atmosphere impoverished in oxygen (O2). On average, up to 10% of the population living at altitude develop diseases linked to insufficient O2 or hypoxia [2]. Hypoxia may occur even at sea level, contributing to morbidity and mortality in numberless patients with acute respiratory diseases (ARDS), chronic pulmonary diseases (COPD), emphysema, lung tumors, anemia, heart failure, and hundreds other disorders that stem from unmatched O2 supply with respect to needs. Such diseases take an intolerable toll on humanity in terms of sufferance, life loss, and economic distress: the burden imposed in one single year (2010) in the US only by COPD alone was USD49.9 billion [3]. Among all the factors that afflict humanity, hypoxia has the unique feature of being both physiological (i.e., due to altitude), and pathological (i.e., due to a pre-existing disease). When physiological, hypoxia may raise some form of adaptation, and the ability to adapt to environmental stress factors represents a key feature in the Darwinian theory of evolution of species. However, it is not yet clear whether humans can ever adapt to hypoxia, given the high incidence of the clinical manifestations associated with both physiological and pathological hypoxia.
The aim of this review is to focus on the capacity to adapt to the challenge represented by insufficient O2. To this purpose, we will examine the mechanisms underlying the responses to hypoxia at several levels spanning from genes to the respiratory and circulatory features, focusing on those that are most relevant in terms of adaptation. To accomplish this goal, we will reexamine the O2 cascade as a tool to understand the genesis and consequences of physiological and pathological hypoxia. We will focus on the paths that impact hypoxia adaptation neglecting the contributions that describe the simple responses to hypoxia. When possible, we will privilege contributions referring to humans rather than animals or in vitro. To indicate the degree of hypoxia, we will use the double notion of altitude in meters above sea level and the corresponding effective %O2, where 21.0 %O2 is equivalent to sea level.
2. Oxygen and Hypoxia
Three 18th century scientists participated in the discovery of O2. Carl Wilhelm Scheele (1742–1786), a Swedish–German pharmaceutical chemist, was the first to discover O2, but his low academic profile and lack of international competitiveness caused him the loss of priority for this discovery [4]. Joseph Priestley (1733–1804), a polyhedric English chemist, natural philosopher, separatist theologian, grammarian, multi-subject educator, and liberal political theorist, was able to intertwine his scientific work with the ongoing debate on phlogiston within a conflict with the Church of England that led him to migrate to the US. His publication, although delayed with respect to that of Scheele, gained him the priority as discoverer of O2 [5]. Finally, Antoine Lavoisier (1743–1794), a French nobleman and chemist, coined, together with Marie-Anne Lavoisier, the term “oxygen”, based on the misleading concept of a substance that generates acidity (-ὀξύς (oxys, acid) + -γενής (-genēs, generator)), during the years of the French Revolution that led Antoine to execution under the guillotine [6]. The contributions by two other 17th century scientists, the English John Mayhow (1661–1679) and the Polish Michael Sendivogius (1566–1636) should also be mentioned, although their extraordinary work probably was not robust enough to assign them the role of discoverers of O2 [7].
2.1. Oxygen at Altitude
Dalton’s law enables the expression of the partial pressure of O2 (PO2) as a function of %O2, the barometric pressure (BP), and the aqueous vapor pressure (PH2O):
BP and altitude are the main determinant of atmospheric PO2 (Figure 1), as PH2O depends uniquely on temperature (47 mmHg at 37 °C) and %O2 is remarkably constant (20.9460%) at any latitude on Earth, except Antarctica, where physical factors slightly decrease the %O2. At Dome C Concordia, which is located at a geographical altitude of 3233 m/14.3 %O2, the slightly lowered %O2 in air (20.82–20.90) [8] decreases the effective %O2 to 3800 m/13.2% [9].
Decreasing the %O2 in breathed air has dramatic effects on exercise capacity. Figure 2 shows the resting and maximal O2 uptake (VO2) with altitude. The resting VO2, not affected by altitude, remains ~3.5 mL O2/kg/min throughout, or 1 metabolic equivalent of training (MET). The maximal VO2, by contrast, decreases progressively from ~18 MET in exceptionally well-trained subjects at sea level to 3–4 MET near the summit of Mt. Everest (8848 m/6.6 %O2), which corresponds to activities such as walking, machine tooling, standing tasks, or golfing. Thus, decreased O2 availability at altitude would enable subjects to perform very light tasks only—let alone climbing and fighting the extremely low temperatures at those altitudes. As the incidence of resting VO2 on maximal VO2 increases from 5% at sea level to ~30% at 8848 m/6.6 %O2, at extreme altitudes a significant portion of inhaled O2 is diverted just to sustain the basal needs of the organism. The first BP measurement taken on the Mt. Everest summit at 8848 m was 253 mmHg [10], perhaps sufficient to sustain basic metabolism. However, measurements taken with stratospheric balloons showed relevant BP variability depending on latitude and seasons [11].
2.2. Hypoxia
Hypoxia is defined as an imbalance between O2 supply and O2 demand. This definition applies at all organization levels from whole organisms to cells in both physiological and pathological contexts. Although the basic concepts related to altitude hypoxia are now found in several human diseases, until recently hypoxia was recognized almost exclusively in altitude contexts, in inhabited balloons flights, as well as in occasional or permanent dwellers in the mountains (Table 1).
Although the hypoxia severity may range from mild to severe depending on arterial PO2 and exposure duration [15], the threshold PO2 that triggers the hypoxia condition is rather undefined as it depends on pathophysiological factors such as the cell metabolic rate, the VO2, the cardiac output, and the O2 extraction, just to name a few, that cause extreme variability in the threshold PO2. Some patients with COPD, heart failure, and anemia are indeed hypoxic even at 0 m/21.0 %O2, while healthy untrained subjects may dwell for short times at >3300 m/14 %O2 without developing appreciable signs of sickness. By contrast, extremely well-trained subjects may survive at 7500 m/8.0 %O2 for hours/days. As for cells, current good practice [16] suggests exposing cultured cells to >20,000 m/1.0 %O2 to simulate hypoxia.
Even the various organs in the same organism vary remarkably in terms of VO2, capillary blood flow, and O2 extraction. This translates to different sensitivities to the same degree of hypoxia depending on the specific organ’s physiological status. In rats exposed to the same degree of hypoxia, various organs respond differently in terms of triggering the O2-sensing machinery and of activation of apoptosis [17]. It is thus expected that at every degree of hypoxia, the organs with a high metabolic rate, such as the brain, heart, and exercising muscles, become hypoxic before those with a low metabolic rate, where the O2 consumption is better matched with perfusion.
2.3. Clinical Manifestations of Hypoxia
How to establish if an organism becomes adapted to a challenge? In principle, wherever the challenged population is free from the diseases inferred by that challenge, can survive, and can reproduce, then it may be considered adapted. When hypoxia constitutes the challenge, the diseases may be acute or chronic, giving rise to the complex pathology addressed by acute (AMS) and chronic mountain sickness (CMS), respectively. Up to 10% of the world population living at altitude develop CMS [2,18], although with marked geographical differences.
The onset of CMS, first described by Carlos Monge Medrano [19], is considered a valuable marker of the adaptation to chronic hypoxia. An elusive disease caused by sustained insufficiency of O2, its major pathogenic etiology is the excessive erythropoietic response, or polycythemia, which originates outcomes collectively known as CMS [20]. Thus, tracking hematological changes enables monitoring of hypoxia adaptation [21,22]. Alternate valid markers of maladaptation include the development of pulmonary hypertension (PH), a frequent condition at altitude, which may be accompanied by right ventricle hypertrophy and failure. Neuropsychological dysfunctions are also of concern in CMS as they may affect school performance in children [23]. To partially compensate such effects, the “oxygen conditioning” paradigm was proposed to enrich %O2 with the use of synthetic zeolite, which adsorbs air nitrogen, based on the observation that a 1 %O2 increase corresponds to a decrease in altitude of 300 m [24].
3. The Oxygen Cascade
Despite important limitations [25], the O2 cascade remains a good representation of the path of O2 from atmosphere to mitochondria, the final O2 users (Figure 3). The blue line represents the progressive stepwise decreases in PO2 along the path in normoxic healthy conditions. The establishment of O2 gradients at the interface between adjacent compartments ensures the correct flow of O2 along the cascade. Each of these gradients must be wide enough to grant a sufficient O2 supply to the mitochondria [26]. The red line represents the situation established when atmospheric PO2 is reduced by one-third, equivalent to 3400 m/13.9 %O2. Such a drop translates into a reduced width of each gradient step, which corresponds to a decreased supply of O2 to the mitochondria. Thus, all the interfaces must cooperate to ensure the correct O2 supply. The same reduction in O2 supply to mitochondria occurs when the environmental PO2 is normal, but some sites of resistance to the O2 flux emerge in the correspondence of one of the interfaces. This situation corresponds to wider PO2 gradients between adjacent compartments. Such sites of resistance are usually associated with diseases that transform a normoxic situation into a hypoxic one downstream of the block in the O2 cascade. This determines the establishment of pathological hypoxia, which differs from the physiological one by being often accompanied by factors such as inflammation and redox imbalance, just to name a few.
The ability to adapt to environmental changes is a key feature in the evolution of species. While the mechanisms underlying the cardiac, nervous, and pulmonary tissues’ initial adaptation, or acclimatization, to hypoxic conditions have recently been worked out [27], the rules for recognizing the onset of long-term adaptation should include the appearance of complex characters that are too well-fitted to the environment for the fit to have arisen by chance and that help their bearers to survive and reproduce. The concept of adaptation physiology greatly depends on semantic problems related to the meaning of this term [28]. Although adaptation is believed to involve the establishment of compensatory responses to environmental challenges, not all the responses to a challenge may be adaptive. Thus, there may be three levels of response to challenges depending on the gain: inadequate (gain < 100%), ideal (gain = 100%), and excessive (gain > 100%) (Figure 4). This review will analyze the responses to the hypoxic challenges in the various resistance sites represented in Figure 3 in the search of an answer to the question of whether humans can ever adapt to hypoxia.
3.1. Cell-to-Mitochondria Interface
3.1.1. Hypoxia-Inducible Factors
Understanding how cells sense and adapt to O2 availability was worth the 2019 Nobel Prize in Physiology and Medicine to William Kaelin, Peter Ratcliffe, and Gregg Semenza, as well as robust worldwide reconnaissance. This ubiquitarian mechanism constitutes the O2-sensing function that orchestrates most, although not all, the cell responses to O2 shortage. The transcription factor family of hypoxia-inducible factors (HIF), a fundamental part of the O2-sensing system, exploits its function by inducing or repressing the expression of hundreds of genes encoding for proteins that target a relevant number of metabolic (e.g., glycolysis), morphological (e.g., angiogenesis), cell proliferation and survival (e.g., apoptosis, cell cycle control), and molecular (e.g., production of erythropoietin) processes.
HIF activity is only in small part regulated by gene expression, with the bulk of the control entrusted to prolyl hydroxylases (PHD) and the factor inhibiting HIF (FIH), which, in the presence of O2, hydroxylate HIF at either the two prolyl or the asparaginyl residues, respectively [29]. When hydroxylated, HIF undergoes ubiquitination and cannot be active in its task of controlling the gene expression. While the β subunit that composes HIF is constitutive and does not respond to O2, at least three isoforms of the O2-sensitive α subunit have been identified. The best known of them, HIF-1α and HIF-2α, share 48% sequence homology but are differentially expressed in various tissues. For example, HIF-2α, also known as the endothelial PAS domain protein 1 (EPAS-1), is principally expressed in endothelial cells [30], brain, heart, lung, kidney, liver, pancreas, and intestine tissues [31]. HIF-2α regulates fewer genes than HIF-1α yet plays an important role in erythropoiesis [32,33]. Whereas HIF-1α appears to be more expressed in response to short periods of intense hypoxia, HIF-2α seems more active during prolonged mild hypoxia [34].
The regulation of HIF activity as a function of O2 availability relies on the sensitivity of PHD and FIH to PO2 changes, which is expressed by the value of the Michaelis–Menten constant (KM), or the PO2 at which the reaction rate is half-maximal. Given the average intracellular PO2 in the 7–20 mmHg/1.0–2.5 %O2 range, FIH (KM = 50–80 mmHg/6.5–10.5 %O2) appears dynamically more effective than PHD (KM = 120–210 mmHg/15.7–27.6 %O2) as a physiological O2 sensor [29,35,36]. However, KM values are to be taken with caution because they are markedly affected by the length of the substrate peptides necessary to perform the measurement. In addition, the slope of the curve representing HIF activity vs. PO2 needs to be assessed to fully appreciate the role of HIF in hypoxia adaptation, but no data are yet available on this key information.
Understanding the rules of the O2-sensing mechanisms of HIF and appreciating HIF as a key factor for hypoxia adaptation requires not only the availability of kinetic data addressing the O2 affinity of PHD and FIH, but also HIF stability in prolonged and chronic hypoxia. In vitro experiments, mostly performed in cultured tumor cells because the problem of prolonged hypoxia is particularly compelling in cancer growth contexts, may help address this issue in part, but data are quite contradictory. In neuroblastoma cells, both HIF-1α and HIF-2α proteins become destabilized after >72 h hypoxia [37]. It was proposed that during cancer development, acute hypoxia first stabilizes HIF-1α, which decreases at about 24 h and is replaced by HIF-2α, which in turn slowly decreases and is replaced by other hypoxia-associated factors [38]. One study followed human microvascular endothelial cells during 14-day hypoxia, highlighting the messenger RNA (mRNA) expression response associated with HIF-2α as more prolonged than that associated with HIF-1α, but HIF protein data, which are more suitable to address the role of HIF in hypoxia adaptation, are lacking [39]. Acute and chronic hypoxia can lead to completely different hypoxia-related responses. While acute hypoxia affected the expression of 196 genes, chronic hypoxia resulted in changes in 4149 transcripts, with only 144 genes common to both [40]. Such marked changes in the transcriptome, potentially able to address the role of HIF in hypoxia adaptation, may stem from the activation of different downstream transcriptional effects originating from the various HIF isoforms [41]. In facts, the mRNA expression of the HIF isoform HIF-3α during 48 h hypoxia revealed that while the HIF-1α mRNA peaked at 4 h, HIF-3α mRNA expression increased on HIF-1α decrease [42].
Spontaneous PO2 fluctuations in tumors in vivo further complicate assessing the role of HIF in prolonged hypoxia, and thus its involvement in hypoxia adaptation, also due to the great heterogeneity in experimental protocols for hypoxia levels and exposure times, which may range from 30 min to several weeks [43]. In healthy rats exposed to 5900 m/10.0 %O2 chronic hypoxia without reoxygenation for 14 days, HIF-1α protein increased in the brain [44,45], muscle [46], and heart [47], but heart mRNA was unaffected [48].
In summary, as HIFs may regulate the expression of several hundred genes, perhaps up to 2.6% of the human genome [49], it remains difficult at present to assess if all such activities translate to useful hypoxia adaptation patterns, leaving the question whether HIFs participate to the adaptation processes relatively unanswered.
3.1.2. Antioxidant Defense
Besides fueling oxidative phosphorylation, O2 may also form highly reactive species (ROS) either through non-enzymatic (e.g., mitochondrial uncoupling) or enzymatic [50] processes. When elevated, ROS are toxic due to their high reactivity, but at physiological levels they modulate a wide range of pathways that control virtually all cell functions. ROS are directly linked to the genesis of oxidative stress, which is the outcome of the imbalance between aggressive species formation and antioxidant defenses.
Mitochondrial uncoupling is the main, but not exclusive, way through which hypoxia causes ROS formation [51]. The crosstalk between dysfunctional mitochondria and NADPH oxidase in the plasma membrane [52] engages this enzyme as an additional ROS source [53]. NADPH oxidase 4 responds directly to the O2 lack [54], thus constituting a major ROS source in hypoxia [55,56], and nearly doubles in the brains of mice exposed to 5900 m/10.0 %O2 for 4 weeks [57]. NADPH oxidase 4 is antagonized by the nuclear factor erythroid 2-related factor 2 (Nrf2), which in rat brains responds to 12 h hypoxia [58] and upregulates the antioxidant defenses [59]. Another protective factor, protein kinase B or Akt, is a serine/threonine-specific protein kinase that plays a key role in neuroprotection by inhibiting apoptosis [60,61] and revealed protective features even in chronic situations, possibly overriding the protection elicited by Nrf2 [57]. Mild 5900 m/10.0 %O2 hypoxia increases both ROS production and defense mechanisms, but since the first is stronger, at least for 4-week hypoxia, the redox imbalance results worsened with the onset of clear signs of oxidative stress [57].
The antioxidant glutathione, a tripeptide thiol that participates to the cell defense against ROS, may also play a relevant role. The ratio of oxidized/reduced glutathione (GSSG/GSH) reflects the cell redox status [62], and the high vulnerability of neurons to ROS may be linked to the relatively low activity of glutathione peroxidase in the brain [63]. As breathing at 8500 m/7.0 %O2 for 6 h reduces GSH and glutathione peroxidase activity in brain extracts, hypoxia clearly impairs the antioxidant defense constituted by glutathione [64].
Exposure to altitude is widely known to enhance ROS generation that overwhelms the cell defenses and leads to lipid, protein, and DNA damage, thereby causing or exacerbating the outcome of several diseases including CMS [65]. The adaption to the oxidative challenge requires relatively long periods of time, with physical exercise usually exacerbating the severity of this challenge [66]. In fact, in humans exposed to 3800 m/13.2 %O2 the indirect markers of oxidative stress peak by the 20th day of hypoxia, followed by a sustained increase for 10 months [67].
Neurons and the brain are highly vulnerable to ROS [68,69] because of high VO2 and low antioxidant defense [70]. Consequently, one of the most remarkable side effects of hypoxia-induced oxidative stress is the frequently observed phenomena related to memory and cognitive impairment. This outcome was observed not only in neurological studies [71], but even in animal models [72]. Thus, the cognitive impairment may be directly linked to the onset of oxidative stress caused by insufficient antioxidant buildup as the increased ROS levels in rats exposed to 6100 m/9.8 %O2 for 7–14 days correlated with nNOS expression, neurodegeneration, and DNA fragmentation in the hippocampus, cortex, and striatum [73]. Likewise, 2-week 5900 m/10.0 %O2 hypoxia was reflected in marked neuronal apoptosis, a hallmark of overt brain damage [44,45].
In summary, there are two aspects to address the ox–redox challenge: the increased hypoxia-induced oxidative stress and the buildup of endogenous antioxidant defenses, which might not be fully exploited, especially for longer hypoxia durations. In terms of hypoxia adaptation, it might appear that the potentially adaptive response represented by the antioxidant defenses is insufficient to meet its target.
3.1.3. Bioenergetics
The great majority, perhaps 97%, of breathed O2 is employed in the final steps of long chains of reactions that oxidize substrates, mainly glucose, fatty acids, and in certain organs, ketone bodies (Figure 5). The O2 vehiculated in the cell, either through simple diffusion or, in contractile cells, via binding with myoglobin (Mb), is finally used up by cytochrome oxidases to yield biological energy in the form of adenosine triphosphate (ATP). We neglect here the contribution of alactic anaerobic pathways, i.e., the biological energy derived from the phosphocreatine shuttle and the adenylate kinase reaction, which provide ATP only for limited periods of time. Clearly, any change in mitochondria morphology and density is unavoidably reflected in changes in the bioenergetic paths.
The effect of hypoxia on the mitochondrial volume and density in skeletal muscle is controversial, mainly due to the presence of two superimposing effects: exercise training, which is expected to increase mitochondrial volume, and hypoxia, which instead antagonizes the effects of training with respect to mitochondria [74]. Clearly, variables linked to hypoxia duration and intensity and to the degree of exercise come into play [75]. However, animal studies have shown that the activity of muscle cytochrome oxidase falls markedly in both chronic and acute hypoxia, which depresses energy production by the mitochondria [76]. In humans, in situations not disturbed by the acute onset of exercise programs, lower muscle mitochondrial contents have been reported in Sherpas [77] and lowland-dwelling Tibetans [78] compared with lowlander populations. Furthermore, the capacity for fatty acid oxidation is reduced in altitude Sherpas, due to downregulated muscle expression of the gene that encodes peroxisome proliferator-activated receptor alpha, a transcriptional regulator of fatty acid metabolism [79]. Decreased mitochondrial volume with a loss in oxidative capacity is indeed a constant finding in physiological hypoxia studies related to sea-level individuals brought to altitude [80].
With impairment of mitochondrial function, glycolysis should surge as pivotal to supplying anaerobic ATP to the cell, the so-called glycolytic switch or Warburg effect. Originally identified in hypoxic cancer cells, the Warburg effect describes the increasing reliance of the cell metabolism on glycolysis to spare the mitochondrial function [81]. The discovery that the Warburg effect is activated by HIF-1α [82] provides the structural basis to identify the glycolytic switch as a response to hypoxia. Although less efficient in building biological energy, glycolysis may nevertheless supply ATP, and human red blood cells (RBCs) rely entirely on glycolysis to produce energy. As well, strongly exercising muscles occasionally rely on anerobic mechanisms, although with lower energy conversion efficiency.
Glycolysis releases lactate and H+. Although high H+ produces reversible effects in isolated perfused hearts, high lactate causes irreversible damage the O2 utilization paths by at least two mechanisms: (a) impairment of phosphorylation coupling efficiency due to the dissipation of the H+ gradient across the inner mitochondrial membrane via upregulation of lactate–H+ cotransport, and (b) lactate-induced increased amplitude of the Ca++ transients, which leads to an increased intracellular Ca++ load with higher costs required for pumping out Ca++ [83]. The limiting factors for lactate exportation out of the cell will be discussed below. Exported lactate is then re-converted to glucose by liver gluconeogenesis at the cost of 6 ATP/mole, while converting glucose to lactate in the glycolysis yields only 2 ATP/mole. Actually, however, the liver ATP necessary for this process, or the Cori cycle, which compensates for the side-products of anaerobic mechanisms in peripheral hypoxic cells, is produced by aerobic mechanisms, which addresses O2 as a non-replaceable substrate for life. The intracellular lactate buildup consequent to the Warburg effect acidifies the cell milieu to a much larger extent than CO2, the product of oxidative phosphorylation. Increased cell acidity conveys side effects that are usually considered noxious. The Warburg effect is commonly observed in carcinogenesis, as wells as in hypoxic lungs in all their components, i.e., smooth muscle cells, endothelial cells, and fibroblasts [84]. Studies of human PH [85] as well as a meta-analysis of PH experimental models [86] confirmed that the changes in substrate utilization in the right ventricle that correlate with disease severity are compatible with the insurgence of the Warburg effect.
Recently, the favorable effect of erythropoietin (EPO) on mitochondrial functionality was examined in both in vivo and in vitro models of Parkinson’s disease, in which augmented oxidative stress leads to mitochondrial disfunction, neuronal apoptosis, and cell toxicity [87,88]. In these models, the administration of EPO could not rescue the mitochondrial functionality, yet it accelerated the glycolytic rate, contributing to the partial restoration of the ATP levels, i.e., in a way similar to that exerted by hypoxia adaptation [89]. It is, however, a matter of fact that the EPO levels usually peak a few days after the onset of hypoxia but gradually return to baseline in the following weeks, rendering it difficult to identify a role of EPO in protecting the mitochondria from prolonged hypoxia and hypoxia adaptation.
In summary, the bioenergetic changes associated with hypoxia, the logical consequences of the aerobic-to-anaerobic switch, may favor hypoxia adaptation, but the unavailability of methods to directly measure the O2 delivery in vivo at the mitochondrial level without the use of indirect mathematical calculations prevents an appreciation of how the bioenergetic changes may impact hypoxia adaptation. In addition, the role of pleiotropic factors that may modulate aerobic and anaerobic energy production, such as EPO, although already hypothesized [90], is still to be worked out.
3.1.4. Lactate Shuttle
An outcome of the Warburg effect, the aerobic-to-anaerobic switch may become costly for cells not only for the impaired energy production, but also because of lactate overproduction. Lactate toxicity may be contrasted, at least in part, by favoring the export of lactate out of the cell. The lactate shuttle hypothesis covers the intracellular and cell–cell movements of lactate produced by glycolysis [91]. While often perceived as a toxic product of anoxic cell metabolism and a determinant of muscle fatigue, lactate mediates several processes spanning from wound repair and regeneration to the mechanisms related to astrocyte–neuron synergy, the lactate–alanine conversion into glucose in the liver via the Cori cycle, and the peroxisome and spermatogenic functions [92].
A key factor in the lactate shuttle is the monocarboxylate transporter (MCT), a ubiquitously expressed family of at least 14 membrane transporters that carry lactate, pyruvate, and ketones across the cell membrane [93]. The most-studied MCT isoforms possess different KM values for lactate [94]: while MCT2 has a high affinity for lactate (KM = 0.5–0.75 mM), MCT1 has a lower affinity (KM = 3.5–10 mM) [95]. MCT4, initially believed to be a low-affinity transporter, was revealed instead to be a high-affinity lactate–H+ symport capable of exporting lactate against significant gradients [96].
The role of MCTs in hypoxia stems principally from the observation that high expression of MCTs correlates with tumor aggressiveness and malignancy [97]. In triple-negative breast cancer, increased expression of MCT4 correlates with the clinical outcome [98], while upregulation of MCT1 supports the glycolytic phenotype of glioblastomas [99]. Both MCT1 and MCT4 are critical for the growth of glycolytic tumors [100] and even MCT2 was found to respond to microenvironmental hypoxia and acidosis to maintain a high glycolytic flux in glioblastomas [101]. Such observations addressed the rationale of targeting MCT-mediated lactate transport for the treatment of some types of cancer [102]. Indeed, augmented intracellular acidosis due to MCT4 inhibition with depressed lactate export decreased cancer cell survival [103]. Likewise, targeting MCT1 in endothelial cells inhibited tumor angiogenesis secondary to lactate-induced HIF-1 activation [104]. By binding and stabilizing a protein that triggers signals for cell growth and angiogenesis, lactate may promote a hypoxic response independently of HIF [105]. However, MCTs are also clinically relevant in diseases involving immunosuppression, inflammation, insulin resistance, and brain function. Almost all MCTs are expressed in red gastrocnemius muscle at the mRNA level and their expression is regulated differently under hypoxia preconditioning and exercise conditions [106]. Prolonged hypoxia induces MCT4 expression in mesenchymal stem cells, which results in a secretome that is deleterious to cardiovascular repair [107].
It may be intuitive that the expression of MCTs and lactate dehydrogenase would increase in physiological hypoxia to facilitate the disposal of glycolytic lactate, thereby preventing acidification of the cell milieu. In rats exposed to 4500 m/12.1 %O2 for 8 weeks, the response was, however, extremely tissue-specific: MCT4 increased by 34% in hearts but decreased by 47% in plantaris muscle, while MCT1 decreased in plantaris muscle, indicating a minor role for MCTs in hypoxic contractile tissues [108]. By contrast, 48 h hypoxia increased MCT1 mRNA and protein (×8.5 and ×2.7, respectively), as well as MCT4 mRNA (×14.3), but not the protein in human adipocytes in culture [109]. Thoroughbred horses trained for 2 weeks under mild hypoxia (1390 m/18.0 %O2) displayed increased MCT4 protein levels and phosphofructokinase activity, vs. constant MCT1 [110]. Horses subjected to more severe hypoxia (3000 m/14.7 %O2) did not show changes in MCT1 protein, but MCT4 protein increased by 13%, which highlights that hypoxia may improve exercise performance and glycolytic capacity of skeletal muscle at least in horses [111]. Finally, in rat soleus and EDL muscles, high-intensity interval training, a model of simulated hypoxia, elevated the mRNA levels of MCT4, PGC-1α, and HIF-1α, but no protein data are available [112]. Such data may thus indicate that MCT1 is insensitive to O2 changes, while MCT4 is dependent on O2 levels.
In summary, in the absence of targeted studies addressing the role of MCT to favor lactate export in somatic cells, there is little support for a relevant role for MCT in hypoxia adaptation.
3.2. Capillary-to-Cell Interface
3.2.1. Nitric Oxide
Despite its short half-life in the circulation of a few milliseconds [113,114], NO, a critical mediator of the circulatory responses to hypoxia, explicates its action through vasodilation, which rescues the O2 supply/demand ratio that is disrupted by hypoxia, especially in acute scenarios [115]. NO acts as a master regulator of the vascular tone and blood pressure [116]. Other important NO functions are also mediated by nitrosylation of proteins such as HIF-1α, which is then stabilized not only by hypoxia but also by NO-dependent S-nitrosylation [117]. A small free radical, NO is produced in the NO synthase (NOS) reaction, with O2 being one of the substrates:
2 L-Arginine + 3 NADPH + 3 H+ + 4 O2 → 2 citrulline + 2 NO + 4 H2O + 3 NADP+
This reaction may be catalyzed by at least three distinct NOS isoforms, each coded in different chromosomes: (1) the Ca++-sensitive, constitutive neuronal NOS (nNOS), located mainly in the nervous tissue and in smooth and skeletal muscles, produces the bulk of the NO that acts as a neurotransmitter; (2) the Ca++-insensitive, inducible NOS (iNOS), located mainly in the immune and cardiovascular systems, in vessel walls and macrophages, in response to inflammatory stimuli and hypoxia; and (3) the Ca++-sensitive, constitutive endothelial NOS (eNOS), expressed in the endothelium of virtually all organs, is subjected to genetic polymorphism [118,119] and is responsible for smooth muscle relaxation.
The regulation of NOS activity by hypoxia has the ability to fine-tune the O2 delivery to cells [120]. The KM value of NOS for O2 is the key to appreciate the O2 sensitivity of the NO-producing machinery. Briefly, the NO synthesis rate in the brain and endothelium is tightly linked to the onset of hypoxia because the KM for O2 of the overall NOS activity (3.6–14.3 mmHg/0.5–1.9 %O2 [121]) is near the expected tissue PO2, which renders the PO2 variations dynamically linked to NO production. The nNOS isoform (KM = 158 mmHg/20.8 %O2 [122]) is expected to respond to PO2 changes, but with less capacity as the microenvironmental PO2 is much less than the KM. By contrast, eNOS displays a high affinity for O2 (KM = 16.6 mmHg/2.2 %O2 and 5.5 mmHg/0.7 %O2 for brain and endothelium, respectively [121]), suggesting that eNOS operates in an environment that already saturates the enzyme, unless in extreme hypoxia conditions. Thus, in non-extreme situations, the changes in PO2 sensed by eNOS are not expected to be tightly linked to changes in NO production.
After release into the blood stream, NO establishes equilibria with at least three components (Figure 6): (1) an inorganic reservoir constituted by plasma NO2− + NO3−, (2) RBC hemoglobin (Hb) to form nitrosylated or nitrosothiolated Hb [123], and (3) soluble guanylate cyclase in smooth muscle cells, which generates cyclic guanosine monophosphate (cGMP), which induces muscle relaxation through the activation of protein kinase G and reduction in intracellular Ca++ [124].
High circulating NO levels are toxic because NO is itself a free radical and its reaction with O2 generates peroxynitrite, a most dangerous reactive nitrogen species. This results in nitrosative stress, which plays an important role in the pathology of various diseases such as heart failure [125]. On the other hand, when present at physiological or slightly supraphysiological levels, NO protects the hypoxic body in several ways.
Besides its main function as a vasorelaxant to enhance tissue perfusion and oxygenation, NO restores peripheral blood mononuclear cell adhesion, which is considerably reduced in hypoxia. This function is explicated via an interaction with the mechanisms that affect remodeling and the extracellular matrix. First, hypoxia-induced overexpression of eNOS tends to rescue basal NO levels, which recovers cell–matrix adhesion via cGMP-dependent protein kinase [126]. This favors the cell’s ability to adhere to the extracellular matrix, an essential step to maintain normal homeostasis, which is hampered by hypoxia, which reduces NO bioavailability (see below). In addition, while hypoxia-induced over-perfusion of the pulmonary bed destabilizes the endothelial cells by increasing their leakiness via depolymerization of F-actin stress fibers (which impairs the permeability of the pulmonary vascular endothelial bed, potentially leading to high-altitude pulmonary edema), activating the NO/cGMP pathway with NO donors or sildenafil can revert this process [127].
NO is tightly linked to hypoxia. In acute contexts (hours–days), hypoxia decreases pulmonary NO and causes vasoconstriction, which may ultimately lead to hypoxic pulmonary vasoconstriction [128,129,130]. In patients with high-altitude pulmonary edema, these symptoms are alleviated by inhaled NO with improvement of the arterial O2 saturation [131]. By contrast, dietary nitrate supplementation in healthy adult volunteers residing at 4559 m/12.0 %O2 for 1 week increased pulmonary NO availability, as marked by higher salivary NO and exhaled NO, but failed to improve hemodynamics, O2 saturation, and AMS development [132]. Animal models gave controversial results. On one hand, plateau pika (Ochotona curzoniae), a hypoxia-tolerant mammal that lives at 3000–5000 m/14.7–11.3 %O2 on the Qinghai-Tibet Plateau, show reduced NOx compared to lower-altitude controls, indicating that NO production is suppressed at altitude [133]. On the other hand, newborn highland lambs show enhanced NO function compared to lowland controls, corresponding to increased RhoA expression in the lungs, which suggests that NO-mediated vasodilation is important to maintain the pulmonary vascular resistance [134]. The general consensus, however, is that the NO level in lungs and plasma falls within 2 h following the hypoxia onset, after which it tends to return toward baseline levels by 48 h, followed by a further increase above baseline by 5 days [135]. Interestingly, melatonin, a pleiotropic hormone secreted in the pineal gland with antioxidant capacity that favors HIF-1α expression [136], depresses the hypoxia-induced increase in nNOS, eNOS, iNOS, and nitrotyrosine in hypoxic rats [137]. The estrogen receptors 1 and 2 seem to be associated with the activation of the NO–cGMP pathway during acute (7 days) acclimatization to 3500 m/13.8 %O2, being correlated to higher protein levels of eNOS, higher plasma NO2− + NO3−, and lower levels of endogenous eNOS, an inhibitor of asymmetric dimethylarginine [138].
As for long-term hypoxia adaptation, a review of 32 articles published before 2012 revealed that Tibetans had the highest NO levels in the lung, plasma, and RBC of any highland populations [135]. Improved NO bioavailability thus appears to favor human adaptation to altitude and underlies the Tibetans’ response to altitude hypoxia [139]. This is compatible with both the increased plasma L-arginine observed in dwellers at 3800 m/13.3 %O2 for 10 months [140] and the beneficial effects of supplementing L-arginine [141] or sildenafil [142] at altitude. Remarkably, Amhara native highlanders in Ethiopia, who do not display erythrocytosis but have normal Hb–O2 saturation, display increased NO and cGMP, suggesting an adaptive pattern via vasodilation with cerebral circulation sensitive to NO but not to hypoxia [143]. In Ladakhi highlanders residing at 3500 m, circulating estrogen receptor 2 was higher than in sea-level controls with lower testosterone and higher eNOS protein level and plasma NO2− + NO3− [144].
In summary, NO handling appears to play an important role in hypoxia adaptation, with this feature particularly relevant for very long (generations) hypoxia exposure.
3.2.2. Capillary Density
The Krogh capillary model is widely used to simulate the PO2 variation along a cylindrical capillary and to describe the O2 supply to the surrounding tissue [145]. The Krogh model helped understand the microcirculation in situations related to exercise and cerebrovascular dysfunction [146]. Despite its limitations, this model enables the appreciation that tissue oxygenation is regulated principally by capillary recruitment, with important effects of changes in the O2 gradient, i.e., hypoxia, in hematocrit and in the RBC transit time [147]. Because the hypoxia-induced reduction in the O2 gradient across the endothelium intuitively has a major role in determining O2 diffusion across the circulation-to-cell interface, and because the efficient flux of O2 from the circulation to the cell may represent a major bottleneck especially for high-O2 consumers such as muscle and brain, we will discuss separately these cases in terms of hypoxia adaptation.
As for the muscle, near-infrared spectroscopy enabled non-invasive assessment that the O2 diffusion in the muscle decreases with the hypoxia-induced fall of the O2 gradient, but this may be compensated by enhanced capillary density [148]. Therefore, augmenting the capillary density, or in the case of muscle the capillary/fiber ratio, or angio-adaptation, may help hypoxia adaptation through matching the O2 delivery to the varying metabolic needs of the myocytes [149]. In a pioneer study in guinea pigs native to the Andes, the capillary supply to skeletal muscle was 30% higher than in animals raised at sea level, highlighting that greater oxidative capacity necessitates increased capillarity, shorter diffusion distances for O2, and higher muscle myoglobin concentration [150]. Another study in Andean coot (Fulica americana peruviana) native to 4200 m/122.6 %O2 documented that, despite lower muscle fiber diameter, the number of capillaries per unit area and the capillary/fiber ratio were higher in all muscles compared with animals born at sea level [151]. Such data obtained in animals, perhaps adapted to altitude, however, were not confirmed in humans. Morphological studies in the skeletal muscle of humans exposed to 5000 m/11.3 %O2 for 8 weeks showed that the increase in the capillary/fiber ratio was not due to an increase in angiogenesis as for the maintained capillary network, but rather to the reduction in muscle cross-sectional area and muscle fiber size, both attributed to the loss of myofibrillar proteins [152]. The inadequate mammalian skeletal muscle angiogenesis observed after 8 weeks at 4500 m/12.0 %O2 was also addressed in biochemical studies that showed decreased markers of angiogenesis, e.g., vascular endothelial growth factor (VEGF), flt-1, and flk-1 mRNA [153]. The same trend was also observed in patients with chronic heart failure, where no appreciable changes in capillarity and fiber area were reported [154].
Theoretical analyses to assess whether the capillary network growth may represent an adaptive response to hypoxia in muscle predicted that increasing the capillary/fiber ratio above 2 may not be of benefit for tissue oxygenation [155]. Thus, capillary density might not appear as an adaptive response to hypoxia. This apparently counterintuitive feature could be justified by focusing on some limits of the Krogh model. Most important, the architecture of the microcirculation based on the Krogh cylinder may become insufficient to describe the O2 delivery to tissues because of the non-exclusive role of capillaries as suppliers of O2 to tissues. Indeed, the arterioles may represent a significant source of O2, as evident from data obtained with phosphorescence decay techniques for the measurement of local intra- and extravascular PO2 [156]. Findings on vascular longitudinal gradients converge in establishing that arterioles exert autocrine O2-driven effects in regulating the blood flow in the tissue [157], and that the blood viscosity, the blood O2-carrying properties, and the slope of the Hb–O2 dissociation curve are variables that should be accounted for to fully describe the O2 transport to tissues [158]. Additional variables such as local heterogeneities in RBC transit time and in hematocrit along the capillary have also been highlighted as critical to match the O2 availability to the varying metabolic demand [159]. As a matter of fact, a dramatically increased capillary density with reduced diffusion distance after short-term immobilization did not appreciably improve the O2 uptake in working skeletal muscle [146].
As for the brain, to defend its function in the face of O2 supply fluctuations, it triggers mechanisms that increase the capillary density and cerebral blood flow through vascular remodeling driven by HIF-1α overexpression with concomitant activation of downstream genes, especially VEGF [160]. Sustained hypoxia (28 days at 4500 m/12.0 %O2) causes remodeling of capillary vessels by increasing their diameter and length [161], and 28 days at 5900 m/10.0%O2 increases the vascular markers CD34 and PECAM-1 [57]. Increasing evidence points at O2-dependent regulation of the brain capillary network by neural stem cells that reside in niches within the brain, whose proliferation and multipotency is enhanced by mild hypoxia [162]. The role of HIF-1α in this mechanism is unclear, as in some studies it appears to be required [163], while in others it only facilitates the signal transduction pathways that promote self-renewal of neural stem cells with inhibition of apoptosis [164]. Alternative mechanisms may involve Wnt/β-catenin signaling [165], the calcium-regulated calcineurin-NFATc4 [166], and ROS [167].
After the initial discovery that hypoxia regulates VEGF gene expression in endothelial cells promoting mechanisms that impact the permeability and proliferation of the endothelium in an autocrine manner [168], a plethora of studies addressed the hypoxia-driven regulation of angiogenesis through an ever-expanding number of pathways in multiple cell types [169]. Today, hypoxia-derived VEGF overexpression is a globally recognized critical factor in malignant tumor expansion and vascular function [170], which is subjected to HIF regulation as an angiogenic master switch [171,172]. Indeed, of the several targets of HIF [49], the most involved here is VEGF along with several angiogenesis markers [173,174].
In summary, although changes in the capillary density do not appear pivotal in determining hypoxia adaptation in heavy-O2-consumer tissues such as muscle and brain, the scenario is different in tumor tissue, especially solid tumors, where the growth of O2-consuming malignant cells exceeds the growth of the capillary network, which finally decreases the capillary/cell ratio, inducing local hypoxia.
3.3. Arterial Blood-to-Capillary Interface
3.3.1. Erythropoiesis
EPO, a glycoprotein synthesized in the kidney interstitial fibroblasts, has long been recognized to stimulate erythropoiesis in the bone marrow [175] in response to hypoxia [176]. The discovery of the underlying mechanisms was related to the discovery of HIF, as the effects of O2 deficiency on HIF’s stabilization and those of HIF on EPO production were the key to understand the pleiotropic mechanisms of the O2 sensors [177]. Once produced, EPO binds to the EPO receptors expressed in the cells of multiple tissues, including bone marrow and neurons, that activate JAK-2, which turns on the STAT, PI3K/Akt, and Erk pathways with multiple outcomes including erythropoiesis [178].
A plethora of studies converged in pointing out that EPO acutely responds to the onset of hypoxia within a few hours, peaks in the first 1–2 days, then progressively returns to baseline values in 1–2 weeks. The EPO peak is followed by a slower weeklong increase in the RBC mass [179]. However, despite the return of EPO to baseline values within a week after the onset of hypoxia, the erythropoietic stimulus persists for at least 10 months at altitude [180]. An important player in this mechanism, the soluble form of the EPO receptor, an endogenous antagonist of EPO, was observed to mirror the EPO response to hypoxia, decreasing by 19%, remaining below baseline values for at least 72 h at 4340 m/12.4 %O2, and then slowly returning to baseline values [181]. This highlights the EPO-to-EPO receptor ratio as the key to understand the long-term relationship between EPO and the RBC mass.
Further insight into the mechanism underlying the path hypoxia→HIF→EPO→erythropoiesis comes from the comparative analysis of permanent altitude dwellers. Tibetans do not have marked erythrocytosis [182]. Accordingly, high- and low-living Tibetans display the same EPO levels [183]. Furthermore, the EPO level is low in Ladakhi highlanders, who are ethnically linked to Tibetans [184]. Likewise, Ethiopian Amharas, who live at comparable altitudes, have no erythrocytosis compared to their sea level counterparts [185]. By contrast, Andean altitude dwellers are well-known to have excessive erythrocytosis [186], which in most cases foreplays the onset of CMS. One would expect that their EPO level would be higher, but this is not the case. Indeed, in South American dwellers at 4340 m/12.0 %O2, circulating EPO does not appear as a discriminating factor for CMS, but the EPO-to-EPO receptor ratio may be the factor that determines excessive erythropoiesis [187]. Likewise, the EPO level was only marginally higher in Andean highlanders with high and low Hb [188].
Augmenting the production of RBCs is perhaps the first physiological response ever recognized to participate in hypoxia adaptation [189]. This response, which was later demonstrated to be mediated by the hyperproduction of EPO [190], was considered adaptive because it translates into a proportionally higher O2 carriage to tissues [191]. The main reason for the injurious effects of polycythemia on blood O2 transport resides in the increased shear rate due to high blood viscosity, which critically determines local blood flow [192]. Along with factors such as fibrinogen, RBC deformability, and aggregation, the hematocrit is indeed the single most important determinant of blood viscosity [193]. In healthy humans, hematocrit variation within the normal 33–45% range results in modest alterations in cerebral blood flow, but beyond this range, hematocrit correlates with decreased blood flow [194].
In summary, the observation that adapted populations exhibit a blunted erythropoietic response, in contrast with the exaggerated response often associated with the adverse clinical features observed in non-adapted populations, suggests that erythropoiesis might not be an adaptative response to hypoxia.
3.3.2. Iron Handling
Augmented erythropoiesis inevitably leads to iron stress. As shown in Figure 7, Hb synthesis implies the intervention of at least two factors: the stimulus driven by hypoxia and EPO discussed above, and the supply of iron, which must thus be linked to the intensity/duration of hypoxia [195]. In physiological hypoxia, the greater need for iron to build up Hb is met through increased absorption from the gut or from the liver, which acts as an iron storage organ. The main iron transporter across the basolateral membrane of enterocytes and hepatocytes, ferroportin, is under tight regulation by hepcidin, which internalizes ferroportin, decreasing the capacity to export iron into the circulation [196,197]. During acute hypoxia (days–weeks), hypoxia downregulates hepcidin, thereby favoring the entry of iron into the bloodstream and hence erythropoiesis [198].
In pathologic hypoxia, however, another scenario occurs. COPD is a disease in which airflow obstruction causes a significant decrease in endothelial function over time and hypoxia [199]. While 40–50% of COPD patients undergo physiological erythropoietic adaptation to hypoxia, 5–30% of them instead develop iron deficiency with anemia [200,201]. Remarkably, anemia surges as a predictive risk factor for worse outcomes [200,202,203]. In ARDS, which is often characterized by severe hypoxia [204], the development of anemia in patients represents a recognized comorbidity. This divergent response to physiological and pathological hypoxia has been attributed to different regulation of hepcidin driven by the redox imbalance, which is higher in ARDS and COPD patients and represents an additional factor that regulates erythropoiesis in the presence of strong inflammatory stimuli, such as the level of C-reactive protein [205]. Remarkably, patients affected by COVID-19, especially the first waves, were often characterized by severe hypoxia as in ARDS patients. It is debated whether COVID-19 resembles ARDS in respiratory failure development, lung compliance, and endothelial inflammation [206], but the redox imbalance and the cytokine storm seem to be even higher in COVID-19 than in ARDS [205]. Sphingolipid synthesis is selectively stimulated by iron-driven inflammation [207], especially sphingosine-1-phosphate, a mediator in COVID-19 syndrome [208].
In summary, the iron-handling response to physiological hypoxia, at least in the absence of inflammatory factors, increases erythropoiesis, which may not be seen as a favorable factor. Therefore, in physiological hypoxia the downregulation of hepcidin may be interpreted as maladaptive.
3.3.3. Hemoglobin Oxygen Affinity
The O2-carrying capacity of blood depends mainly on the blood Hb concentration (discussed elsewhere) and the Hb–O2 affinity, which determines the Hb–O2 saturation at any given PO2 as a function of pH, PCO2, the intraerythrocytic level of 2,3-diphosphoglycerate (DPG, a glycolytic intermediate that binds to the T-form of Hb and acts as an effector of the Hb function), temperature, and the level of carbon-monoxide-bound Hb [209]. The P50, i.e., the PO2 at 50% Hb–O2 saturation, is a useful index of the Hb–O2 affinity. Diminished Hb–O2 affinity, or higher P50, is believed to be favorable at altitudes ranging from sea level to moderate because it augments the arteriovenous O2 difference by decreasing the venous Hb–O2 saturation [210]. From Fick’s equation, which relates VO2 to the cardiac output and the arteriovenous O2 content difference, it can be argued that a lower Hb–O2 affinity or higher P50 may translate either to a better tissue oxygenation (venous PO2 = +3.2 mmHg for a P50 change of 1 mmHg) or to a lower circulatory load (cardiac output = −5.8% for a P50 change of 1 mmHg). By contrast, at extreme altitudes, the O2 delivery to tissues is favored by a high Hb–O2 affinity, or lower P50, because it compensates the compromised arterial Hb saturation [211]. An arterial pH shift of 0.1 units increases the Hb–O2 saturation by up to 5% at 6450 m [212]. It is thus expected that, at moderate altitude, alkalosis, which increases the Hb–O2 affinity and whose genesis will be discussed below, is deleterious because it lowers the P50, while increased DPG is instead favorable for the opposite reason. As shown in Table 2, the changes in the two factors that were measured up to 6450 m altitude appear to offset each other almost perfectly, leaving the P50 value nearly unchanged, in agreement with early hypotheses [213].
It should, however, be highlighted that two seldom-analyzed factors must be accounted for a full appreciation of the relevance of the blood O2 transport picture at altitude. First, the fluctuations in temperature, which may be non-trivial especially in extreme environments: a change of 1 °C shifts the P50 by about 1.5 mmHg [214], a relevant value with respect to those indicated in Table 2. Second, the altitude-induced erythropoietic stimulus admits in the circulation an increasing fraction of newly synthesized, “young” RBCs [215]. When RBCs emerge from the bone marrow, they have a higher DPG/Hb ratio than the “old” ones (0.96 ± 0.13 vs. 0.57 ± 0.13 mole/mole) [216], which corresponds to a P50 change from 29.1 to 24.8, i.e., a 4.3 mmHg change, which is much higher than those reported in Table 2. Thus, in circulating blood with a normal average DPG/Hb, the RBC distribution in terms of O2 affinity may be very wide. Unfortunately, it is not possible to predict which type of RBC actively participates in the O2 exchange in the capillary or is preferentially diverted to the arteriovenous shunt. However, if flexible RBCs are predicted to undergo the tortuous path along the capillary, then it is likely that “young” RBCs with a lower mean corpuscular Hb concentration and lower O2 affinity because of their higher DPG/Hb ratio [215] are those that actively release O2 with respect to old, sticky (due to their higher mean corpuscular Hb concentration), RBCs with a low DPG/Hb ratio and high O2 affinity. We recognize that the lack of any data in this respect may render this discussion highly speculative.
In summary, although the changes in Hb–O2 affinity may in principle participate in hypoxia adaptation, it appears that they, if any, are very small and may not be considered pivotal to postulate a relevant role during hypoxia adaptation.
3.4. The Alveoli-to-Arterial Blood Interface and the Circulatory Responses
3.4.1. Cardiac Output
The responses to acute hypoxia encompass increased cardiac output in an attempt to deliver more O2 to tissues by a mechanism that occurs mainly through a compensatory increase in heart rate [217]. At the base of this response, the adrenergic G protein system emerges, as evident from elevated levels of catecholamines and adrenergic fiber activity [218]. The role of stroke volume alterations in modifying high-altitude changes in cardiac output is controversial, as most studies did not observe relevant changes in stroke volume after acute exposure to hypoxia [219]. Another mechanism behind cardiac output, the reduction in systemic vascular resistance, was recently observed in healthy individuals in hypoxic chambers simulating high-altitude exposure [220]. Peripheral arteriole dilatation [221] further helps matching the tissue O2 demand/supply ratio [222]. Despite such a mechanism, a decreased stroke volume at high altitude was observed in several studies. Impaired systolic contractile function secondary to reduced coronary PO2 has been investigated as a potential cause [223]. At the same time, as the basal cardiac output is increased, there is less chance for an exercise-related cardiac output increase in response to exercise [224].
The cardiac output, on the other hand, is not as much modified by persistent hypoxia. In the long term, the β-adrenergic receptors are downregulated [225], while muscarinic receptors are upregulated with alterations in the expression and activity of both inhibitory and stimulatory G proteins and a decreased response of the adenylate cyclase system [218]. Cardiac chronotropic function could be controlled by a local mechanism linked to myocardial PO2 [226], which leads to a decrease in compensatory tachycardia in order to reduce VO2 further.
In summary, the acutely observed changes in cardiac output, combining heart rate and stroke volume, do not appear to sustain long-term hypoxia and hypoxia adaptation.
3.4.2. The Red Blood Cell Pulmonary Capillary Transit Time
RBCs might not have enough time in the capillary to uptake all O2. The binding of Hb with O2 has a finite reaction rate, but the rate of this reaction, which is accomplished in <10 msec [227], is much faster than the diffusion rate. The limiting factors that influence the O2 diffusion in the lung, after assuming irrelevant impacts of the O2 diffusion coefficient, temperature, fluid viscosity, chemical properties of the membranes, and surface area of the pulmonary gas exchange, thus are (a) the alveolar-to-arterial PO2 difference, (b) the blood–gas barrier thickness, and (c) the RBC capillary transit time. Both acute and chronic hypoxia may have substantial effects on these factors.
The alveolar-to-arterial PO2 difference is assessed through the ventilation/perfusion (V/Q) ratio, which is known to be impaired by hypoxia, especially in acute situations. Indeed, relative hypoventilation mismatches V/Q, but this is compensated by pulmonary vasoconstriction that redirects the blood flow from poorly ventilated areas to well-ventilated areas in the lung, resulting in better oxygenation. This phenomenon is understood to be the primary active regulator of V/Q matching. A recently developed computational model summarizes ongoing research in this field, as it predicts that hypoxic pulmonary vasoconstriction matches perfusion to ventilation, homogenizes regional alveolar–capillary O2 flux, and increases O2 loading in the circulation by improving V/Q [228].
The thickness of the basal lamina, a layer of extracellular matrix formed by epithelial cells that acts as an attachment point for cells, may constitute a major resistance element in the pulmonary blood–gas barrier, representing a compromise between two needs: to provide mechanical resistance against excessive pressure and to facilitate gas diffusion across the alveolar barrier [229]. This compromise may break in pulmonary pathologies linked to hypoxia-induced pulmonary edema due to stress failure of pulmonary capillaries in PH [230] as well as in COPD [231]. Experimental data obtained in rats exposed to 5900 m/10.0 %O2 for 2 weeks show that the thickness of the basal lamina nearly doubles, but this increase is efficiently prevented by phosphodiesterase 5 inhibitors in parallel with other parameters highlighting the development of PH and right-heart failure [232]. Indeed, the HIF-1α pathway has been shown to be activated in alveolar type II cells after lung injury, promoting proliferation and spreading during repair [233].
A higher cardiac output in acute hypoxia translates into proportionally shorter RBC transit times. In normal conditions, the average transit time of RBCs in the alveolar capillaries is ∼0.75 s, despite a very wide distribution of transit times in the lungs [234]. In excised rabbit lungs, no limitation to oxygenation were observed for a sixfold increase in blood flow [235]. The competition between shortening RBC transit time and full RBC equilibration with alveolar gasses was examined [236] with the Fick diffusion equation in a simple boundary analysis to estimate the external resistance to O2 diffusion for RBCs [237]. While the rate of the Hb–O2 loading is not a critical parameter at sea level because of the steep alveolar-to-arterial PO2 difference, this may not hold true at altitude, where low alveolar O2 limits O2 diffusion and may impair the equilibration of RBCs with alveolar O2. In fact, O2-demanding conditions such as working at 3840 m/13.2 %O2 imply a lack of full equilibration due, at least in part, to shortened RBC transit times [238].
Sustained hypoxia is expected to determine pulmonary remodeling, the basis for the onset of PH [239]. This process involves the mobilization of bone-marrow-derived progenitor cells that express the transmembrane tyrosine kinase receptor c-kit [240,241]. In a model of chronic 2-week hypoxia at 10 %O2, rat lungs exhibited a tenfold increase in c-kit-positive cells, accompanied by marked vessel muscularization and redistribution of pulmonary capillary size, with a great increase in small (<50 μm) capillaries vs. an unchanged pattern of larger capillaries [242]. This pattern was observed nearly unaltered for double hypoxia times, indicating that it might be part of hypoxia adaptation [243]. However, it is not yet possible to predict the impact of varying pulmonary capillary size on the RBC transit time and the role of the alveoli-to-arterial blood interface on hypoxia adaptation.
What was discussed above for the RBC O2 loading in the lungs may also apply in reverse to the O2-unloading process in the peripheral tissues but is complicated by the relatively slow rate with respect to O2 binding by two orders of magnitude [237]. This might render the RBC O2 unloading problematic in the case of short transit times, low PO2 gradients across the endothelium, altered viscosity due to excessive erythropoiesis, Hb–O2 affinity changes, modifications in the acid–base status, and other factors. However, few data are presently available on this topic. However, an analysis showed that the diffusing O2 does not encounter resistance in the passage through the RBC membrane but rather in the unstirred layer on the RBC surface [237].
In summary, the RBC pulmonary capillary transit time, and perhaps to a minor degree the peripheral capillary transit time, may represent two important features that determine the response to acute and chronic hypoxia. However, more detailed studies on the regenerative capacity of the pulmonary capillary network are needed to understand the relevance of this phenomenon in terms of hypoxia adaptation.
3.5. The Atmosphere-to-Alveoli Interface and the Ventilatory Responses
3.5.1. Carotid Bodies
With progressive hypoxia, the ventilatory rate augments in an attempt to favor blood oxygenation in the alveoli. The carotid body (CB), a cluster of cells located in the adventitia at the fork of the common carotid artery, is the main chemoreceptor that senses the insufficiency of O2 and turns on this reflex [244]. A series of mechanisms enables the CB glomus type I cells to perform the O2-sensing function. A major one involves to the inhibition of the O2-sensitive K+ channels [245], which leads to cell depolarization, increased Ca++ entry, and the release of intracellular vesicles containing neurotransmitters such as dopamine, acetylcholine, ATP, and catecholamines [246] that increase the ventilatory rate [247]. This mechanism requires a functional electron transport chain with intact coupling between mitochondrial complexes I and IV [248]. The downregulation of mitochondrial complex IV activity increases the production of NADH and ROS, which negatively modulate the membrane ion channel activity [249,250].
Alternative mechanisms may affect the O2 sensing by the CB glomus cells as well. First is the release of gaseous messengers not stored in vesicles such as NO, carbon monoxide, and hydrogen sulfide that antagonize the response to hypoxia by inhibiting CB excitation with depression of the ventilatory responses [251]. By contrast, EPO at low concentrations (<0.5 IU/mL) agonizes the CB response to hypoxia [252]. Indeed, EPO is emerging as an effective prophylactic treatment not only for the treatment of AMS, but also for a variety of acute respiratory and non-respiratory symptoms of SARS-CoV-2 infection [253].
Clearly, the mechanisms underlying the O2-sensing activity in the CB still need more insight to justify a few recent observations. For example, there is an attenuated CB sensitivity to hypoxia in naked mole rats, which exhibit a blunted hypoxic ventilatory response despite larger CBs with more glomus cells than in mice, perhaps due to the interruption of gas-messenger signaling by heme oxygenase-1 [254]. In addition, high-altitude deer mice display attenuated ventilatory sensitivity and CB growth, perhaps associated with a genetic variation in HIF-2α [255]. Finally, the transient receptor potential ankyrin 1 is a cation channel broadly expressed in the trigeminal and vagus nerves that can detect irritants in inspired gasses and is activated by mild (4000–2500 m/13.0–15.0 %O2) but not severe (5900 m/10.0 %O2) hypoxia [256]. Intriguingly, SARS-CoV-2 CB infection could be the cause of the “silent hypoxemia” observed in COVID-19 patients, which may warrant the use of CB activators as respiratory stimulants in COVID-19 patients [257]. The mechanisms illustrated for the CB are shared in principle by other O2 sensors such as the aortic bodies and astrocytes [258].
In summary, the CB response to hypoxia is a recognized pattern with a potentially strong impact. While moderate increases in the ventilation rate might appear protective and useful to compensate the effects of hypoxia, strong increases may by contrast appear maladaptive, such as for the risk to cause hyperventilation-driven alkalosis, as discussed in the next sub-chapter.
3.5.2. Hyperventilation and Alkalosis
As a consequence of hypoxia-driven hyperventilation, arterial PCO2 decreases due to excessive CO2 washout from the lungs, causing alkalemia. Alkalosis is in part compensated by the kidney through a metabolic adjustment that establishes a negative base excess. Different patterns were observed at 6450 m/9.3 %O2 in unacclimatized Caucasians and better-acclimatized Sherpas [212]. It is hard to believe that Sherpas may ever adapt to that altitude despite their long-time, perhaps millennia-long [259], exposure to 4000–5000 m/12.9–11.3 %O2, but their degree of acclimatization to 6450 m/9.3 %O2 is almost surely better than that of Caucasians independently of the training degree. Despite the same arterial PO2 and O2 saturation (Figure 8), arterial PCO2 is lower in Caucasians than in Sherpas, perhaps due to a blunted respiratory sensitivity to hypoxia in Sherpas. An irreversible blunted respiratory drive was observed in high-altitude natives [260] and was confirmed in preterm infants at altitude [261] and in obesity hypoventilation syndrome [262] and has been linked to the development of the so-called “happy hypoxemia” [263]. Blunting the respiratory drive appears protective in Sherpas exposed at extreme altitudes because it depresses the need for excessive ventilation, which reduces alkalosis. Remarkably, the base excess was −6 mEq/L in both Caucasians and Sherpas, compared to the −10 mEq/L that would have elicited full compensation of alkalosis, highlighting that the metabolic compensation operates to the same extent for both. However, depressed ventilation in Sherpas compared to Caucasians produced a lower arterial pH. The apparently small changes in the pH values should not be misleading: while at pH 7.4 [H+] = 3.98 × 10−8, at pH 7.45 and 7.5 [H+] = 3.54 × 10−8 (−10%) and [H+] = 3.16 × 10−8 (−20%) of the initial value, respectively, which is a remarkable decrease. AMS sufferers displayed even greater alkalosis: pH 7.57–7.63, or [H+] = 2.51 × 10−8, i.e., −37% of the normal value.
Thus, correcting hypoxemia through hyperventilation might represent a costly option because of the onset of alkalosis. The effect of persistent alkalosis on brain function has not yet been dissected in detail, but it may pave the road to at least two undesirable events associated with high plasma pH: (a) promoted binding of Ca++ to albumin, which reduces blood ionized Ca++ at constant total calcium levels and keeps neurons and muscles in an irritable state (tetany); and (b) vasoconstriction of the brain’s blood vessels [265], which impairs brain function. Direct data on cognitive impairment at altitude is scarce but two meta-analyses revealed negative effects of hypoxia on cognition [266,267], a finding further confirmed in lowlanders exposed for 3 days at 3269 m [268]. The effect of hypoxia on cognitive function in humans residing at altitude is now recognized [269,270,271,272], and O2 supplementation in high-altitude schools has been recommended to prevent the impairment of learning processes [24].
In summary, hyperventilation appears as an excessive response to hypoxia in consideration of CO2 depletion with alkalosis, and may conflict with wellness, which fits the definition of maladaptation.
4. Adaptation vs. Maladaptation
4.1. Ability of Sea Level Populations to Adapt to High Altitude
Most literature data are obtained in the days or weeks immediately following the onset of hypoxia, where the signs of maladaptation are addressed in terms of AMS insurgence. Relatively little data are available to document the effects of longer (months, years) exposures, which may give clues for the ability of low-altitude dwellers to adapt to hypoxia.
The sojourners in the Antarctica plateau are fit and healthy individuals who are specifically trained to sojourn for up to 10 months at an equivalent altitude of 3800 m/13.2 %O2 in an environment that excludes the presence of disturbing variables related to cold and changes in altitude, with the only probable exception the rupture of circadian rhythms [180]. In these subjects, the expected changes in the acid–base status are maintained for 10 months without any appreciable modification [180]. Remarkably, even the metabolome changes remain unaffected for that period of time, with the exception of a slow return toward baseline of the non-polar metabolome after 6 months of hypoxia [140]. Likewise, the variables linked to the redox imbalance, including ROS, oxidative stress biomarkers, NO, and proinflammatory cytokines, peak by the 20th day of hypoxia but do not return to baseline levels [67]. Such data highlight a missed capacity of adaptation to hypoxia in Caucasians born at sea level, at least for one year. Longer permanence of sea-level dwellers at altitude, such as immigrant Han Chinese in Tibet, gives rise to up an to 18% incidence of CMS [273]. Thus, to the best of our knowledge, available data indicate that sea-level-dwellers exposed to hypoxia for years have missed the ability to adapt to this challenge.
4.2. Generations-Long Adaptation to Hypoxia
About 80 million people live permanently at >2500 m altitude in the Andes, South-East Asia, and Ethiopia [1]. These populations have developed diverging responses to altitude hypoxia. Andeans go the hematological route [274] with elevated Hb that enables carrying more O2 in blood, but high CMS incidence, which occurs in 16% of the adult male population, with increasing prevalence with age, rising up to 30% by the fifth decade of age [275], highlighting poor adaptation to hypoxia. Tibetans go the respiratory route [274]: they inhale more air with each breath and breathe more rapidly with high plasma NO2− + NO3−, a biomarker of the NO-storing capacity. This results in sporadic CMS in altitude-native Tibetans [273,276]. Ethiopians Amhara highlanders display lower CMS rates [277], reduced erythropoiesis, higher plasma NO2− + NO3− and cGMP, and lower diastolic blood pressure. These features translate into a marked vasodilatory response to hypoxia with respect to related lowlanders at altitude, who instead display an elevated erythropoietic response [278]. Although not properly an altitude population, the Kyrghyz commuters are of interest because 14–20% of them show signs of altitude PH and are characterized by a higher-than-normal fraction of hyper-responders to acute hypoxia [279]. A case–control study identified in healthy Kyrghyz highlanders the presence of genetic traits that discriminate hyper-responders to hypoxia who develop PH [280]. It was pointed out [280] that Kyrghyz commuters have unique patterns with respect to other altitude populations because they do not present other features linked to chronic hypoxia besides PH, unlike the Andean population, where polycythemia may have confounding characteristics, or Tibetans, where PH is practically absent.
It is therefore tempting to state that high-altitude populations born and residing in the Himalayas (Tibetans, Sherpas, and to a lesser extent Ladakhis) are “adapted” to altitude because they suffer altitude-related diseases to a lesser extent and display a reduced erythropoietic response. By contrast, Andean populations (Aymaras and Quechuas) reveal an exaggerated erythropoietic response with hematocrit values exceeding 50% and an increased risk of dysfunction due to high blood viscosity [259,281,282]. A hypothesis explaining the Tibetan–Andean differences is the longer altitude residence for Tibetans (about 30,000 years), with enough time to adapt genetically, than for Andeans (about 10,000 years) [283]. These figures are, however, complicated by the relatively recent massive migration into the Andes in the XVIIth century, as opposed to the relatively conserved population distribution in the Himalayas.
4.3. Ability of High-Altitude Populations to Adapt to Sea Level
Although uncommon, in some instances altitude-adapted subjects are forced, mostly for political reasons, to flee their native habitats in search of more suitable environments, often at lower altitudes. Several thousand Tibetan refugees born at altitude but residing at sea level provide the unique opportunity to test the reversibility of the processes that may have driven altitude adaptation. In this instance, the hypoxic stimulus is broken, and the subjects are exposed to a condition of relative hyperoxia. Figure 9 (Samaja et al., unpublished observations) shows the blood Hb concentration in individuals born in Tibet who have fled to <500 m/>20.1 %O2, and in Indian-ancestry residents in the same area as a control. Clearly, missing the hypoxic stimulus remarkably depresses erythropoiesis, at least in males, who face the risk of becoming anemic. Another large-scale study revealed a lower RBC count, hematocrit, and Hb levels in Tibetans living long-term at low altitude compared to their high-altitude counterparts [183]. In a further study, it was found that the Hb concentration was lower in Tibetans living at sea level than in Han Chinese individuals, along with a higher minute ventilation and blunted pulmonary vascular responses to acute (minutes) and sustained (8 h) hypoxia [284]. The same study also shows a lower hypoxic induction of HIF-regulated genes in peripheral blood lymphocytes as well as a significant correlation between EPAS1 and EGLN1 genotypes and induction of EPO by hypoxia in Tibetans compared with Han Chinese, highlighting the less-vigorous response to hypoxic challenge [284]. This evidence converges in indicating the occurrence of outcomes compatible with a response to a relatively hyperoxic environment in subjects adapted to live at altitude. It remains to be established if such a hematological response to relative hyperoxia is harmful.
5. A Glance to the Future: The -Omics
5.1. Genomics
Marked discordant responses to altitude provide a unique opportunity for genome-wide association studies (GWAS) to identify the gene patterns that determine different responses to hypoxia across the world. More than 1000 genes appear to be potentially involved in the response to hypoxia in the three altitude populations [285]. These genes are related to the development of the nervous, lymphoid, and cardiovascular systems, as well as with cell growth, survival, and death, with only four genes (PIK3CB, HLA-DQB1, CNTNAP2, and DLG2) commonly altered in all the populations [285]. Such findings highlight that hypoxia adaptation is polygenic and impacts many biochemical paths, each of which in turn affects many physiological responses. Despite substantial convergence on a few physiological mechanisms (RBC homeostasis, O2 transport, circulatory adjustments, and O2 sensing) as key paths that affect hypoxia adaptation, other GWAS could not distinguish the roles played by certain genes believed to be associated with excessive erythropoiesis [286]. This emphasizes a major limitation of GWAS, i.e., the presence in the analyzed populations of confounding factors such as population migrations, geographical differences, and diet restrictions that may introduce disturbing signals not related to hypoxia.
In a powerful GWAS performed on >3000 Tibetans and >7000 non-Tibetan individuals of Eastern Asian ancestry, signals of hypoxia adaptation were detected at nine genomic loci, one of which corresponds to EPAS1 [287]. Another GWAS of high-altitude populations identified the O2-sensing machinery as responsible for the phenotypic adjustments leading to adaptation [288]. EGLN1 and EPAS1 play an important role in determining the variability in the response to hypobaric hypoxia at altitude and have been linked to the development of polycythemia and associated abnormalities [289]. Overall, it seems that when gene expression overresponds to the hypoxic challenge, maladaptive patterns occur. For example, exaggerated stimulation of the O2-sensing system leads to HIF overexpression, which in turn leads to excessive erythropoiesis and CMS-related pathology. Remarkably, suppression of HIF-2α in North American deer mice exposed to chronic hypoxia (4 weeks at arterial PO2 = 90 mmHg, or 4630 m) attenuates carotid body growth and glomus cell hyperplasia without effects on hematology [255].
5.2. Metabolomics and Amino Acids
Amino acids are not only simple building blocks for proteins but may also have relevant functions as carriers of signals. Despite the scarcity of studies correlating hypoxia with amino acids, they may be highly involved in the responses to hypoxia and perhaps adaptation. Undoubtedly, the availability of -omics techniques may enable us in the future to take a deeper look at the potential role played by amino acids during hypoxia adaptation.
A metabolomic study performed in healthy lowlanders exposed for up to 10 months to 3800 m/13.2 %O2 altitude revealed a modest decrease in major lipids, but also marked alterations in amino acid metabolism, with increases in plasma arginine, glutamine, phenylalanine, tryptophan, and tyrosine [140]. Similar increases in plasma tryptophan and serotonin were also found in acutely hypoxic (19 days up to 6885 m/8.8 %O2) subjects [290]. Much work demonstrates higher plasma levels of some, but not all amino acids in the plasma of humans exposed to prolonged physiological and pathological hypoxia. This may be attributed to hypoxia-linked protein catabolism [291], but as the pool of proteogenic amino acids is not altered by hypoxia nor varied with time [140], it suggests insufficient hypoxia-induced protein catabolism, and the above changes in plasma amino acids indicate the occurrence of pathways that involve amino acids not as building blocks of proteins but as messengers.
Among the paths involving amino acids, the glutamine/glutamate balance is relevant for its implications in regulating the brain cognitive function and the ventilatory rate. Indeed, low glutamate stimulates ventilation to compensate lower O2 saturation [292], and high glutamine may affect the brain cognitive function by disrupting the ammonia vs. glutamate balance [293]. Such a disruption with decreased N-acetylputrescine, spermine, and ornithine also affects angiogenesis and reproductive physiology [294].
High levels of plasma tryptophan may reflect an impaired activity of tryptophan hydroxylase, which requires O2 and is the committing step in the synthesis of serotonin [295]. Downregulation of brain serotonin results in depression, reduced appetite, and disruption of sleep patterns, as commonly found in altitude residents [296,297]. Altered metabolisms of glutamate, threonine, taurine, lysine, arginine, and proline are frequently observed in pathological hypoxia such as in ARDS [298] and asthma patients [299]. Upregulation of amino-acid-related pathways was also observed in experimental perinatal asphyxia and neonatal hypoxia-ischemia encephalopathy [300], while in fetal growth restriction pregnancies the kynurenine pathway is enhanced [301]. This pathway is relevant because the linked nicotinamide pathway promotes pulmonary vascular remodeling [302], a common finding in chronic hypoxia [303]. Nicotinamide [304] and niacin [305] are being investigated as drugs to ameliorate the progression of hypoxia-induced pulmonary vascular remodeling.
5.3. Limitations of the Study
We omitted the issues related to biological systems in which the adaptation process does not yet emerge with clarity such as, for example, the immunological system and the microbiome. The rationale underlying the references to tumor tissue as a responder to pathological hypoxia is based on the paradigm that tumors may represent an example of successful adaptation to hypoxia, which translates into better viability and growth of tumor cells with respect to healthy cells. However, we avoided the extensive use of this paradigm in this study because this possibility has not yet been fully demonstrated and may appear speculative.
Several geographically isolated high-altitude mammals such as yaks [306,307], llamas [308], pikas [309], and many others have been considered models for the evolutionary adaptation to hypoxia. Comparison studies using such models may indeed constitute an appreciable experimental bench to test the credibility and suitability of the adaptation patterns observed in humans. However, each animal displays species-specific characteristics that would deserve separate analyses for each animal type and each genetic trait to enable meaningful comparisons with the layouts observed in humans. Therefore, despite being aware of this limitation, in this review we opted to focus on data obtained in human populations, as well as in controlled in vitro and in vivo laboratory contexts, thereby excluding valuable studies conducted on altitude-dwelling animals, especially for -omics studies.
We regret neglecting many precious contributions for reasons of space.
6. Conclusions
In the past eras, %O2 has fluctuated within the 0–30% range [310]. Although various forms of life could adapt successfully from time to time to the varying O2 level, it remains uncertain whether humans have lost the ability to adapt to lower-than-normal %O2. The analysis of the bottlenecks during the cascade of O2 from the atmosphere to the mitochondria might address the sites where the human organism succeeds or fails to adapt to hypoxia. Remarkably, many of the analyzed sites are practically superimposable to what was anticipated in an article published 70 years ago [311]. Extreme variability in targets, methods of analysis, as well as the presence of disturbing factors in the analyzed individuals or populations might have blurred to a considerable degree the judgement on how the responses at the sites along the O2 cascade concur in building up hypoxia adaptation. Some of the responses appear compatible with progressive adaptation, for example, the O2-sensing mechanism, the HIFs, the buildup of antioxidant defenses, the bioenergetic adjustment, and NO-mediated responses. By contrast, exaggerated CB sensitivity to O2 changes, with excessive hyperventilation, uncontrolled erythropoiesis, and iron handling, might appear as signs of maladaptation. Other responses, such as the changes in cardiac output and Hb–O2 affinity, appear irrelevant in terms of hypoxia adaptation. The observation that adaptation to hypoxia seems to be accomplished only after thousands of years of continuous hypoxia indicates that humans are animals not particularly suitable for living at altitude. As has already been pointed out [312], this should readdress high-altitude research, now based upon lowlanders from Western, educated, industrialized, and rich countries ascending to high altitude, toward understanding to a greater extent the principles of genes, cells, and molecular physiology in permanent high-altitude residents.
Author Contributions
Both authors participated equally to the conceptualization, literature search, methodology, writing, reviewing, and editing of this article. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
%O2, effective O2 percentage; AMS, acute mountain sickness; ARDS, acute respiratory distress syndrome; ATP, adenosine triphosphate; BP, barometric pressure; Ca++, calcium ion; CB, carotid body; cGMP, cyclic guanosine monophosphate; CMS, chronic mountain sickness; CO2, carbon dioxide; COPD, chronic obstructive pulmonary disease; COVID-19, coronavirus disease 2019; DPG, 2,3-diphosphoglycerate; EPAS1, endothelial PAS domain protein 1 or HIF-2α; EPO, erythropoietin; FIH, factor inhibiting HIF; GWAS, genome-wide association studies; H+, hydrogen ion; Hb, hemoglobin; HIF, hypoxia-inducible factor; KM, Michaelis–Menten constant; MCT, monocarboxylate transporter; MET, metabolic equivalent of task; mRNA, messenger RNA; NO, nitric oxide; NOS, NO synthase; O2, oxygen; P50, PO2 at 50% Hb–O2 saturation; PCO2, CO2 partial pressure; PDE5, phosphodiesterase 5; PH, pulmonary hypertension; PH2O, aqueous vapor pressure; PHD, prolyl hydroxylase; PO2, O2 partial pressure; RBC, erythrocyte or red blood cell; ROS, reactive O2 species; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; V/Q, ventilation/perfusion ratio; VEGF, vascular endothelial growth factor; VO2, O2 uptake.
References
- Beall, C.M. Adaptation to High Altitude: Phenotypes and Genotypes. Annu. Rev. Anthropol. 2014, 43, 251–272. [Google Scholar] [CrossRef]
- Leon-Velarde, F.; Maggiorini, M.; Reeves, J.T.; Aldashev, A.; Asmus, I.; Bernardi, L.; Ge, R.L.; Hackett, P.; Kobayashi, T.; Moore, L.G.; et al. Consensus statement on chronic and subacute high altitude diseases. High Alt. Med. Biol. 2005, 6, 147–157. [Google Scholar] [CrossRef]
- Guarascio, A.J.; Ray, S.M.; Finch, C.K.; Self, T.H. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clin. Outcomes Res. CEOR 2013, 5, 235–245. [Google Scholar] [CrossRef]
- West, J.B. Carl Wilhelm Scheele, the discoverer of oxygen, and a very productive chemist. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L811–L816. [Google Scholar] [CrossRef]
- West, J.B. Joseph Priestley, oxygen, and the enlightenment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L111–L119. [Google Scholar] [CrossRef] [PubMed]
- West, J.B. The collaboration of Antoine and Marie-Anne Lavoisier and the first measurements of human oxygen consumption. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L775–L785. [Google Scholar] [CrossRef]
- Sternbach, G.L.; Varon, J. The discovery and rediscovery of oxygen. J. Emerg. Med. 2005, 28, 221–224. [Google Scholar] [CrossRef]
- Kanwisher, J. Oxygen in Antarctic Air. Tellus 1957, 9, 137–138. [Google Scholar]
- Venemans-Jellema, A.; Schreijer, A.J.; Le Cessie, S.; Emmerich, J.; Rosendaal, F.R.; Cannegieter, S.C. No effect of isolated long-term supine immobilization or profound prolonged hypoxia on blood coagulation. J. Thromb. Haemost. 2014, 12, 902–909. [Google Scholar] [CrossRef] [PubMed]
- West, J.B.; Boyer, S.J.; Graber, D.J.; Hackett, P.H.; Maret, K.H.; Milledge, J.S.; Peters, R.M.J.; Pizzo, C.J.; Samaja, M.; Sarnquist, F.H. Maximal exercise at extreme altitudes on Mount Everest. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 688–698. [Google Scholar] [CrossRef]
- Garrido, E.; Sibila, O.; Viscor, G. Breathing at extreme altitudes—Scientific projects “EVEREST” (Second part). Arch. Med. Deporte 2017, 34, 338–344. [Google Scholar]
- Fulco, C.S.; Rock, P.B.; Cymerman, A. Maximal and submaximal exercise performance at altitude. Aviat. Space Env. Med. 1998, 69, 793–801. [Google Scholar]
- Wylie, A. Notes on the Western regions. J. R. Anthropol. Inst. 1881, 10, 36–38. [Google Scholar]
- West, J.; Alexander, M. Kellas and the physiological challenge of Mt. Everest. J. Appl. Physiol. 1987, 63, 3–11. [Google Scholar] [CrossRef]
- Jahan, E.; Barua, T.; Salma, U. An Overview on Heart Rate Monitoring and Pulse Oximeter System; MNK Publications: Haryana, India, 2014. [Google Scholar]
- Klein, S.G.; Alsolami, S.M.; Steckbauer, A.; Arossa, S.; Parry, A.J.; Ramos Mandujano, G.; Alsayegh, K.; Izpisua Belmonte, J.C.; Li, M.; Duarte, C.M. A prevalent neglect of environmental control in mammalian cell culture calls for best practices. Nat. Biomed. Eng. 2021, 5, 787–792. [Google Scholar] [CrossRef]
- Bianciardi, P.; Fantacci, M.; Caretti, A.; Ronchi, R.; Milano, G.; Morel, S.; von Segesser, L.; Corno, A.; Samaja, M. Chronic in vivo hypoxia in various organs: Hypoxia-inducible factor-1 alpha and apoptosis. Biochem. Biophys. Res. Commun. 2006, 342, 875–880. [Google Scholar] [CrossRef]
- Villafuerte, F.C.; Corante, N. Chronic Mountain Sickness: Clinical Aspects, Etiology, Management, and Treatment. High Alt. Med. Biol. 2016, 17, 61–69. [Google Scholar] [CrossRef]
- Monge, C. Life in the Andes and chronic mountain sickness. Science 1942, 95, 79–84. [Google Scholar] [CrossRef]
- Winslow, R.; Monge, C. Hypoxia, polycythemia, and chronic mountain sickness. In Baltimore and London; Johns Hopkins University Press: Baltimore, MD, USA, 1987. [Google Scholar]
- Zubieta-Calleja, G.R.; Paulev, P.E.; Zubieta-Calleja, L.; Zubieta-Castillo, G. Altitude adaptation through hematocrit changes. J. Physiol. Pharm. 2007, 58 (Suppl. 5), 811–818. [Google Scholar]
- Villafuerte, F.C.; Simonson, T.S.; Bermudez, D.; León-Velarde, F. High-Altitude Erythrocytosis: Mechanisms of Adaptive and Maladaptive Responses. Physiology 2022, 37, 175–186. [Google Scholar] [CrossRef]
- West, J.B. Cognitive Impairment of School Children at High Altitude: The Case for Oxygen Conditioning in Schools. High Alt. Med. Biol. 2016, 17, 203–207. [Google Scholar] [CrossRef] [PubMed]
- West, J.B. Oxygen Conditioning: A New Technique for Improving Living and Working at High Altitude. Physiology 2016, 31, 216–222. [Google Scholar] [CrossRef]
- Poole, D.C.; Musch, T.I.; Colburn, T.D. Oxygen flux from capillary to mitochondria: Integration of contemporary discoveries. Eur. J. Appl. Physiol. 2022, 122, 7–28. [Google Scholar] [CrossRef] [PubMed]
- Dominelli, P.B.; Wiggins, C.C.; Roy, T.K.; Secomb, T.W.; Curry, T.B.; Joyner, M.J. The Oxygen Cascade During Exercise in Health and Disease. Mayo Clin. Proc. 2021, 96, 1017–1032. [Google Scholar] [CrossRef]
- Mallet, R.T.; Burtscher, J.; Pialoux, V.; Pasha, Q.; Ahmad, Y.; Millet, G.P.; Burtscher, M. Molecular Mechanisms of High-Altitude Acclimatization. Int. J. Mol. Sci. 2023, 24, 1698. [Google Scholar] [CrossRef] [PubMed]
- Monge, C.; Leon-Velarde, F. Physiological adaptation to high altitude: Oxygen transport in mammals and birds. Physiol. Rev. 1991, 71, 1135–1172. [Google Scholar] [CrossRef]
- Volkova, Y.L.; Pickel, C.; Jucht, A.E.; Wenger, R.H.; Scholz, C.C. The asparagine hydroxylase FIH—A unique oxygen sensor. Antioxid. Redox Signal. 2022, 37, 913–935. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 271–273. [Google Scholar] [CrossRef]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, A.; Gradin, K.; et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Winning, S.; Fandrey, J. Oxygen Sensing in Innate Immune Cells: How Inflammation Broadens Classical Hypoxia-Inducible Factor Regulation in Myeloid Cells. Antioxid. Redox Signal. 2022, 37, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Mallet, R.T.; Pialoux, V.; Millet, G.P.; Burtscher, M. Adaptive Responses to Hypoxia and/or Hyperoxia in Humans. Antioxid. Redox Signal. 2022, 37, 887–912. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Cong, X.; Yun, Z. Differential hypoxic regulation of hypoxia-inducible factors 1alpha and 2alpha. Mol. Cancer Res. 2011, 9, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Nauta, T.D.; van den Broek, M.; Gibbs, S.; van der Pouw-Kraan, T.C.; Oudejans, C.B.; van Hinsbergh, V.W.; Koolwijk, P. Identification of HIF-2alpha-regulated genes that play a role in human microvascular endothelial sprouting during prolonged hypoxia in vitro. Angiogenesis 2017, 20, 39–54. [Google Scholar] [CrossRef]
- Lendahl, U.; Lee, K.L.; Yang, H.; Poellinger, L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet. 2009, 10, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Bartoszewska, S.; Kochan, K.; Piotrowski, A.; Kalinowski, L.; Kamysz, W.; Ochocka, R.J.; Bartoszewski, R.; Collawn, J.F. miR-429 regulates the transition between Hypoxia-Inducible Factor (HIF)1A and HIF3A expression in human endothelial cells. Sci. Rep. 2016, 6, 22775. [Google Scholar] [CrossRef]
- Bayer, C.; Vaupel, P. Acute versus chronic hypoxia in tumors: Controversial data concerning time frames and biological consequences. Strahlenther. Und Onkol. Organ Der Dtsch. Rontgenges. 2012, 188, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Caretti, A.; Bianciardi, P.; Ronchi, R.; Fantacci, M.; Guazzi, M.; Samaja, M. Phosphodiesterase-5 inhibition abolishes neuron apoptosis induced by chronic hypoxia independently of hypoxia-inducible factor-1 alpha signaling. Exp. Biol. Med. 2008, 233, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Fantacci, M.; Bianciardi, P.; Caretti, A.; Coleman, T.R.; Cerami, A.; Brines, M.; Samaja, M. Carbamylated erythropoietin ameliorates the metabolic stress induced in vivo by severe chronic hypoxia. Proc. Natl. Acad. Sci. USA 2006, 103, 17531–17536. [Google Scholar] [CrossRef] [PubMed]
- De Palma, S.; Ripamonti, M.; Vigano, A.; Moriggi, M.; Capitanio, D.; Samaja, M.; Milano, G.; Cerretelli, P.; Wait, R.; Gelfi, C. Metabolic modulation induced by chronic hypoxia in rats using a comparative proteomic analysis of skeletal muscle tissue. J. Proteome Res. 2007, 6, 1974–1984. [Google Scholar] [CrossRef]
- Caretti, A.; Morel, S.; Milano, G.; Fantacci, M.; Bianciardi, P.; Ronchi, R.; Vassalli, G.; von Segesser, L.K.; Samaja, M. Heart HIF-1 alpha and MAP kinases during hypoxia: Are they associated in vivo? Exp. Biol. Med. 2007, 232, 887–894. [Google Scholar]
- Vigano, A.; Vasso, M.; Caretti, A.; Bravata, V.; Terraneo, L.; Fania, C.; Capitanio, D.; Samaja, M.; Gelfi, C. Protein modulation in mouse heart under acute and chronic hypoxia. Proteomics 2011, 11, 4202–4217. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox signaling during hypoxia in mammalian cells. Redox. Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharm. 2017, 174, 1670–1689. [Google Scholar] [CrossRef]
- Infanger, D.W.; Sharma, R.V.; Davisson, R.L. NADPH oxidases of the brain: Distribution, regulation, and function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar] [CrossRef]
- Diebold, I.; Flugel, D.; Becht, S.; Belaiba, R.S.; Bonello, S.; Hess, J.; Kietzmann, T.; Gorlach, A. The hypoxia-inducible factor-2alpha is stabilized by oxidative stress involving NOX4. Antioxid. Redox Signal. 2010, 13, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Radermacher, K.A.; Wingler, K.; Langhauser, F.; Altenhofer, S.; Kleikers, P.; Hermans, J.J.; Hrabe de Angelis, M.; Kleinschnitz, C.; Schmidt, H.H. Neuroprotection after stroke by targeting NOX4 as a source of oxidative stress. Antioxid. Redox Signal. 2013, 18, 1418–1427. [Google Scholar] [CrossRef]
- Kuroda, J.; Ago, T.; Nishimura, A.; Nakamura, K.; Matsuo, R.; Wakisaka, Y.; Kamouchi, M.; Kitazono, T. Nox4 is a major source of superoxide production in human brain pericytes. J. Vasc. Res. 2014, 51, 429–438. [Google Scholar] [CrossRef]
- Terraneo, L.; Paroni, R.; Bianciardi, P.; Giallongo, T.; Carelli, S.; Gorio, A.; Samaja, M. Brain adaptation to hypoxia and hyperoxia in mice. Redox. Biol. 2017, 11, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Sethy, N.K.; Singh, M.; Kumar, R.; Ilavazhagan, G.; Bhargava, K. Upregulation of transcription factor NRF2-mediated oxidative stress response pathway in rat brain under short-term chronic hypobaric hypoxia. Funct. Integr. Genom. 2011, 11, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ren, C.; Chen, X.; Shen, J. From rapid to delayed and remote postconditioning: The evolving concept of ischemic postconditioning in brain ischemia. Curr. Drug Targets 2012, 13, 173–187. [Google Scholar] [CrossRef]
- Yan, W.; Chen, Z.; Chen, J.; Chen, H. Isoflurane preconditioning protects rat brain from ischemia reperfusion injury via up-regulating the HIF-1alpha expression through Akt/mTOR/s6K activation. Cell. Mol. Biol. 2016, 62, 38–44. [Google Scholar]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J. 2003, 17, 64–66. [Google Scholar] [CrossRef]
- Power, J.H.; Blumbergs, P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009, 117, 63–73. [Google Scholar] [CrossRef]
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox. Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Pena, E.; El Alam, S.; Siques, P.; Brito, J. Oxidative Stress and Diseases Associated with High-Altitude Exposure. Antioxidants 2022, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Dosek, A.; Ohno, H.; Acs, Z.; Taylor, A.W.; Radak, Z. High altitude and oxidative stress. Respir. Physiol. Neurobiol. 2007, 158, 128–131. [Google Scholar] [CrossRef]
- Mrakic-Sposta, S.; Montorsi, M.; Porcelli, S.; Marzorati, M.; Healey, B.; Dellanoce, C.; Vezzoli, A. Effects of Prolonged Exposure to Hypobaric Hypoxia on Oxidative Stress: Overwintering in Antarctic Concordia Station. Oxidative Med. Cell. Longev. 2022, 2022, 4430032. [Google Scholar] [CrossRef]
- Benderro, G.F.; Sun, X.; Kuang, Y.; Lamanna, J.C. Decreased VEGF expression and microvascular density, but increased HIF-1 and 2alpha accumulation and EPO expression in chronic moderate hyperoxia in the mouse brain. Brain Res. 2012, 1471, 46–55. [Google Scholar] [CrossRef]
- Lee, E.S.; Smith, W.E.; Quach, H.T.; Jones, B.D.; Santilli, S.M.; Vatassery, G.T. Moderate hyperoxia (40%) increases antioxidant levels in mouse tissue. J. Surg. Res. 2005, 127, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y. Effects of Long-Term Exposure to High Altitude Hypoxia on Cognitive Function and Its Mechanism: A Narrative Review. Brain Sci. 2022, 12, 808. [Google Scholar] [CrossRef]
- Koester-Hegmann, C.; Bengoetxea, H.; Kosenkov, D.; Thiersch, M.; Haider, T.; Gassmann, M.; Schneider Gasser, E.M. High-Altitude Cognitive Impairment Is Prevented by Enriched Environment Including Exercise via VEGF Signaling. Front. Cell Neurosci. 2018, 12, 532. [Google Scholar] [CrossRef]
- Maiti, P.; Singh, S.B.; Ilavazhagan, G. Nitric oxide system is involved in hypobaric hypoxia-induced oxidative stress in rat brain. Acta Histochem. 2010, 112, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.A.; Lundby, A.K.; Fenk, S.; Gehrig, S.; Siebenmann, C.; Fluck, D.; Kirk, N.; Hilty, M.P.; Lundby, C. Twenty-eight days of exposure to 3454 m increases mitochondrial volume density in human skeletal muscle. J. Physiol. 2016, 594, 1151–1166. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.A.; Boushel, R.; Wright-Paradis, C.; Calbet, J.A.; Robach, P.; Gnaiger, E.; Lundby, C. Mitochondrial function in human skeletal muscle following high-altitude exposure. Exp. Physiol. 2013, 98, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Ripamonti, M.; Vigano, A.; Moriggi, M.; Milano, G.; von Segesser, L.K.; Samaja, M.; Gelfi, C. Cytochrome c oxidase expression in chronic and intermittent hypoxia rat gastrocnemius muscle quantitated by CE. Electrophoresis 2006, 27, 3897–3903. [Google Scholar] [CrossRef]
- Kayser, B.; Hoppeler, H.; Claassen, H.; Cerretelli, P. Muscle structure and performance capacity of Himalayan Sherpas. J. Appl. Physiol. 1991, 70, 1938–1942. [Google Scholar] [CrossRef]
- Kayser, B.; Hoppeler, H.; Desplanches, D.; Marconi, C.; Broers, B.; Cerretelli, P. Muscle ultrastructure and biochemistry of lowland Tibetans. J. Appl. Physiol. 1996, 81, 419–425. [Google Scholar] [CrossRef]
- Murray, A.J.; Montgomery, H.E.; Feelisch, M.; Grocott, M.P.W.; Martin, D.S. Metabolic adjustment to high-altitude hypoxia: From genetic signals to physiological implications. Biochem. Soc. Trans. 2018, 46, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Hoppeler, H.; Vogt, M. Muscle tissue adaptations to hypoxia. J. Exp. Biol. 2001, 204, 3133–3139. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef]
- Samaja, M.; Allibardi, S.; Milano, G.; Neri, G.; Grassi, B.; Gladden, L.B.; Hogan, M.C. Differential depression of myocardial function and metabolism by lactate and H+. Am. J. Physiol. 1999, 276, H3–H8. [Google Scholar] [CrossRef]
- Suliman, H.B.; Nozik-Grayck, E. Mitochondrial Dysfunction: Metabolic Drivers of Pulmonary Hypertension. Antioxid. Redox Signal. 2019, 31, 843–857. [Google Scholar] [CrossRef]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef]
- Koop, A.C.; Bossers, G.P.L.; Ploegstra, M.J.; Hagdorn, Q.A.J.; Berger, R.M.F.; Sillje, H.H.W.; Bartelds, B. Metabolic Remodeling in the Pressure-Loaded Right Ventricle: Shifts in Glucose and Fatty Acid Metabolism-A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2019, 8, e012086. [Google Scholar] [CrossRef]
- Umeno, A.; Biju, V.; Yoshida, Y. In vivo ROS production and use of oxidative stress-derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes. Free Radic. Res. 2017, 51, 413–427. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Rey, F.; Ottolenghi, S.; Giallongo, T.; Balsari, A.; Martinelli, C.; Rey, R.; Allevi, R.; Giulio, A.M.D.; Zuccotti, G.V.; Mazzucchelli, S.; et al. Mitochondrial Metabolism as Target of the Neuroprotective Role of Erythropoietin in Parkinson’s Disease. Antioxidants 2021, 10, 121. [Google Scholar] [CrossRef]
- Ottolenghi, S.; Milano, G.; Cas, M.D.; Findley, T.O.; Paroni, R.; Corno, A.F. Can Erythropoietin Reduce Hypoxemic Neurological Damages in Neonates With Congenital Heart Defects? Front. Pharmacol. 2021, 12, 770590. [Google Scholar] [CrossRef]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharm. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef]
- Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Transport function, regulation, and biology of human monocarboxylate transporter 1 (hMCT1) and 4 (hMCT4). Pharmacol. Ther. 2021, 226, 107862. [Google Scholar] [CrossRef]
- Doyen, J.; Trastour, C.; Ettore, F.; Peyrottes, I.; Toussant, N.; Gal, J.; Ilc, K.; Roux, D.; Parks, S.K.; Ferrero, J.M.; et al. Expression of the hypoxia-inducible monocarboxylate transporter MCT4 is increased in triple negative breast cancer and correlates independently with clinical outcome. Biochem. Biophys. Res. Commun. 2014, 451, 54–61. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef] [Green Version]
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.P.; Pouyssegur, J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [CrossRef]
- Caruso, J.P.; Koch, B.J.; Benson, P.D.; Varughese, E.; Monterey, M.D.; Lee, A.E.; Dave, A.M.; Kiousis, S.; Sloan, A.E.; Mathupala, S.P. pH, Lactate, and Hypoxia: Reciprocity in Regulating High-Affinity Monocarboxylate Transporter Expression in Glioblastoma. Neoplasia 2017, 19, 121–134. [Google Scholar] [CrossRef]
- Marchiq, I.; Pouysségur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef]
- Wang, Z.H.; Peng, W.B.; Zhang, P.; Yang, X.P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Shukla, D.; Bansal, A. Expression of Monocarboxylate Transporter Isoforms in Rat Skeletal Muscle Under Hypoxic Preconditioning and Endurance Training. High Alt. Med. Biol. 2016, 17, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Saraswati, S.; Guo, Y.; Atkinson, J.; Young, P.P. Prolonged hypoxia induces monocarboxylate transporter-4 expression in mesenchymal stem cells resulting in a secretome that is deleterious to cardiovascular repair. Stem Cells 2015, 33, 1333–1344. [Google Scholar] [CrossRef]
- McClelland, G.B.; Brooks, G.A. Changes in MCT 1, MCT 4, and LDH expression are tissue specific in rats after long-term hypobaric hypoxia. J. Appl. Physiol. (1985) 2002, 92, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Perez de Heredia, F.; Wood, I.S.; Trayhurn, P. Hypoxia stimulates lactate release and modulates monocarboxylate transporter (MCT1, MCT2, and MCT4) expression in human adipocytes. Pflug. Arch. 2010, 459, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mukai, K.; Takahashi, K.; Ohmura, H.; Takahashi, T.; Hatta, H.; Kitaoka, Y. Short-term hypoxic training increases monocarboxylate transporter 4 and phosphofructokinase activity in Thoroughbreds. Physiol. Rep. 2020, 8, e14473. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Kitaoka, Y.; Takahashi, Y.; Takahashi, T.; Takahashi, K.; Ohmura, H. Moderate-intensity training in hypoxia improves exercise performance and glycolytic capacity of skeletal muscle in horses. Physiol. Rep. 2021, 9, e15145. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Sheikholeslami-Vatani, D.; Ghaeeni, S.; Baazm, M. The effects of different training modalities on monocarboxylate transporters MCT1 and MCT4, hypoxia inducible factor-1α (HIF-1α), and PGC-1α gene expression in rat skeletal muscles. Mol. Biol. Rep. 2021, 48, 2153–2161. [Google Scholar] [CrossRef]
- Rand, M.J. Nitrergic transmission: Nitric oxide as a mediator of non-adrenergic, non-cholinergic neuro-effector transmission. Clin. Exp. Pharm. Physiol. 1992, 19, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Janczuk, A.; Wang, P.G. Current trends in the development of nitric oxide donors. Curr. Pharm. Des. 1999, 5, 417–441. [Google Scholar] [CrossRef] [PubMed]
- Umbrello, M.; Dyson, A.; Feelisch, M.; Singer, M. The key role of nitric oxide in hypoxia: Hypoxic vasodilation and energy supply-demand matching. Antioxid. Redox Signal. 2013, 19, 1690–1710. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharm. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef]
- Ahsan, A.; Mohd, G.; Norboo, T.; Baig, M.A.; Pasha, M.A. Heterozygotes of NOS3 polymorphisms contribute to reduced nitrogen oxides in high-altitude pulmonary edema. Chest 2006, 130, 1511–1519. [Google Scholar] [CrossRef]
- Wang, P.; Ha, A.Y.; Kidd, K.K.; Koehle, M.S.; Rupert, J.L. A variant of the endothelial nitric oxide synthase gene (NOS3) associated with AMS susceptibility is less common in the Quechua, a high altitude Native population. High Alt. Med. Biol. 2010, 11, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Ottolenghi, S.; Attanasio, U.; Tocchetti, C.G.; Paroni, R.; Pagliaro, P.; Samaja, M. Janus, or the inevitable battle between too much and too little oxygen. Antioxid. Redox Signal. 2022, 37, 972–989. [Google Scholar] [CrossRef]
- Rengasamy, A.; Johns, R.A. Determination of Km for oxygen of nitric oxide synthase isoforms. J. Pharmacol. Exp. Ther. 1996, 276, 30–33. [Google Scholar]
- Elayan, I.M.; Axley, M.J.; Prasad, P.V.; Ahlers, S.T.; Auker, C.R. Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brain. J. Neurophysiol. 2000, 83, 2022–2029. [Google Scholar] [CrossRef]
- Gladwin, M.; Ognibene, F.; Pannell, L.; Nichols, J.; Pease-Fye, M.; Shelhamer, J.; Schechter, A. Relative role of heme nitrosylation and beta-cysteine 93 nitrosation in the transport and metabolism of nitric oxide by hemoglobin in the human circulation. Proc. Natl. Acad. Sci. USA 2000, 97, 9943–9948. [Google Scholar] [CrossRef]
- Murad, F.; Leitman, D.C.; Bennett, B.M.; Molina, C.; Waldman, S.A. Regulation of guanylate cyclase by atrial natriuretic factor and the role of cyclic GMP in vasodilation. Am. J. Med. Sci. 1987, 294, 139–143. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Behera, J.; Nagarajan, S.; Saran, U.; Kumar, R.; Keshri, G.K.; Suryakumar, G.; Chatterjee, S. Nitric oxide restores peripheral blood mononuclear cell adhesion against hypoxia via NO-cGMP signalling. Cell Biochem. Funct. 2020, 38, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Tamilarasan, K.P.; Rajkumar, A.S.; Geetha Priya, S.; Rajaram, M.; Saleem, N.K.; Majumder, S.; Jaffar Ali, B.M.; Illavazagan, G.; Chatterjee, S. Nitric oxide/cGMP protects endothelial cells from hypoxia-mediated leakiness. Eur. J. Cell Biol. 2008, 87, 147–161. [Google Scholar] [CrossRef]
- Galkin, A.; Higgs, A.; Moncada, S. Nitric oxide and hypoxia. Essays Biochem. 2007, 43, 29–42. [Google Scholar] [CrossRef]
- Moncada, S. The L-arginine:nitric oxide pathway. Acta Physiol. Scand 1992, 145, 201–227. [Google Scholar] [CrossRef]
- Rees, D.; Palmer, R.; Moncada, S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar] [CrossRef] [Green Version]
- Scherrer, U.; Vollenweider, L.; Delabays, A.; Savcic, M.; Eichenberger, U.; Kleger, G.R.; Fikrle, A.; Ballmer, P.E.; Nicod, P.; Bartsch, P. Inhaled nitric oxide for high-altitude pulmonary edema. N. Engl. J. Med. 1996, 334, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Cumpstey, A.F.; Hennis, P.J.; Gilbert-Kawai, E.T.; Fernandez, B.O.; Poudevigne, M.; Cobb, A.; Meale, P.; Mitchell, K.; Moyses, H.; Pohnl, H.; et al. Effects of dietary nitrate on respiratory physiology at high altitude—Results from the Xtreme Alps study. Nitric Oxide 2017, 71, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhang, X.; Qi, D.; Guo, X.; Pang, B.; Du, Y.; Zou, X.; Guo, S.; Zhao, X. Inhibition of inducible nitric oxide synthase expression and nitric oxide production in plateau pika (Ochotona curzoniae) at high altitude on Qinghai-Tibet Plateau. Nitric Oxide 2014, 38, 38–44. [Google Scholar] [CrossRef]
- Herrera, E.A.; Ebensperger, G.; Hernandez, I.; Sanhueza, E.M.; Llanos, A.J.; Reyes, R.V. The role of nitric oxide signaling in pulmonary circulation of high- and low-altitude newborn sheep under basal and acute hypoxic conditions. Nitric Oxide 2019, 89, 71–80. [Google Scholar] [CrossRef]
- Beall, C.M.; Laskowski, D.; Erzurum, S.C. Nitric oxide in adaptation to altitude. Free Radic. Biol. Med. 2012, 52, 1123–1134. [Google Scholar] [CrossRef]
- Paroni, R.; Terraneo, L.; Bonomini, F.; Finati, E.; Virgili, E.; Bianciardi, P.; Favero, G.; Fraschini, F.; Reiter, R.J.; Rezzani, R.; et al. Antitumor activity of melatonin in a mouse model of human prostate cancer: Relationship with hypoxia signaling. J. Pineal. Res. 2014, 57, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Lai, C.J.; Tsai, M.H.; Wu, Y.C.; Chen, K.T.; Jou, M.J.; Fu, P.I.; Wu, C.H.; Wei, I.H. Effects of melatonin on the nitric oxide system and protein nitration in the hypobaric hypoxic rat hippocampus. BMC Neurosci. 2015, 16, 61. [Google Scholar] [CrossRef] [PubMed]
- Pooja; Sharma, M.; Singh, K.; Himashree, G.; Bhaumik, G.; Kumar, B.; Sethy, N.K. Estrogen receptor (ESR1 and ESR2)-mediated activation of eNOS-NO-cGMP pathway facilitates high altitude acclimatization. Nitric Oxide 2020, 102, 12–20. [Google Scholar] [CrossRef]
- Erzurum, S.C.; Ghosh, S.; Janocha, A.J.; Xu, W.; Bauer, S.; Bryan, N.S.; Tejero, J.; Hemann, C.; Hille, R.; Stuehr, D.J.; et al. Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans. Proc. Natl. Acad. Sci. USA 2007, 104, 17593–17598. [Google Scholar] [CrossRef] [PubMed]
- Cas, M.D.; Morano, C.; Ottolenghi, S.; Dicasillati, R.; Roda, G.; Samaja, M.; Paroni, R. Inside the Alterations of Circulating Metabolome in Antarctica: The Adaptation to Chronic Hypoxia. Front Physiol. 2022, 13, 819345. [Google Scholar] [CrossRef]
- Schneider, J.C.; Blazy, I.; Dechaux, M.; Rabier, D.; Mason, N.P.; Richalet, J.P. Response of nitric oxide pathway to L-arginine infusion at the altitude of 4350 m. Eur. Respir. J. 2001, 18, 286–292. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Y.; Liu, J.; Qian, G. Meta-analysis of clinical efficacy of sildenafil, a phosphodiesterase type-5 inhibitor on high altitude hypoxia and its complications. High Alt. Med. Biol. 2014, 15, 46–51. [Google Scholar] [CrossRef]
- Getu, A. Ethiopian Native Highlander’s Adaptation to Chronic High-Altitude Hypoxia. Biomed. Res. Int. 2022, 2022, 5749382. [Google Scholar] [CrossRef] [PubMed]
- Pooja; Sharma, V.; Sharma, M.; Varshney, R.; Kumar, B.; Sethy, N.K. Association Between 17beta-Estradiol Receptors and Nitric Oxide Signaling Augments High-Altitude Adaptation of Ladakhi Highlanders. High Alt. Med. Biol. 2021, 22, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A. The supply of oxygen to the tissues and the regulation of the capillary circulation. J. Physiol. 1919, 52, 457–474. [Google Scholar] [CrossRef]
- Poole, D.C.; Kano, Y.; Koga, S.; Musch, T.I. August Krogh: Muscle capillary function and oxygen delivery. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2021, 253, 110852. [Google Scholar] [CrossRef] [PubMed]
- Angleys, H.; Østergaard, L. Krogh’s capillary recruitment hypothesis, 100 years on: Is the opening of previously closed capillaries necessary to ensure muscle oxygenation during exercise? Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H425–H447. [Google Scholar] [CrossRef]
- Pilotto, A.M.; Adami, A.; Mazzolari, R.; Brocca, L.; Crea, E.; Zuccarelli, L.; Pellegrino, M.A.; Bottinelli, R.; Grassi, B.; Rossiter, H.B.; et al. Near-infrared spectroscopy estimation of combined skeletal muscle oxidative capacity and O(2) diffusion capacity in humans. J. Physiol. 2022, 600, 4153–4168. [Google Scholar] [CrossRef]
- Lemieux, P.; Birot, O. Altitude, Exercise, and Skeletal Muscle Angio-Adaptive Responses to Hypoxia: A Complex Story. Front. Physiol. 2021, 12, 735557. [Google Scholar] [CrossRef]
- Valdivia, E. Total capillary bed in striated muscles of guinea pigs native to the Peruvian mountains. Am. J. Physiol. 1958, 194, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Leon-Velarde, F.; Sanchez, J.; Bigard, A.X.; Brunet, A.; Lesty, C.; Monge, C. High altitude tissue adaptation in Andean coots: Capillarity, fibre area, fibre type and enzymatic activities of skeletal muscle. J. Comp. Physiol. B 1993, 163, 52–58. [Google Scholar] [CrossRef]
- Hoppeler, H.; Kleinert, E.; Schlegel, C.; Claassen, H.; Howald, H.; Kayar, S.R.; Cerretelli, P. Morphological adaptations of human skeletal muscle to chronic hypoxia. Int. J. Sport. Med. 1990, 11 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef]
- Olfert, I.M.; Breen, E.C.; Mathieu-Costello, O.; Wagner, P.D. Chronic hypoxia attenuates resting and exercise-induced VEGF, flt-1, and flk-1 mRNA levels in skeletal muscle. J. Appl. Physiol. (1985) 2001, 90, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Mathieu-Costello, O.; Entin, P.L.; Wagner, P.D.; Richardson, R.S. The skeletal muscle VEGF mRNA response to acute exercise in patients with chronic heart failure. Growth Factors 2010, 28, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Snyder, G.K. Model analyses of capillary growth and tissue oxygenation during hypoxia. J. Appl. Physiol. (1985) 1988, 65, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.G.; Intaglietta, M. Local tissue oxygenation during constant red blood cell flux: A discrete source analysis of velocity and hematocrit changes. Microvasc. Res. 1989, 37, 308–322. [Google Scholar] [CrossRef]
- Secomb, T.W.; Hsu, R. Simulation of O2 transport in skeletal muscle: Diffusive exchange between arterioles and capillaries. Am. J. Physiol. 1994, 267, H1214–H1221. [Google Scholar] [CrossRef] [PubMed]
- Intaglietta, M.; Johnson, P.; Winslow, R. Microvascular and tissue oxygen distribution. Cardiovasc. Res. 1996, 32, 632–643. [Google Scholar] [CrossRef]
- Østergaard, L. Blood flow, capillary transit times, and tissue oxygenation: The centennial of capillary recruitment. J. Appl. Physiol. (1985) 2020, 129, 1413–1421. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Boero, J.A.; Ascher, J.; Arregui, A.; Rovainen, C.; Woolsey, T.A. Increased brain capillaries in chronic hypoxia. J. Appl. Physiol. (1985) 1999, 86, 1211–1219. [Google Scholar] [CrossRef]
- Santilli, G.; Lamorte, G.; Carlessi, L.; Ferrari, D.; Rota Nodari, L.; Binda, E.; Delia, D.; Vescovi, A.L.; De Filippis, L. Mild hypoxia enhances proliferation and multipotency of human neural stem cells. PLoS ONE 2010, 5, e8575. [Google Scholar] [CrossRef]
- Li, L.; Candelario, K.M.; Thomas, K.; Wang, R.; Wright, K.; Messier, A.; Cunningham, L.A. Hypoxia inducible factor-1alpha (HIF-1alpha) is required for neural stem cell maintenance and vascular stability in the adult mouse SVZ. J. Neurosci. 2014, 34, 16713–16719. [Google Scholar] [CrossRef] [PubMed]
- Panchision, D.M. The role of oxygen in regulating neural stem cells in development and disease. J. Cell. Physiol. 2009, 220, 562–568. [Google Scholar] [CrossRef]
- Braunschweig, L.; Meyer, A.K.; Wagenfuhr, L.; Storch, A. Oxygen regulates proliferation of neural stem cells through Wnt/beta-catenin signalling. Mol. Cell. Neurosci. 2015, 67, 84–92. [Google Scholar] [CrossRef]
- Moreno, M.; Fernandez, V.; Monllau, J.M.; Borrell, V.; Lerin, C.; de la Iglesia, N. Transcriptional Profiling of Hypoxic Neural Stem Cells Identifies Calcineurin-NFATc4 Signaling as a Major Regulator of Neural Stem Cell Biology. Stem. Cell. Rep. 2015, 5, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Vieira, H.L.; Alves, P.M.; Vercelli, A. Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Prog. Neurobiol. 2011, 93, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ. Res. 1995, 77, 638–643. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Grunstein, J.; Roberts, W.G.; Mathieu-Costello, O.; Hanahan, D.; Johnson, R.S. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res. 1999, 59, 1592–1598. [Google Scholar]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Shibasaki, F. Hypoxia-inducible factor as an angiogenic master switch. Front. Pediatr. 2015, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharm. 2014, 9, 142–160. [Google Scholar] [CrossRef]
- Xu, Y.; Kong, X.; Li, J.; Cui, T.; Wei, Y.; Xu, J.; Zhu, Y.; Zhu, X. Mild Hypoxia Enhances the Expression of HIF and VEGF and Triggers the Response to Injury in Rat Kidneys. Front. Physiol. 2021, 12, 690496. [Google Scholar] [CrossRef] [PubMed]
- Carnot, P.; Deflandre, C. Sur l’activite hematopoietique du serum au cours de la regeneration du sang. Compt. Rend. Acad. Sci. 1906, 143, 384–386. [Google Scholar]
- Jelkmann, W.; Kurtz, A.; Seidl, J.; Bauer, C. Temporal pattern of erythropoietin titers in kidney tissue during hypoxic hypoxia. Pflug. Arch. 1982, 393, 88–91. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Siebenmann, C.; Cathomen, A.; Hug, M.; Keiser, S.; Lundby, A.K.; Hilty, M.P.; Goetze, J.P.; Rasmussen, P.; Lundby, C. Hemoglobin mass and intravascular volume kinetics during and after exposure to 3454-m altitude. J. Appl. Physiol. (1985) 2015, 119, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, S.; Marzorati, M.; Healey, B.; Terraneo, L.; Vezzoli, A.; Bella, S.D.; Dicasillati, R.; Samaja, M. Lack of acclimatization to chronic hypoxia in humans in the Antarctica. Sci. Rep. 2017, 7, 18090. [Google Scholar] [CrossRef] [PubMed]
- Vizcardo-Galindo, G.; Leon-Velarde, F.; Villafuerte, F.C. High-Altitude Hypoxia Decreases Plasma Erythropoietin Soluble Receptor Concentration in Lowlanders. High Alt. Med. Biol. 2020, 21, 92–98. [Google Scholar] [CrossRef]
- Beall, C.M.; Brittenham, G.M.; Strohl, K.P.; Blangero, J.; Williams-Blangero, S.; Goldstein, M.C.; Decker, M.J.; Vargas, E.; Villena, M.; Soria, R.; et al. Hemoglobin concentration of high-altitude Tibetans and Bolivian Aymara. Am. J. Phys. Anthropol. 1998, 106, 385–400. [Google Scholar] [CrossRef]
- Basak, N.; Norboo, T.; Mustak, M.S.; Thangaraj, K. Heterogeneity in Hematological Parameters of High and Low Altitude Tibetan Populations. J. Blood Med. 2021, 12, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, U.; Senee, H.; Yanamadra, S.; Das, S.K.; Bhattachar, S.A.; Das, R.; Kumar, S.; Malhotra, P.; Varma, S.; Varma, N.; et al. Erythropoietin and ferritin response in native highlanders aged 4–19 years from the Leh-Ladakh region of India. Br. J. Haematol. 2019, 184, 263–268. [Google Scholar] [CrossRef]
- Beall, C.M.; Decker, M.J.; Brittenham, G.M.; Kushner, I.; Gebremedhin, A.; Strohl, K.P. An Ethiopian pattern of human adaptation to high-altitude hypoxia. Proc. Natl. Acad. Sci. USA 2002, 99, 17215–17218. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.M.; Almasy, L.A.; Blangero, J.; Williams-Blangero, S.; Brittenham, G.M.; Strohl, K.P.; Decker, M.J.; Vargas, E.; Villena, M.; Soria, R.; et al. Percent of oxygen saturation of arterial hemoglobin among Bolivian Aymara at 3900–4000 m. Am. J. Phys. Anthropol. 1999, 108, 41–51. [Google Scholar] [CrossRef]
- Villafuerte, F.C.; Macarlupu, J.L.; Anza-Ramirez, C.; Corrales-Melgar, D.; Vizcardo-Galindo, G.; Corante, N.; Leon-Velarde, F. Decreased plasma soluble erythropoietin receptor in high-altitude excessive erythrocytosis and Chronic Mountain Sickness. J. Appl. Physiol. (1985) 2014, 117, 1356–1362. [Google Scholar] [CrossRef]
- Painschab, M.S.; Malpartida, G.E.; Dávila-Roman, V.G.; Gilman, R.H.; Kolb, T.M.; León-Velarde, F.; Miranda, J.J.; Checkley, W. Association between serum concentrations of hypoxia inducible factor responsive proteins and excessive erythrocytosis in high altitude Peru. High Alt. Med. Biol. 2015, 16, 26–33. [Google Scholar] [CrossRef]
- Viault, F. Sur l’augmentation considerable du nombre des globules rouges dans le sang chez les habitants des hauts plateaux de l’Amerique du Sud. C R Seances Acad. Sci. 1890, 111, 917–918. [Google Scholar]
- Erslev, A. Humoral regulation of red cell production. Blood 1953, 8, 349–357. [Google Scholar] [CrossRef]
- Barcroft, J.; Binger, C.; Bock, A.; Doggart, J.; Forbes, H.; Harrop, G.; Meakins, J.; Redfield, A. Observations upon the effect of high altitude on the physiological processes of human body, carried out in the Peruvian Andes, chiefly at Cerro de Pasco. Philos Trans. Roy Soc. Lond Biol. Sci. 1923, 211, 351–454. [Google Scholar]
- Nader, E.; Skinner, S.; Romana, M.; Fort, R.; Lemonne, N.; Guillot, N.; Gauthier, A.; Antoine-Jonville, S.; Renoux, C.; Hardy-Dessources, M.D.; et al. Blood Rheology: Key Parameters, Impact on Blood Flow, Role in Sickle Cell Disease and Effects of Exercise. Front. Physiol. 2019, 10, 1329. [Google Scholar] [CrossRef]
- Cinar, Y.; Demir, G.; Paç, M.; Cinar, A.B. Effect of hematocrit on blood pressure via hyperviscosity. Am. J. Hypertens. 1999, 12, 739–743. [Google Scholar] [CrossRef]
- Grotta, J.; Ackerman, R.; Correia, J.; Fallick, G.; Chang, J. Whole blood viscosity parameters and cerebral blood flow. Stroke 1982, 13, 296–301. [Google Scholar] [CrossRef] [Green Version]
- Gassmann, M.; Muckenthaler, M.U. Adaptation of iron requirement to hypoxic conditions at high altitude. J. Appl. Physiol. (1985) 2015, 119, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E. Iron regulation and erythropoiesis. Curr. Opin. Hematol. 2008, 15, 169–175. [Google Scholar] [CrossRef]
- Piperno, A.; Galimberti, S.; Mariani, R.; Pelucchi, S.; Ravasi, G.; Lombardi, C.; Bilo, G.; Revera, M.; Giuliano, A.; Faini, A.; et al. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: Data from the HIGHCARE project. Blood 2011, 117, 2953–2959. [Google Scholar] [CrossRef]
- Clarenbach, C.F.; Sievi, N.A.; Kohler, M. Annual progression of endothelial dysfunction in patients with COPD. Respir. Med. 2017, 132, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Mumby, S.; Adcock, I.M.; Choi, A.M.K.; Chung, K.F.; Quinlan, G.J. The “Iron”-y of Iron Overload and Iron Deficiency in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 196, 1103–1112. [Google Scholar] [CrossRef]
- Portillo, K.; Martinez-Rivera, C.; Ruiz-Manzano, J. Anaemia in chronic obstructive pulmonary disease. Does it really matter? Int. J. Clin. Pract. 2013, 67, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.; Christensen, S.; Lenler-Petersen, P.; Johnsen, S.P. Anemia and 90-day mortality in COPD patients requiring invasive mechanical ventilation. Clin. Epidemiol. 2010, 3, 1–5. [Google Scholar] [CrossRef]
- Lima, D.F.; Dela Coleta, K.; Tanni, S.E.; Silveira, L.V.; Godoy, I.; Godoy, I. Potentially modifiable predictors of mortality in patients treated with long-term oxygen therapy. Respir. Med. 2011, 105, 470–476. [Google Scholar] [CrossRef]
- Camporota, L.; Ranieri, V.M. What’s new in the “Berlin” definition of acute respiratory distress syndrome? Minerva. Anestesiol. 2012, 78, 1162–1166. [Google Scholar] [PubMed]
- Duca, L.; Ottolenghi, S.; Coppola, S.; Rinaldo, R.; Dei Cas, M.; Rubino, F.M.; Paroni, R.; Samaja, M.; Chiumello, D.A.; Motta, I. Differential Redox State and Iron Regulation in Chronic Obstructive Pulmonary Disease, Acute Respiratory Distress Syndrome and Coronavirus Disease 2019. Antioxidants 2021, 10, 1460. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Chiumello, D.; Rossi, S. COVID-19 pneumonia: ARDS or not? Crit. Care 2020, 24, 154. [Google Scholar] [CrossRef] [PubMed]
- Rockfield, S.; Chhabra, R.; Robertson, M.; Rehman, N.; Bisht, R.; Nanjundan, M. Links Between Iron and Lipids: Implications in Some Major Human Diseases. Pharmaceuticals 2018, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Ottolenghi, S.; Zulueta, A.; Caretti, A. Iron and Sphingolipids as Common Players of (Mal)Adaptation to Hypoxia in Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 307. [Google Scholar] [CrossRef]
- Rovida, E.; Niggeler, M.; Carlone, S.; Samaja, M. Carboxyhemoglobin and oxygen affinity of human blood. Clin. Chem. 1984, 30, 1250–1251. [Google Scholar] [CrossRef]
- Samaja, M.; di Prampero, P.; Cerretelli, P. The role of 2,3-DPG in the oxygen transport at altitude. Respir. Physiol. 1986, 64, 191–202. [Google Scholar] [CrossRef]
- Webb, K.L.; Dominelli, P.B.; Baker, S.E.; Klassen, S.A.; Joyner, M.J.; Senefeld, J.W.; Wiggins, C.C. Influence of High Hemoglobin-Oxygen Affinity on Humans During Hypoxia. Front. Physiol. 2021, 12, 763933. [Google Scholar] [CrossRef]
- Samaja, M.; Mariani, C.; Prestini, A.; Cerretelli, P. Acid-base balance and O2 transport at high altitude. Acta Physiol. Scand 1997, 159, 249–256. [Google Scholar] [CrossRef]
- Mairbaurl, H.; Weber, R.E. Oxygen transport by hemoglobin. Compr. Physiol. 2012, 2, 1463–1489. [Google Scholar] [CrossRef] [PubMed]
- Samaja, M.; Melotti, D.; Rovida, E.; Rossi Bernardi, L. Effect of temperature on the p50 value for human blood. Clin. Chem. 1983, 29, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaja, M.; Brenna, L.; Allibardi, S.; Cerretelli, P. Human red blood cell aging at 5050-m altitude: A role during adaptation to hypoxia. J. Appl. Physiol. (1985) 1993, 75, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Samaja, M.; Rovida, E.; Motterlini, R.; Tarantola, M.; Rubinacci, A.; di Prampero, P. Human red cell age, oxygen affinity and oxygen transport. Respir. Physiol. 1990, 79, 69–80. [Google Scholar] [CrossRef]
- Siebenmann, C.; Lundby, C. Regulation of cardiac output in hypoxia. Scand J. Med. Sci. Sport 2015, 25 (Suppl. 4), 53–59. [Google Scholar] [CrossRef]
- Richalet, J.P. Physiological and Clinical Implications of Adrenergic Pathways at High Altitude. Adv. Exp. Med. Biol. 2016, 903, 343–356. [Google Scholar] [CrossRef]
- Naeije, R. Physiological adaptation of the cardiovascular system to high altitude. Prog. Cardiovasc. Dis. 2010, 52, 456–466. [Google Scholar] [CrossRef]
- Theunissen, S.; Balestra, C.; Bolognési, S.; Borgers, G.; Vissenaeken, D.; Obeid, G.; Germonpré, P.; Honoré, P.M.; De Bels, D. Effects of Acute Hypobaric Hypoxia Exposure on Cardiovascular Function in Unacclimatized Healthy Subjects: A “Rapid Ascent” Hypobaric Chamber Study. Int. J. Env. Res. Public Health 2022, 19, 5394. [Google Scholar] [CrossRef]
- Phillips, B.A.; McConnell, J.W.; Smith, M.D. The effects of hypoxemia on cardiac output. A dose-response curve. Chest 1988, 93, 471–475. [Google Scholar] [CrossRef]
- Casey, D.P.; Joyner, M.J. Local control of skeletal muscle blood flow during exercise: Influence of available oxygen. J. Appl. Physiol. (1985) 2011, 111, 1527–1538. [Google Scholar] [CrossRef]
- Stembridge, M.; Ainslie, P.N.; Shave, R. Mechanisms underlying reductions in stroke volume at rest and during exercise at high altitude. Eur. J. Sport. Sci. 2016, 16, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.D. Reduced maximal cardiac output at altitude--mechanisms and significance. Respir. Physiol. 2000, 120, 1–11. [Google Scholar] [CrossRef]
- Antezana, A.M.; Kacimi, R.; Trong, J.L.; Marchal, M.; Abousahl, I.; Dubray, C.; Richalet, J.P. Adrenergic status of humans during prolonged exposure to the altitude of 6542m. J. Appl. Physiol. 1994, 76, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Richalet, J.P.; Hermand, E. Modeling the oxygen transport to the myocardium at maximal exercise at high altitude. Physiol. Rep. 2022, 10, e15262. [Google Scholar] [CrossRef]
- Gibson, Q. The kinetics of reactions between hemoglobin and gases. Progr. Biophys. Biophys. Chem. 1959, 9, 1–53. [Google Scholar] [CrossRef]
- Marquis, A.D.; Jezek, F.; Pinsky, D.J.; Beard, D.A. Hypoxic pulmonary vasoconstriction as a regulator of alveolar-capillary oxygen flux: A computational model of ventilation-perfusion matching. PLoS Comput. Biol. 2021, 17, e1008861. [Google Scholar] [CrossRef] [PubMed]
- West, J.B.; Mathieu-Costello, O. Structure, strength, failure, and remodeling of the pulmonary blood-gas barrier. Annu. Rev. Physiol. 1999, 61, 543–572. [Google Scholar] [CrossRef] [PubMed]
- West, J.B.; Colice, G.L.; Lee, Y.J.; Namba, Y.; Kurdak, S.S.; Fu, Z.; Ou, L.C.; Mathieu-Costello, O. Pathogenesis of high-altitude pulmonary oedema: Direct evidence of stress failure of pulmonary capillaries. Eur. Respir. J. 1995, 8, 523–529. [Google Scholar] [CrossRef]
- Liesker, J.J.; Ten Hacken, N.H.; Zeinstra-Smith, M.; Rutgers, S.R.; Postma, D.S.; Timens, W. Reticular basement membrane in asthma and COPD: Similar thickness, yet different composition. Int. J. Chron. Obs. Pulmon. Dis. 2009, 4, 127–135. [Google Scholar]
- Nydegger, C.; Martinelli, C.; Di Marco, F.; Bulfamante, G.; von Segesser, L.; Tozzi, P.; Samaja, M.; Milano, G. Phosphodiesterase-5 Inhibition Alleviates Pulmonary Hypertension and Basal Lamina Thickening in Rats Challenged by Chronic Hypoxia. Front. Physiol. 2018, 9, 289. [Google Scholar] [CrossRef]
- McClendon, J.; Jansing, N.L.; Redente, E.F.; Gandjeva, A.; Ito, Y.; Colgan, S.P.; Ahmad, A.; Riches, D.W.H.; Chapman, H.A.; Mason, R.J.; et al. Hypoxia-Inducible Factor 1α Signaling Promotes Repair of the Alveolar Epithelium after Acute Lung Injury. Am. J. Pathol. 2017, 187, 1772–1786. [Google Scholar] [CrossRef]
- Klocke, R.A.; Schünemann, H.J.; Grant, B.J. Distribution of pulmonary capillary transit times. Am. J. Respir. Crit. Care Med. 1995, 152, 2014–2020. [Google Scholar] [CrossRef]
- Ayappa, I.; Brown, L.V.; Wang, P.M.; Katzman, N.; Houtz, P.; Bruce, E.N.; Lai-Fook, S.J. Effect of blood flow on capillary transit time and oxygenation in excised rabbit lung. Respir. Physiol. 1996, 105, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Samaja, M.; Crespi, T.; Guazzi, M.; Vandegriff, K.D. Oxygen transport in blood at high altitude: Role of the hemoglobin-oxygen affinity and impact of the phenomena related to hemoglobin allosterism and red cell function. Eur. J. Appl. Physiol. 2003, 90, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Vandegriff, K.; Olson, J. The kinetics of O2 release by human red blood cells in the presence of external sodium dithionite. J. Biol. Chem. 1984, 259, 12609–12618. [Google Scholar] [CrossRef] [PubMed]
- Beretta, E.; Grasso, G.S.; Forcaia, G.; Sancini, G.; Miserocchi, G. Differences in alveolo-capillary equilibration in healthy subjects on facing O2 demand. Sci. Rep. 2019, 9, 16693. [Google Scholar] [CrossRef] [PubMed]
- Ramchandran, R.; Pilipenko, E.; Bach, L.; Raghavan, A.; Reddy, S.P.; Raj, J.U. Hypoxic regulation of pulmonary vascular smooth muscle cyclic guanosine monophosphate-dependent kinase by the ubiquitin conjugating system. Am. J. Respir. Cell Mol. Biol. 2012, 46, 323–330. [Google Scholar] [CrossRef]
- Frid, M.G.; Brunetti, J.A.; Burke, D.L.; Carpenter, T.C.; Davie, N.J.; Reeves, J.T.; Roedersheimer, M.T.; van Rooijen, N.; Stenmark, K.R. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006, 168, 659–669. [Google Scholar] [CrossRef]
- Davie, N.J.; Crossno, J.T., Jr.; Frid, M.G.; Hofmeister, S.E.; Reeves, J.T.; Hyde, D.M.; Carpenter, T.C.; Brunetti, J.A.; McNiece, I.K.; Stenmark, K.R. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: Contribution of progenitor cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L668–L678. [Google Scholar] [CrossRef]
- Favre, S.; Gambini, E.; Nigro, P.; Scopece, A.; Bianciardi, P.; Caretti, A.; Pompilio, G.; Corno, A.F.; Vassalli, G.; von Segesser, L.K.; et al. Sildenafil attenuates hypoxic pulmonary remodelling by inhibiting bone marrow progenitor cells. J. Cell. Mol. Med. 2017, 21, 871–880. [Google Scholar] [CrossRef]
- Nydegger, C.; Corno, A.F.; von Segesser, L.K.; Beghetti, M.; Samaja, M.; Milano, G. Effects of PDE-5 Inhibition on the Cardiopulmonary System After 2 or 4 Weeks of Chronic Hypoxia. Cardiovasc. Drugs Ther. 2019, 33, 407–414. [Google Scholar] [CrossRef]
- Hornbein, T.F.; Severinghaus, J.W. Carotid chemoreceptor response to hypoxia and acidosis in cats living at high altitude. J. Appl. Physiol. 1969, 27, 837–839. [Google Scholar] [CrossRef]
- Lopez-Barneo, J.; Pardal, R.; Ortega-Saenz, P. Cellular mechanism of oxygen sensing. Annu. Rev. Physiol. 2001, 63, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Leonard, E.M.; Salman, S.; Nurse, C.A. Sensory Processing and Integration at the Carotid Body Tripartite Synapse: Neurotransmitter Functions and Effects of Chronic Hypoxia. Front. Physiol. 2018, 9, 225. [Google Scholar] [CrossRef]
- Del Rio, R.; Andrade, D.C.; Toledo, C.; Diaz, H.S.; Lucero, C.; Arce-Alvarez, A.; Marcus, N.J.; Schultz, H.D. Carotid Body-Mediated Chemoreflex Drive in The Setting of low and High Output Heart Failure. Sci. Rep. 2017, 7, 8035. [Google Scholar] [CrossRef]
- Gao, L.; Ortega-Sáenz, P.; Moreno-Domínguez, A.; López-Barneo, J. Mitochondrial Redox Signaling in O(2)-Sensing Chemoreceptor Cells. Antioxid. Redox Signal. 2022, 37, 274–289. [Google Scholar] [CrossRef] [PubMed]
- López-Barneo, J.; Ortega-Sáenz, P. Mitochondrial acute oxygen sensing and signaling. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 205–225. [Google Scholar] [CrossRef]
- Cabello-Rivera, D.; Ortega-Saenz, P.; Gao, L.; Munoz-Cabello, A.M.; Bonilla-Henao, V.; Schumacker, P.T.; Lopez-Barneo, J. Oxygen regulation of breathing is abolished in mitochondrial complex III-deficient arterial chemoreceptors. Proc. Natl. Acad. Sci. USA 2022, 119, e2202178119. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Semenza, G.L. Gaseous messengers in oxygen sensing. J. Mol. Med. 2012, 90, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Arias-Reyes, C.; Laouafa, S.; Zubieta-DeUrioste, N.; Joseph, V.; Bairam, A.; Schneider Gasser, E.M.; Soliz, J. Erythropoietin Produces a Dual Effect on Carotid Body Chemoreception in Male Rats. Front. Pharm. 2021, 12, 727326. [Google Scholar] [CrossRef]
- Soliz, J.; Schneider-Gasser, E.M.; Arias-Reyes, C.; Aliaga-Raduan, F.; Poma-Machicao, L.; Zubieta-Calleja, G.; Furuya, W.I.; Trevizan-Bau, P.; Dhingra, R.R.; Dutschmann, M. Coping with hypoxemia: Could erythropoietin (EPO) be an adjuvant treatment of COVID-19? Respir. Physiol. Neurobiol. 2020, 279, 103476. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.J.; Nanduri, J.; Wang, N.; Khan, S.A.; Pamenter, M.E.; Prabhakar, N.R. Carotid body responses to O2 and CO2 in hypoxia-tolerant naked mole rats. Acta Physiol. 2022, 236, e13851. [Google Scholar] [CrossRef] [PubMed]
- Ivy, C.M.; Velotta, J.P.; Cheviron, Z.A.; Scott, G.R. Genetic variation in HIF-2α attenuates ventilatory sensitivity and carotid body growth in chronic hypoxia in high-altitude deer mice. J. Physiol. 2022, 600, 4207–4225. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Takahashi, N.; Chen, C.; Pauli, J.L.; Kuroki, C.; Kaminosono, J.; Kashiwadani, H.; Kanmura, Y.; Mori, Y.; Ou, S.; et al. Transient Receptor Potential Ankyrin 1 Mediates Hypoxic Responses in Mice. Front. Physiol. 2020, 11, 576209. [Google Scholar] [CrossRef]
- Villadiego, J.; Ramirez-Lorca, R.; Cala, F.; Labandeira-Garcia, J.L.; Esteban, M.; Toledo-Aral, J.J.; Lopez-Barneo, J. Is Carotid Body Infection Responsible for Silent Hypoxemia in COVID-19 Patients? Function 2021, 2, zqaa032. [Google Scholar] [CrossRef]
- Fitzgerald, R.S.; Rocher, A. Physiology and Pathophysiology of Oxygen Sensitivity. Antioxidants 2021, 10, 1114. [Google Scholar] [CrossRef]
- Winslow, R.M.; Chapman, K.W.; Gibson, C.C.; Samaja, M.; Monge, C.C.; Goldwasser, E.; Sherpa, M.; Blume, F.D.; Santolaya, R. Different hematologic responses to hypoxia in Sherpas and Quechua Indians. J. Appl. Physiol. (1985) 1989, 66, 1561–1569. [Google Scholar] [CrossRef]
- Lahiri, S.; Edelman, N.; Cherniack, N.; Fishman, A. Blunted hypoxic drive to ventilation in subjects with life-long hypoxemia. Fed. Proc. 1969, 28, 1289–1295. [Google Scholar]
- Mouradian, G.C., Jr.; Lakshminrusimha, S.; Konduri, G.G. Perinatal Hypoxemia and Oxygen Sensing. Compr. Physiol. 2021, 11, 1653–1677. [Google Scholar] [CrossRef]
- Pierce, A.M.; Brown, L.K. Obesity hypoventilation syndrome: Current theories of pathogenesis. Curr. Opin. Pulm. Med. 2015, 21, 557–562. [Google Scholar] [CrossRef]
- Ora, J.; Rogliani, P.; Dauri, M.; O’Donnell, D. Happy hypoxemia, or blunted ventilation? Respir. Res. 2021, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.; Samaja, M.; West, J. Red cell function at extreme altitudes on Mount Everest. J. Appl. Physiol. 1984, 56, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Farnam, A. pH of soul: How does acid-base balance affect our cognition? Bioimpacts 2014, 4, 53–54. [Google Scholar] [CrossRef] [PubMed]
- McMorris, T.; Hale, B.J.; Barwood, M.; Costello, J.; Corbett, J. Effect of acute hypoxia on cognition: A systematic review and meta-regression analysis. Neurosci. Biobehav. Rev. 2017, 74, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Zou, L.; Yu, J.J.; Ryu, S.; Kong, Z.; Yang, L.; Kang, M.; Lin, J.; Li, H.; Smith, L.; et al. Does exercise have a protective effect on cognitive function under hypoxia? A systematic review with meta-analysis. J. Sport Health Sci. 2020, 9, 562–577. [Google Scholar] [CrossRef]
- Falla, M.; Papagno, C.; Dal Cappello, T.; Vögele, A.; Hüfner, K.; Kim, J.; Weiss, E.M.; Weber, B.; Palma, M.; Mrakic-Sposta, S.; et al. A Prospective Evaluation of the Acute Effects of High Altitude on Cognitive and Physiological Functions in Lowlanders. Front. Physiol. 2021, 12, 670278. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.X.; Li, P.; Jiang, C.H.; Liu, C.; Chen, Y.; Chen, L.; Ruan, H.Z.; Gao, Y.Q. Psychological and cognitive impairment of long-term migrators to high altitudes and the relationship to physiological and biochemical changes. Eur. J. Neurol. 2015, 22, 1363–1369. [Google Scholar] [CrossRef]
- Merz, T.M.; Bosch, M.M.; Barthelmes, D.; Pichler, J.; Hefti, U.; Schmitt, K.U.; Bloch, K.E.; Schoch, O.D.; Hess, T.; Turk, A.J.; et al. Cognitive performance in high-altitude climbers: A comparative study of saccadic eye movements and neuropsychological tests. Eur. J. Appl. Physiol. 2013, 113, 2025–2037. [Google Scholar] [CrossRef]
- Phillips, J.B.; Horning, D.; Funke, M.E. Cognitive and perceptual deficits of normobaric hypoxia and the time course to performance recovery. Aerosp. Med. Hum. Perform. 2015, 86, 357–365. [Google Scholar] [CrossRef]
- Yan, X. Cognitive impairments at high altitudes and adaptation. High Alt. Med. Biol. 2014, 15, 141–145. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, J.; Liu, F.; Luo, Y.; Xu, G.; Shen, H.Y.; Gao, Y.; Gao, W. Chronic mountain sickness in Chinese Han males who migrated to the Qinghai-Tibetan plateau: Application and evaluation of diagnostic criteria for chronic mountain sickness. BMC Public Health 2014, 14, 701. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.M. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc. Natl. Acad. Sci. USA 2007, 104 (Suppl. 1), 8655–8660. [Google Scholar] [CrossRef] [PubMed]
- Monge, C.C.; Arregui, A.; Leon-Velarde, F. Pathophysiology and epidemiology of chronic mountain sickness. Int. J. Sport. Med. 1992, 13 (Suppl. 1), S79–S81. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.Y. Chronic mountain sickness on the Qinghai-Tibetan plateau. Chin. Med. J. 2005, 118, 161–168. [Google Scholar]
- Sahota, I.S.; Panwar, N.S. Prevalence of Chronic Mountain Sickness in high altitude districts of Himachal Pradesh. Indian J. Occup. Env. Med. 2013, 17, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, H.I.; Janocha, A.J.; Monocello, L.T.; Garchar, A.C.; Gebremedhin, A.; Erzurum, S.C.; Beall, C.M. Alternative hematological and vascular adaptive responses to high-altitude hypoxia in East African highlanders. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L172–L177. [Google Scholar] [CrossRef]
- Aldashev, A.A.; Sarybaev, A.S.; Sydykov, A.S.; Kalmyrzaev, B.B.; Kim, E.V.; Mamanova, L.B.; Maripov, R.; Kojonazarov, B.K.; Mirrakhimov, M.M.; Wilkins, M.R.; et al. Characterization of high-altitude pulmonary hypertension in the Kyrgyz: Association with angiotensin-converting enzyme genotype. Am. J. Respir. Crit. Care Med. 2002, 166, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Iranmehr, A.; Stobdan, T.; Zhou, D.; Poulsen, O.; Strohl, K.P.; Aldashev, A.; Telenti, A.; Wong, E.H.M.; Kirkness, E.F.; Venter, J.C.; et al. Novel insight into the genetic basis of high-altitude pulmonary hypertension in Kyrgyz highlanders. Eur. J. Hum. Genet 2018, 27, 150–159. [Google Scholar] [CrossRef]
- Santolaya, R.; Lahiri, S.; Alfaro, R.; Schoene, R. Respiratory adaptation in the highest inhabitants and highest Sherpas mountaineers. Respir. Physiol. 1989, 77, 253–262. [Google Scholar] [CrossRef]
- Beall, C.; Brittenham, G.; Macuaga, F.; Barragan, M. Variation in hemoglobin concentration among samples of high-altitude natives in the Andes and the Himalayas. Am. J. Hum. Biol. 1990, 2, 639–651. [Google Scholar] [CrossRef]
- Beall, C.M. Human adaptability studies at high altitude: Research designs and major concepts during fifty years of discovery. Am. J. Hum. Biol. 2013, 25, 141–147. [Google Scholar] [CrossRef]
- Petousi, N.; Croft, Q.P.; Cavalleri, G.L.; Cheng, H.Y.; Formenti, F.; Ishida, K.; Lunn, D.; McCormack, M.; Shianna, K.V.; Talbot, N.P.; et al. Tibetans living at sea level have a hyporesponsive hypoxia-inducible factor system and blunted physiological responses to hypoxia. J. Appl. Physiol. (1985) 2014, 116, 893–904. [Google Scholar] [CrossRef]
- Azad, P.; Stobdan, T.; Zhou, D.; Hartley, I.; Akbari, A.; Bafna, V.; Haddad, G.G. High-altitude adaptation in humans: From genomics to integrative physiology. J. Mol. Med. 2017, 95, 1269–1282. [Google Scholar] [CrossRef]
- Jeong, C.; Witonsky, D.B.; Basnyat, B.; Neupane, M.; Beall, C.M.; Childs, G.; Craig, S.R.; Novembre, J.; Di Rienzo, A. Detecting past and ongoing natural selection among ethnically Tibetan women at high altitude in Nepal. PLoS Genet. 2018, 14, e1007650. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, Z.B.; Chen, J.; Huang, X.F.; Li, X.M.; Liang, Y.B.; Mao, J.Y.; Chen, X.; Zheng, Z.; Bakshi, A.; et al. Genetic signatures of high-altitude adaptation in Tibetans. Proc. Natl. Acad. Sci. USA 2017, 114, 4189–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, J.F. High-Altitude Adaptation: Mechanistic Insights from Integrated Genomics and Physiology. Mol. Biol. Evol. 2021, 38, 2677–2691. [Google Scholar] [CrossRef] [PubMed]
- Simonson, T.S.; Yang, Y.; Huff, C.D.; Yun, H.; Qin, G.; Witherspoon, D.J.; Bai, Z.; Lorenzo, F.R.; Xing, J.; Jorde, L.B.; et al. Genetic evidence for high-altitude adaptation in Tibet. Science 2010, 329, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Pichler Hefti, J.; Sonntag, D.; Hefti, U.; Risch, L.; Schoch, O.D.; Turk, A.J.; Hess, T.; Bloch, K.E.; Maggiorini, M.; Merz, T.M.; et al. Oxidative stress in hypobaric hypoxia and influence on vessel-tone modifying mediators. High Alt. Med. Biol. 2013, 14, 273–279. [Google Scholar] [CrossRef]
- Pasiakos, S.M.; Berryman, C.E.; Carrigan, C.T.; Young, A.J.; Carbone, J.W. Muscle Protein Turnover and the Molecular Regulation of Muscle Mass during Hypoxia. Med. Sci. Sport. Exerc. 2017, 49, 1340–1350. [Google Scholar] [CrossRef]
- Honda, Y.; Tani, H.; Masuda, A.; Kobayashi, T.; Nishino, T.; Kimura, H.; Masuyama, S.; Kuriyama, T. Effect of prior O2 breathing on ventilatory response to sustained isocapnic hypoxia in adult humans. J. Appl. Physiol. (1985) 1996, 81, 1627–1632. [Google Scholar] [CrossRef]
- Dos Santos Quaresma, M.; Souza, W.; Lemos, V.A.; Caris, A.V.; Thomatieli-Santos, R.V. The Possible Importance of Glutamine Supplementation to Mood and Cognition in Hypoxia from High Altitude. Nutrients 2020, 12, 3627. [Google Scholar] [CrossRef] [PubMed]
- Lenis, Y.Y.; Elmetwally, M.A.; Maldonado-Estrada, J.G.; Bazer, F.W. Physiological importance of polyamines. Zygote 2017, 25, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, P.F. Tetrahydropterin-dependent amino acid hydroxylases. Annu. Rev. Biochem. 1999, 68, 355–381. [Google Scholar] [CrossRef]
- Maa, E.H. Hypobaric hypoxic cerebral insults: The neurological consequences of going higher. NeuroRehabilitation 2010, 26, 73–84. [Google Scholar] [CrossRef]
- DelMastro, K.; Hellem, T.; Kim, N.; Kondo, D.; Sung, Y.H.; Renshaw, P.F. Incidence of major depressive episode correlates with elevation of substate region of residence. J. Affect. Disord. 2011, 129, 376–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswan, A.; Singh, C.; Rai, R.K.; Azim, A.; Sinha, N.; Baronia, A.K. Metabolomics based predictive biomarker model of ARDS: A systemic measure of clinical hypoxemia. PLoS ONE 2017, 12, e0187545. [Google Scholar] [CrossRef]
- Kelly, R.S.; Dahlin, A.; McGeachie, M.J.; Qiu, W.; Sordillo, J.; Wan, E.S.; Wu, A.C.; Lasky-Su, J. Asthma Metabolomics and the Potential for Integrative Omics in Research and the Clinic. Chest 2017, 151, 262–277. [Google Scholar] [CrossRef]
- Denihan, N.M.; Kirwan, J.A.; Walsh, B.H.; Dunn, W.B.; Broadhurst, D.I.; Boylan, G.B.; Murray, D.M. Untargeted metabolomic analysis and pathway discovery in perinatal asphyxia and hypoxic-ischaemic encephalopathy. J. Cereb. Blood Flow Metab. 2019, 39, 147–162. [Google Scholar] [CrossRef]
- Murthi, P.; Wallace, E.M.; Walker, D.W. Altered placental tryptophan metabolic pathway in human fetal growth restriction. Placenta 2017, 52, 62–70. [Google Scholar] [CrossRef]
- Chen, J.; Sysol, J.R.; Singla, S.; Zhao, S.; Yamamura, A.; Valdez-Jasso, D.; Abbasi, T.; Shioura, K.M.; Sahni, S.; Reddy, V.; et al. Nicotinamide Phosphoribosyltransferase Promotes Pulmonary Vascular Remodeling and Is a Therapeutic Target in Pulmonary Arterial Hypertension. Circulation 2017, 135, 1532–1546. [Google Scholar] [CrossRef]
- Gassmann, M.; Cowburn, A.; Gu, H.; Li, J.; Rodriguez, M.; Babicheva, A.; Jain, P.P.; Xiong, M.; Gassmann, N.N.; Yuan, J.X.; et al. Hypoxia-induced pulmonary hypertension-Utilizing experiments of nature. Br. J. Pharm. 2021, 178, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Sztormowska-Achranowicz, K.; Jankowski, Z.; Kocic, I. Protective effect of nicotinamide and L-arginine against monocrotaline-induced pulmonary hypertension in rats: Gender dependence. Pharm. Rep. 2020, 72, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Bai, P.; Wan, N.; Liu, J.; Zhu, Q.; He, Y.; Chen, G.; Wang, J.; Chen, H.; Wang, C.; et al. Niacin Attenuates Pulmonary Hypertension Through H-PGDS in Macrophages. Circ. Res. 2020, 127, 1323–1336. [Google Scholar] [CrossRef]
- Ayalew, W.; Chu, M.; Liang, C.; Wu, X.; Yan, P. Adaptation Mechanisms of Yak (Bos grunniens) to High-Altitude Environmental Stress. Animals 2021, 11, 2344. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraga, F.A.; Reyes, R.V.; Ebensperger, G.; López, V.; Llanos, A.J. Enhanced Vasoconstriction Mediated by α(1)-Adrenergic Mechanisms in Small Femoral Arteries in Newborn Llama and Sheep Gestated at Low and High Altitudes. Front. Physiol. 2021, 12, 697211. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Wei, L.; Xu, B.; Wang, Z.; Gao, C.; Li, J.; Wei, L.; Qi, D.; Shi, P.; Zhang, T.; et al. A homotetrameric hemoglobin expressed in alveolar epithelial cells increases blood oxygenation in high-altitude plateau pika (Ochotona curzoniae). Cell. Rep. 2022, 41, 111446. [Google Scholar] [CrossRef]
- Hashimoto, T.; Laporte, N.; Mawatari, K.; Ellis, R.S.; Inoue, A.K.; Zackrisson, E.; Roberts-Borsani, G.; Zheng, W.; Tamura, Y.; Bauer, F.E.; et al. The onset of star formation 250 million years after the Big Bang. Nature 2018, 557, 392–395. [Google Scholar] [CrossRef]
- Stickney, J.C.; Van Liere, E.J. Acclimatization to low oxygen tension. Physiol. Rev. 1953, 33, 13–34. [Google Scholar] [CrossRef]
- Tremblay, J.C.; Ainslie, P.N. Global and country-level estimates of human population at high altitude. Proc. Natl. Acad. Sci. USA 2021, 118, e2102463118. [Google Scholar] [CrossRef]
Figure 1.
Behavior of the barometric pressure (left Y-axis) and of the effective %O2 (right Y-axis) at varying altitude above sea level in the Earth atmosphere. Data from the International Standard Atmosphere (ISA). The ISA does not contain water.
Figure 1.
Behavior of the barometric pressure (left Y-axis) and of the effective %O2 (right Y-axis) at varying altitude above sea level in the Earth atmosphere. Data from the International Standard Atmosphere (ISA). The ISA does not contain water.

Figure 2.
Behavior of maximal (red) and resting (blue) VO2 at altitude. Data redrawn from [12], which reports the average of 146 studies performed at various real and simulated altitudes in the 0–9000 m range regardless of the training degree. The dashed area represents the O2 available to perform exercise.
Figure 2.
Behavior of maximal (red) and resting (blue) VO2 at altitude. Data redrawn from [12], which reports the average of 146 studies performed at various real and simulated altitudes in the 0–9000 m range regardless of the training degree. The dashed area represents the O2 available to perform exercise.

Figure 3.
The oxygen cascade in normoxia (blue) and in hypoxia (red, which depicts the situation at 3400 m/13.9 %O2). The arrows indicate the sites of resistance to the O2 flux that increase the PO2 gradient. These sites of resistance may convert a normoxic situation into a hypoxic one. Other explanations in the text.
Figure 3.
The oxygen cascade in normoxia (blue) and in hypoxia (red, which depicts the situation at 3400 m/13.9 %O2). The arrows indicate the sites of resistance to the O2 flux that increase the PO2 gradient. These sites of resistance may convert a normoxic situation into a hypoxic one. Other explanations in the text.

Figure 4.
Three ways to respond to a challenge.

Figure 5.
Scheme of the ATP production paths. In normoxia, most of the ATP is produced aerobically with a stringent need for O2 during oxidative phosphorylation, which follows the tricarboxylic acid (TCA) cycle. The utilization of O2 is helped by the presence of myoglobin (Mb) in contractile tissues. The oxidizable substrates that most contribute to aerobic ATP production are glucose, fatty acids, and (in heart and brain) ketone bodies. In acute hypoxia (red arrows and crosses), the O2 supply to oxidative phosphorylation is reduced along with the aerobic ATP production. Reduced disposal of acetyl-CoA by the TCA cycle, which is inhibited by hypoxia because of the hypoxia-induced downregulation of oxidative phosphorylation, in turn blocks the utilization of fatty acids (and ketone bodies in heart and brain). Simultaneous stimulation of glycolysis transiently increases anaerobic ATP production from glucose at the cost of increased lactate synthesis as partial compensation for the depressed aerobic ATP production. For longer hypoxia (brown dotted line), lactate overproduction is not compensated by proportional upregulation of the monocarboxylate transporters (MCT) at least in contractile tissues (see 3.1.4 Lactate shuttle). This leads to intracellular lactate buildup with lactate-driven inhibition of the glycolytic flux and impairment of anaerobic ATP production.
Figure 5.
Scheme of the ATP production paths. In normoxia, most of the ATP is produced aerobically with a stringent need for O2 during oxidative phosphorylation, which follows the tricarboxylic acid (TCA) cycle. The utilization of O2 is helped by the presence of myoglobin (Mb) in contractile tissues. The oxidizable substrates that most contribute to aerobic ATP production are glucose, fatty acids, and (in heart and brain) ketone bodies. In acute hypoxia (red arrows and crosses), the O2 supply to oxidative phosphorylation is reduced along with the aerobic ATP production. Reduced disposal of acetyl-CoA by the TCA cycle, which is inhibited by hypoxia because of the hypoxia-induced downregulation of oxidative phosphorylation, in turn blocks the utilization of fatty acids (and ketone bodies in heart and brain). Simultaneous stimulation of glycolysis transiently increases anaerobic ATP production from glucose at the cost of increased lactate synthesis as partial compensation for the depressed aerobic ATP production. For longer hypoxia (brown dotted line), lactate overproduction is not compensated by proportional upregulation of the monocarboxylate transporters (MCT) at least in contractile tissues (see 3.1.4 Lactate shuttle). This leads to intracellular lactate buildup with lactate-driven inhibition of the glycolytic flux and impairment of anaerobic ATP production.

Figure 6.
Simplified scheme of the mechanisms that modulate the NO–cGMP pathway in the smooth muscle cell. Four compartments are examined: the endothelial cells (green) that generate NO, the RBCs (red) that store NO as nitrosylated Hb (HbNO) or nitrosothiolated Hb (HbSNO), the plasma that stores NO as inorganic nitrites and nitrates (NO2− + NO3−), and the smooth muscle cells (light blue). Here, NO favors the decrease in [Ca++] that is sequestered in the sarcoplasmic reticulum, diminishing the activity of myosin kinase and favoring muscle relaxation.
Figure 6.
Simplified scheme of the mechanisms that modulate the NO–cGMP pathway in the smooth muscle cell. Four compartments are examined: the endothelial cells (green) that generate NO, the RBCs (red) that store NO as nitrosylated Hb (HbNO) or nitrosothiolated Hb (HbSNO), the plasma that stores NO as inorganic nitrites and nitrates (NO2− + NO3−), and the smooth muscle cells (light blue). Here, NO favors the decrease in [Ca++] that is sequestered in the sarcoplasmic reticulum, diminishing the activity of myosin kinase and favoring muscle relaxation.

Figure 7.
Scheme of iron handling and the impact of hypoxia and inflammation.

Figure 8.
Respiratory adjustment to altitude (6450 m/9.3 %O2) in unacclimatized Caucasians (red) and acclimatized Sherpas (blue). Data from [212,264].

Figure 9.
Hemoglobin concentration in the blood of Tibetan and Indian males (n = 37 and 56, respectively) residing at <500 m/>20.1 %O2. Tibetans display a lower Hb level (p < 0.0001, Student’s t-test). The green lines represent the 95% confidence limits.
Figure 9.
Hemoglobin concentration in the blood of Tibetan and Indian males (n = 37 and 56, respectively) residing at <500 m/>20.1 %O2. Tibetans display a lower Hb level (p < 0.0001, Student’s t-test). The green lines represent the 95% confidence limits.


{kind=link}
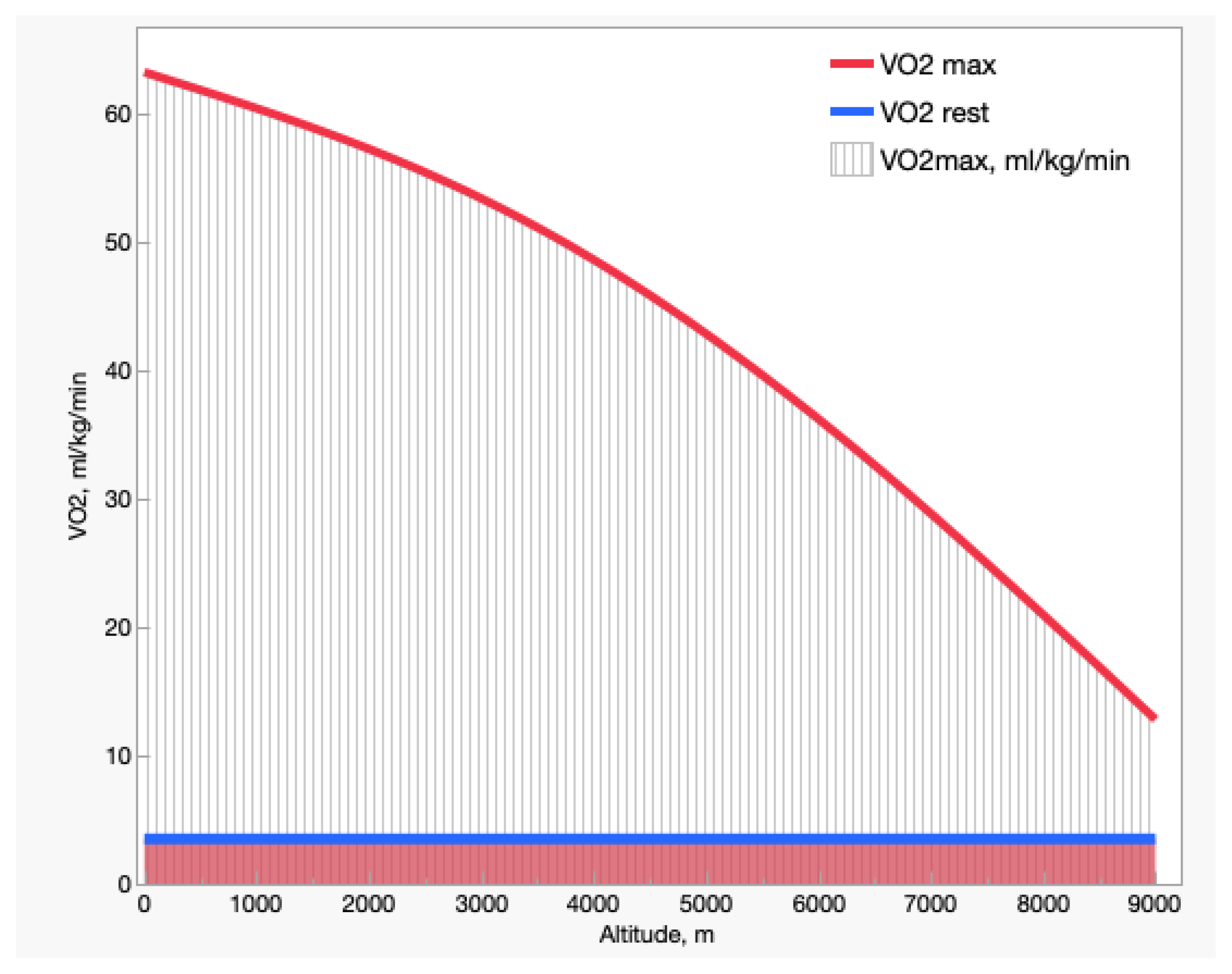{kind=link}
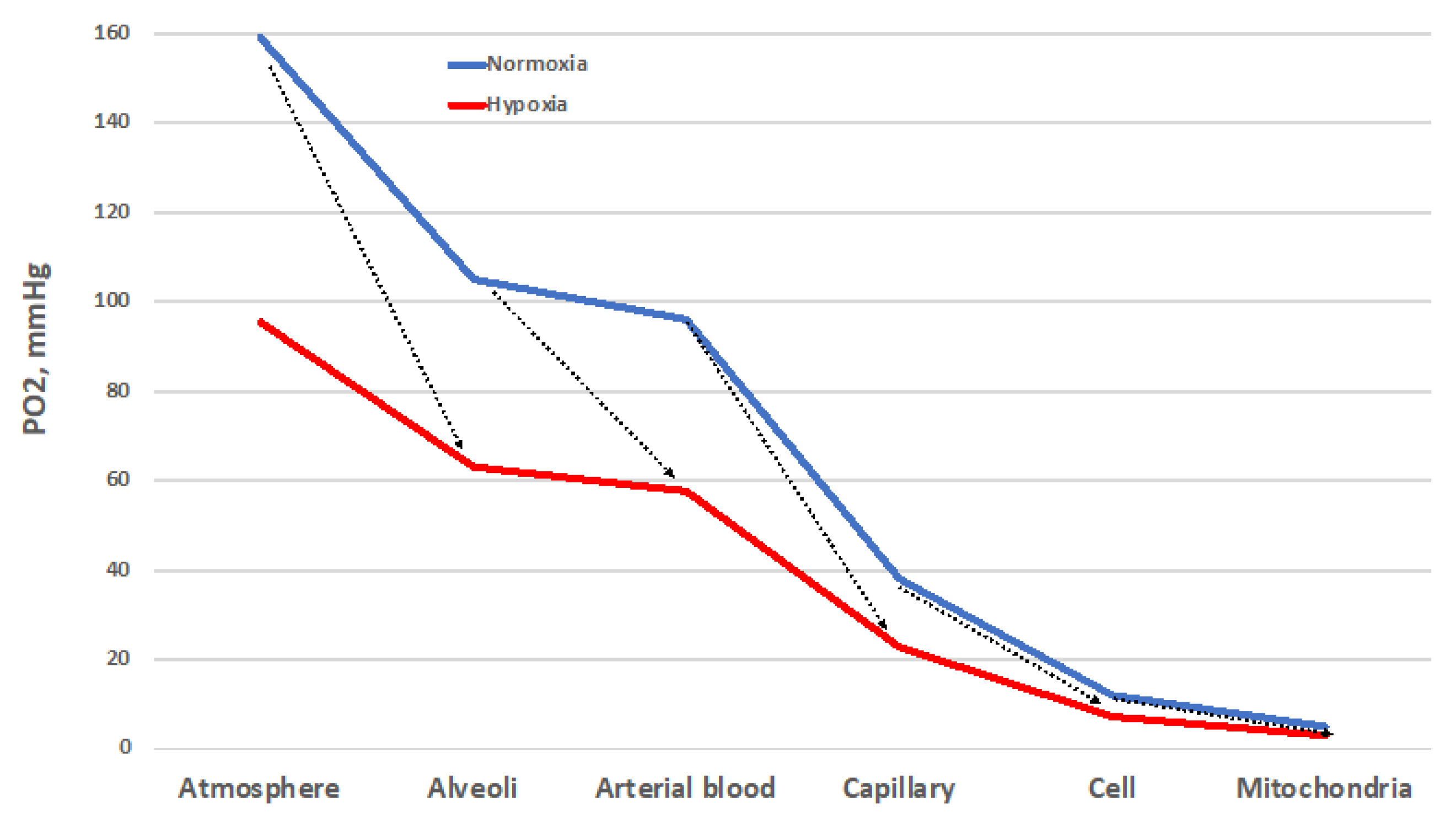{kind=link}
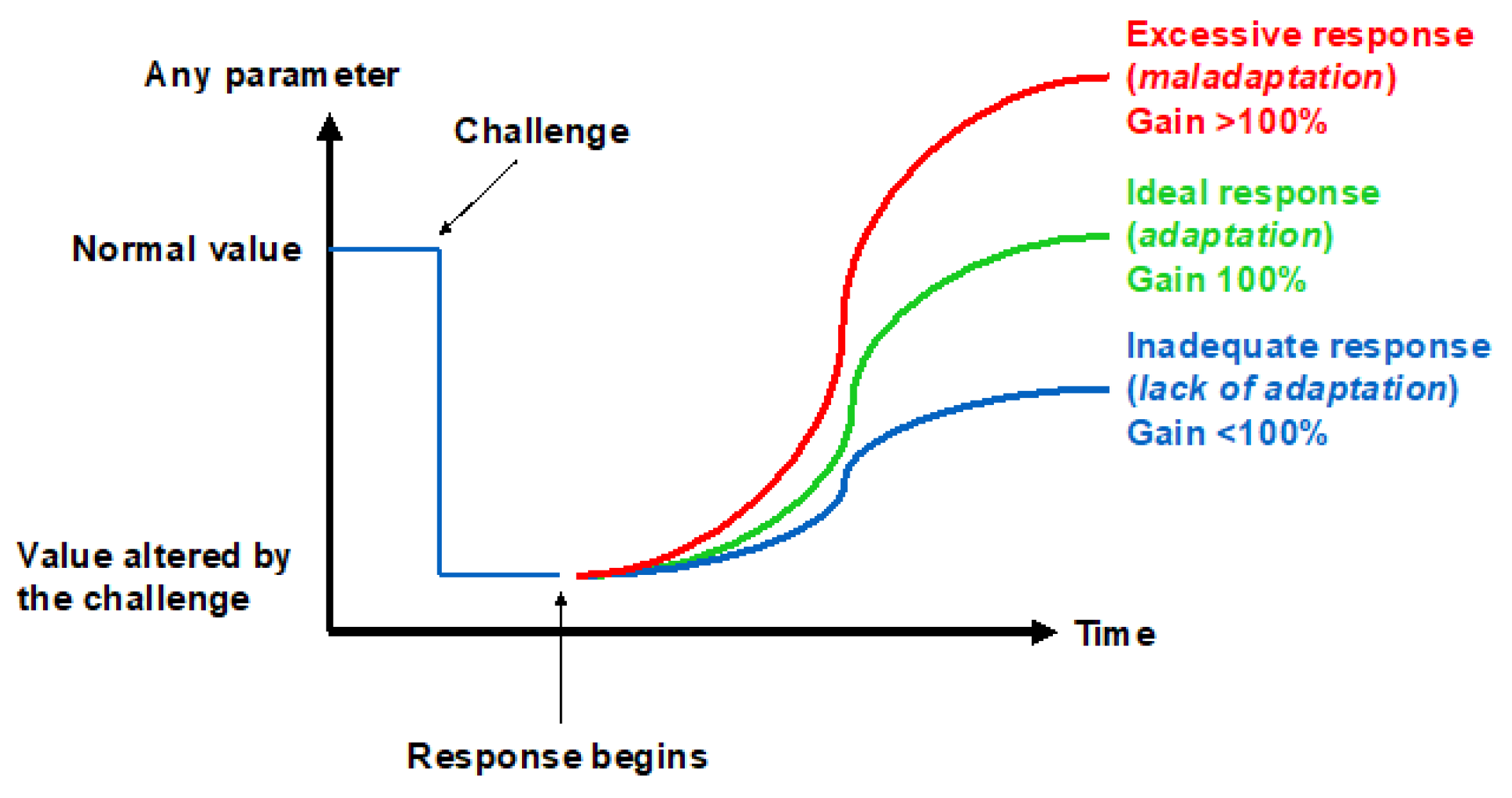{kind=link}
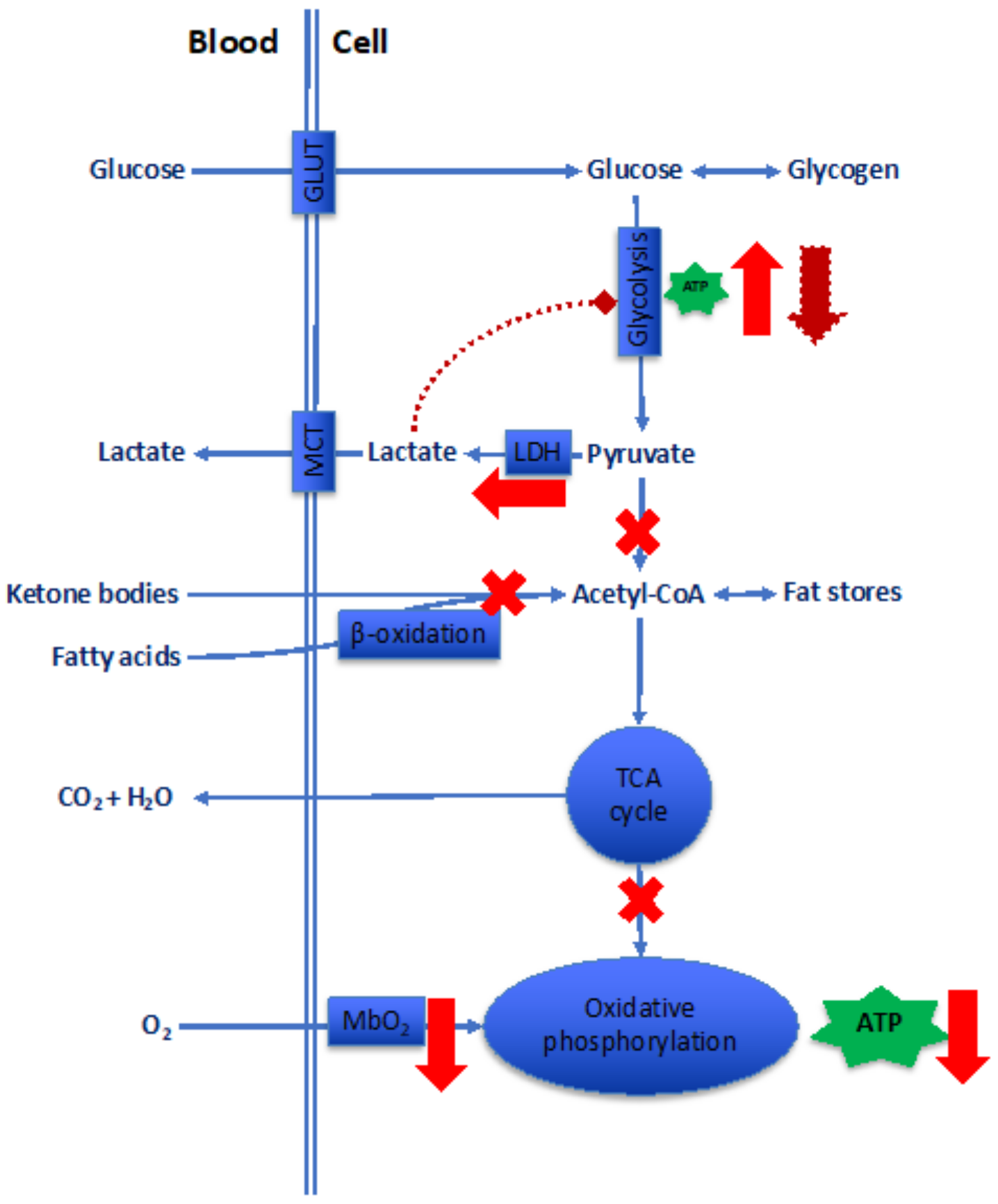{kind=link}
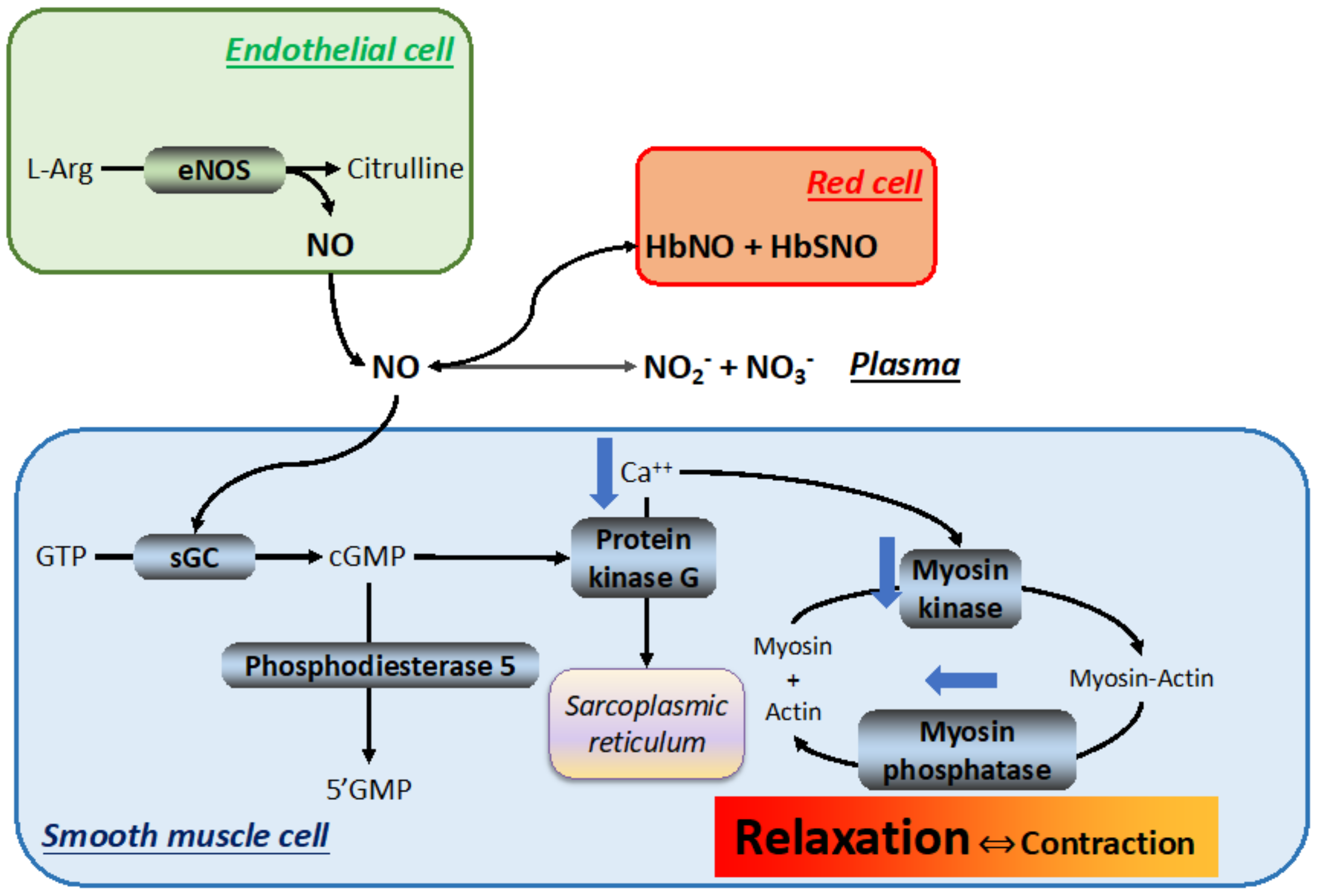{kind=link}
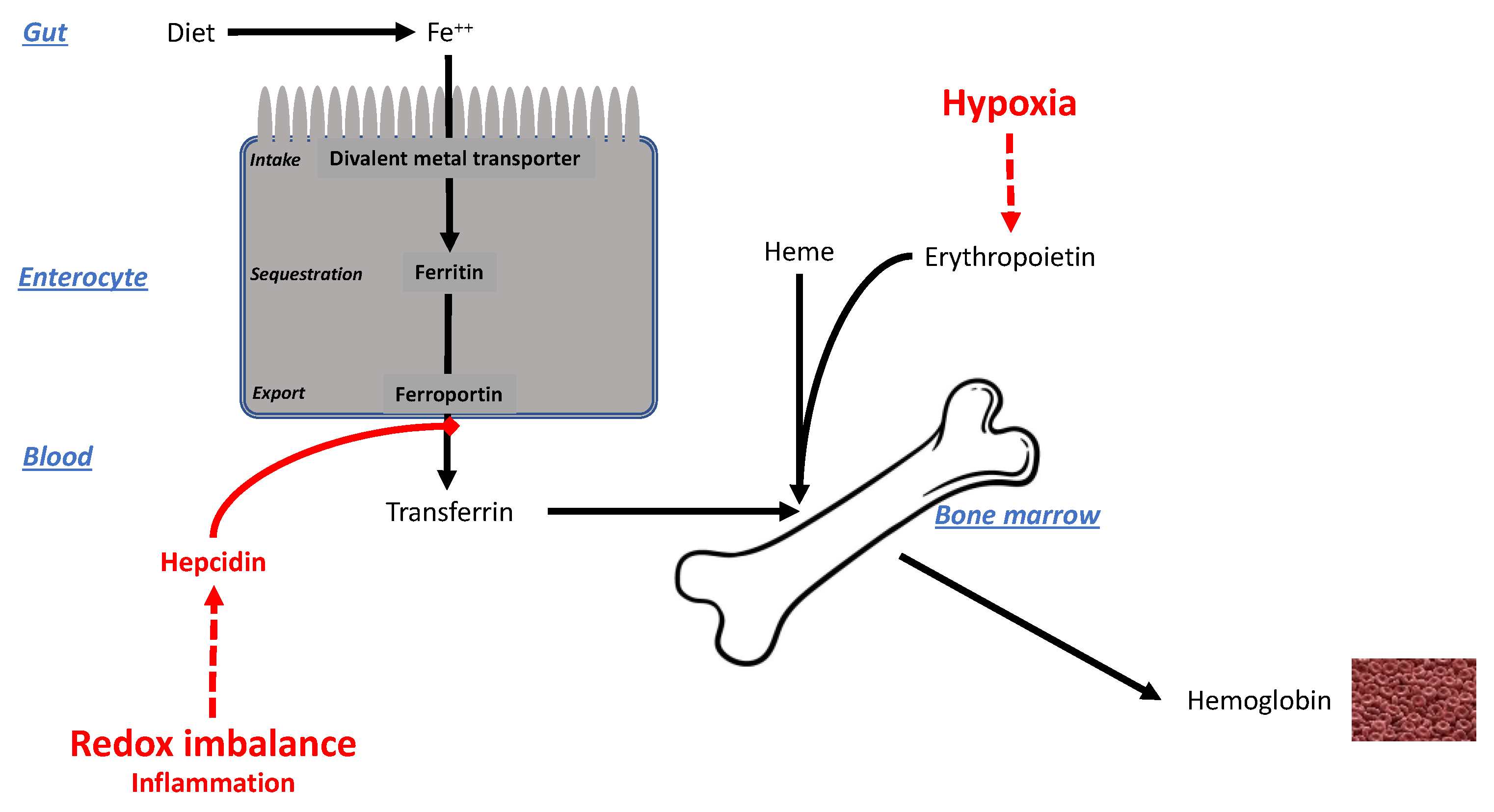{kind=link}
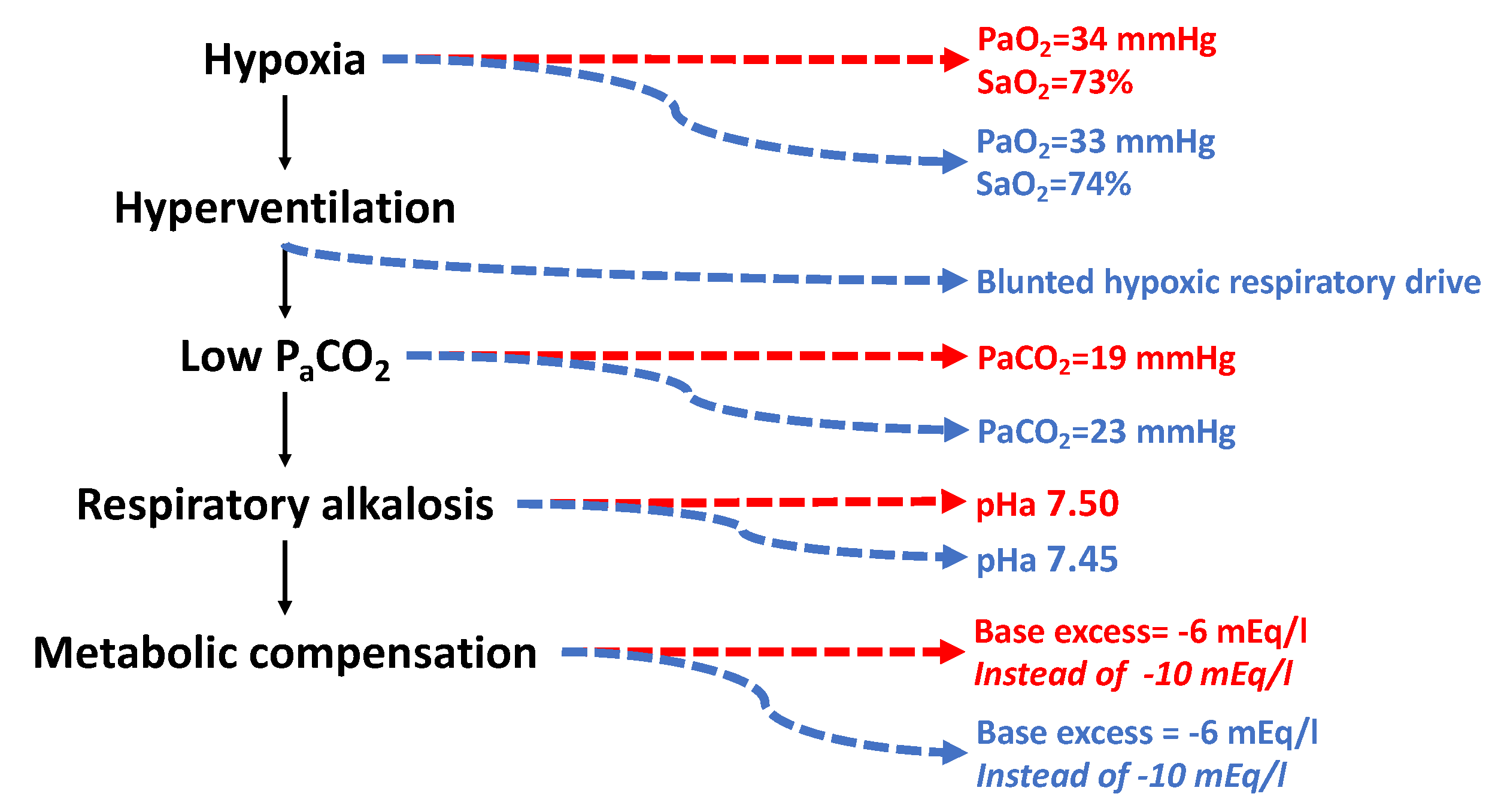{kind=link}
{kind=link}
Table 1.
A few historical excerpts on the impact of hypoxia on mankind.
| 30 BC: First description of the effects of hypoxia by Tseen Han Shoo, while traveling along the Silk Road into Hindu Kush | ”...the Great Headache Mountain, the Little Headache Mountain [… where] men’s bodies become feverish, they lose color, and are attacked with headache and vomiting...” [13] |
| 1590: Padre Jose de Acosta, Spanish Jesuit, while crossing the Andes | “Many soldiers felt ill during the passage through Paso de Pariacaca” (4800 m/11.6 %O2), “… I was surprised by the so strange pain and deathful that I fell on the ground and noticed that air was so thin and not suitable for human breathing…” |
| 1862: Henry Coxwell and James Glaisher, when flying at 8850 m at Wolverhampton | “.. Arms and legs became paralyzed, we felt blindness, difficulty in talking and experienced loss of conscience …” When back, they could recover fully. |
| 1875: Sivel, Tissandier and Croce-Spinelli, in a balloon at 8500 m | They experienced loss of consciousness; Tissandier only recovered but became deaf. |
| 1897: Angelo Mosso (1846–1910) | Published “Fisiologia dell’Uomo sulle Alpi” (Human Physiology in the Alps, Fratelli Treves Editors), where he described experiments performed at Capanna Regina Margherita, 4554 m/12.0 %O2, accounting for muscle strength, respiration, blood circulation, myocardial performance, nutrition, vital capacity, mountain sickness, and neurophysiological issues. |
| 1916: Alexander Kellas (1868–1921) | In an article for the Royal Geographical Society, he analyzed the possibility of reaching the highest peaks of the Himalayas. In a later manuscript, which he failed to publish in life, he concluded that a physically gifted and well-trained man is able to climb Everest without O2 [14]. |
| 1953: Edmund Hillary and Tensing Norkay | First climb of Mt. Everest (8848 m/6.6 %O2) with the aid of supplemental O2. |
| 1978: Reinhold Messner and Peter Habeler | First climb of Mt. Everest without the aid of supplemental O2. |
| 1979–2022: Mt. Everest (8848 m/6.6 %O2) | Up to 270 climbers died, most from causes linked directly or indirectly to hypoxia. |
| 1987: Robert M. Winslow (1941–2009) and Carlos Monge Cassinelli (1921–2006) | Published “Hypoxia, Polycythemia, and Chronic Mountain Sickness” (Johns Hopkins University Press, ISBN 0-8018-3448-1) in which they describe the origins of chronic mountain sickness in high-altitude natives in various parts of the world. |
Table 2.
Oxygen transport characteristics in Caucasians and Sherpas at varying altitudes. All data except P50 are from [212]. The P50 was calculated using the equations reported in [209] assuming carbon-monoxide-bound Hb = 0 and temperature = 37 °C.
| Altitude/%O2 | Arterial pH | Arterial PCO2 | Arterial PO2 | [Hb] | DPG/Hb | P50 | |
|---|---|---|---|---|---|---|---|
| mmHg | mmHg | g/L | mmHg | ||||
| Caucasians | 0/21.0 | 7.38 | 40 | 95 | 160 | 0.80 | 27.9 |
| 3400/13.9 | 7.46 | 22 | 51 | 170 | 1.03 | 27.4 | |
| 5050/11.3 | 7.48 | 21 | 44 | 191 | 1.04 | 26.9 | |
| 6450/9.3 | 7.50 | 19 | 34 | 201 | 1.36 | 28.8 | |
| Sherpas | 3400/13.9 | 7.46 | 29 | 53 | 169 | 1.01 | 27.5 |
| 5050/11.3 | 7.45 | 27 | 42 | 186 | 1.04 | 27.9 | |
| 6450/9.3 | 7.45 | 22 | 33 | NA | 1.20 | 29.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Samaja, M.; Ottolenghi, S. The Oxygen Cascade from Atmosphere to Mitochondria as a Tool to Understand the (Mal)adaptation to Hypoxia. Int. J. Mol. Sci. 2023, 24, 3670. https://doi.org/10.3390/ijms24043670
AMA Style
Samaja M, Ottolenghi S. The Oxygen Cascade from Atmosphere to Mitochondria as a Tool to Understand the (Mal)adaptation to Hypoxia. International Journal of Molecular Sciences. 2023; 24(4):3670. https://doi.org/10.3390/ijms24043670
Chicago/Turabian StyleSamaja, Michele, and Sara Ottolenghi. 2023. "The Oxygen Cascade from Atmosphere to Mitochondria as a Tool to Understand the (Mal)adaptation to Hypoxia" International Journal of Molecular Sciences 24, no. 4: 3670. https://doi.org/10.3390/ijms24043670
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.