

Brugada Syndrome: From Molecular Mechanisms and Genetics to Risk Stratification
 , and
, and
Abstract
:1. Introduction
2. Clinical Characteristics and Diagnostic Criteria of BrS
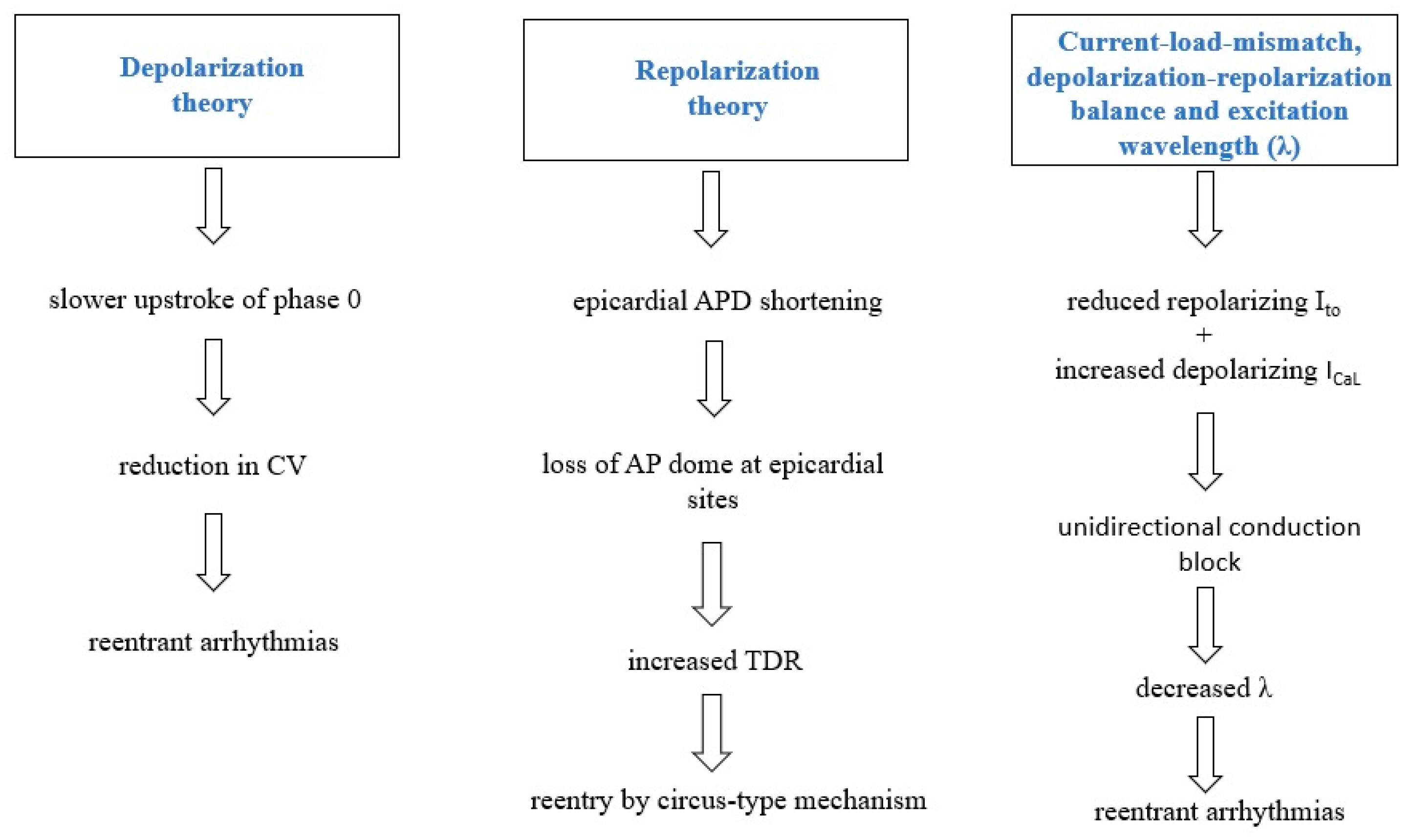
3. Pathophysiology of BrS
3.1. Depolarization Theory
3.2. Repolarization Theory
3.3. Current-Load-Mismatch, Depolarization-Repolarization Balance and Excitation Wavelength (λ)
4. Genetics of BrS
5. Human Induced Pluripotent Stem Cell Models
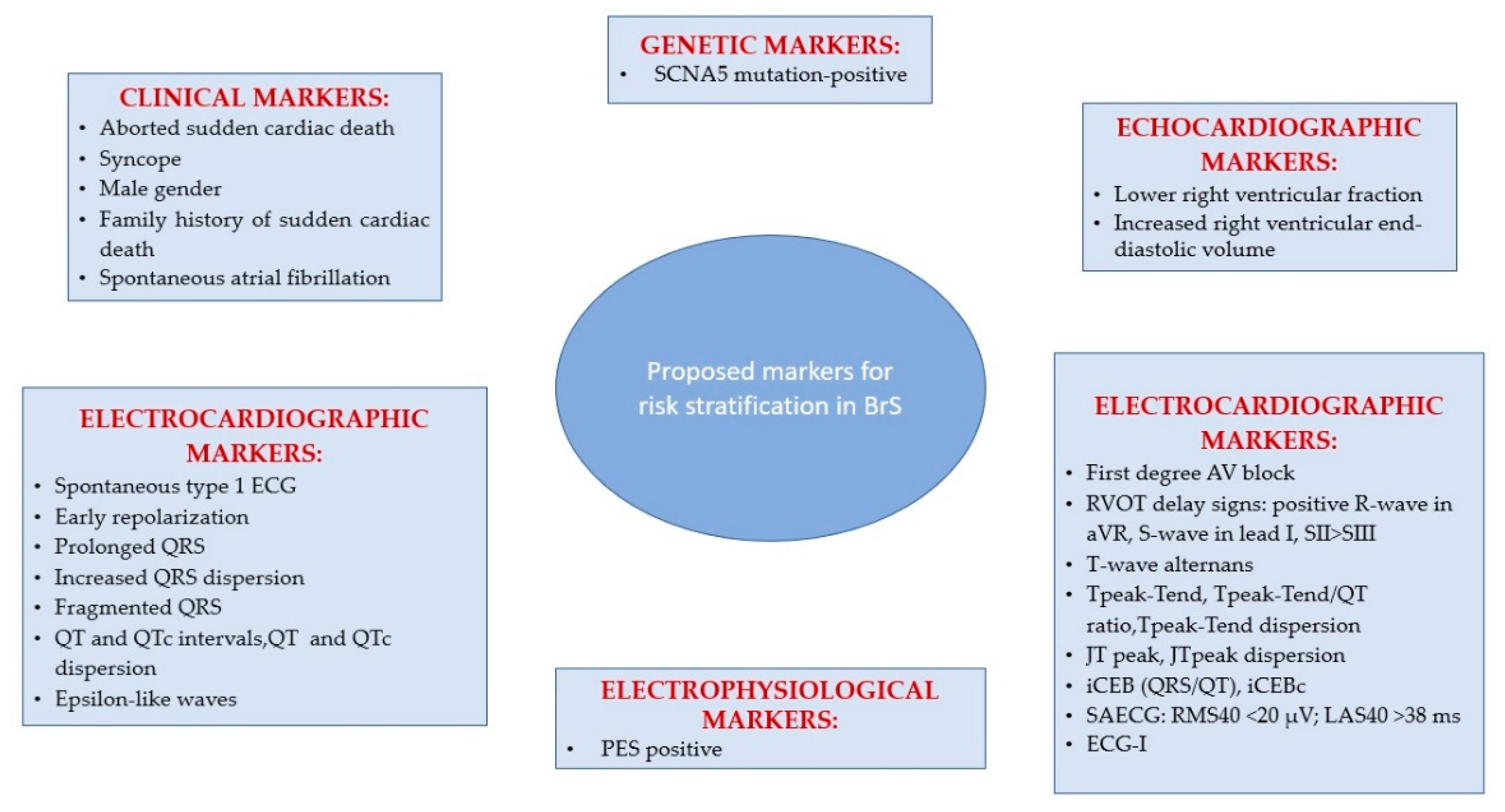
6. Arrhythmias and Sudden Cardiac Death Risk Stratification in BrS
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Juang, J.-M.; Huang, S.K.S. Brugada syndrome—An under-recognized electrical disease in patients with sudden cardiac death. Cardiology 2004, 101, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mittal, S.; Sherrid, M.V. Current review of Brugada syndrome: From epidemiology to treatment. Anadolu Kardiyol. Derg. 2009, 9 (Suppl. 2), 12–16. [Google Scholar] [PubMed]
- Sendfeld, F.; Selga, E.; Scornik, F.S.; Pérez, G.J.; Mills, N.L.; Brugada, R. Experimental Models of Brugada syndrome. Int. J. Mol. Sci. 2019, 20, 2123. [Google Scholar] [CrossRef] [PubMed]
- Gussak, I.; Antzelevitch, C.; Bjerregaard, P.; Towbin, J.A.; Chaitman, B.R. The Brugada syndrome: Clinical, electrophysiologic and genetic aspects. J. Am. Coll. Cardiol. 1999, 33, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef]
- London, B. Letter by London Regarding Article, “Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome”. Circulation 2019, 139, 1758–1759. [Google Scholar] [CrossRef]
- Garcia, J.; Tahiliani, J.; Johnson, N.M.; Aguilar, S.; Beltran, D.; Daly, A.; Decker, E.; Haverfield, E.; Herrera, B.; Murillo, L.; et al. Clinical genetic testing for the cardiomyopathies and arrhythmias: A systematic framework for establishing clinical validity and addressing genotypic and phenotypic heterogeneity. Front. Cardiovasc. Med. 2016, 3, 20. [Google Scholar] [CrossRef]
- Kline, J.; Costantini, O. Inherited cardiac arrhythmias and channelopathies. Med. Clin. N. Am. 2019, 103, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Skinner, J.R.; Winbo, A.; Abrams, D.; Vohra, J.; Wilde, A.A. Channelopathies that lead to sudden cardiac death: Clinical and genetic aspects. Heart Lung Circ. 2019, 28, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Patocskai, B. Brugada syndrome: Clinical, genetic, molecular, cellular, and ionic aspects. Curr. Probl. Cardiol. 2016, 41, 7–57. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, L.; Kirchhof, P.; Johna, R.; Haverkamp, W.; Breithardt, G.; Borggrefe, M. Wolff-Parkinson-White syndrome associated with Brugada syndrome. Pacing Clin. Electrophysiol. 2001, 24, 1423–1424. [Google Scholar] [CrossRef]
- Morita, H.; Fukushima-Kusano, K.; Nagase, S.; Miyaji, K.; Hiramatsu, S.; Banba, K.; Nishii, N.; Watanabe, A.; Kakishita, M.; Takenaka-Morita, S.; et al. Sinus node function in patients with Brugada-type ECG. Circ. J. 2004, 68, 473–476. [Google Scholar] [CrossRef]
- Takehara, N.; Makita, N.; Kawabe, J.; Sato, N.; Kawamura, Y.; Kitabatake, A.; Kikuchi, K. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J. Intern. Med. 2004, 255, 137–142. [Google Scholar] [CrossRef]
- Bordachar, P.; Reuter, S.; Garrigue, S.; Caï, X.; Hocini, M.; Jaïs, P.; Haïssaguerre, M.; Clementy, J. Incidence, clinical implications and prognosis of atrial arrhythmias in Brugada syndrome. Eur. Heart J. 2004, 25, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Antzelevitch, C.; Borggrefe, M.; Brugada, J.; Brugada, R.; Brugada, P.; Corrado, D.; Hauer, R.N.W.; Kass, R.S.; Nademanee, K.; et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation 2002, 106, 2514–2519. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Perez Riera, A.R.; et al. Brugada syndrome: Report of the second consensus conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005, 111, 659–670. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.-E.; Huikuri, H.; et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef]
- Bayés de Luna, A.; Brugada, J.; Baranchuk, A.; Borggrefe, M.; Breithardt, G.; Goldwasser, D.; Lambiase, P.; Riera, A.P.; Garcia-Niebla, J.; Pastore, C.; et al. Current electrocardiographic criteria for diagnosis of Brugada pattern: A consensus report. J. Electrocardiol. 2012, 45, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Executive summary: A report of the american college of cardiology/american heart association task force on clinical practice guidelines and the heart rhythm society. Circulation 2018, 138, e210–e271. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Yan, G.-X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace 2017, 19, 665–694. [Google Scholar] [CrossRef] [PubMed]
- Kawada, S.; Morita, H.; Antzelevitch, C.; Morimoto, Y.; Nakagawa, K.; Watanabe, A.; Nishii, N.; Nakamura, K.; Ito, H. Shanghai score system for diagnosis of brugada syndrome: Validation of the score system and system and reclassification of the patients. JACC Clin. Electrophysiol. 2018, 4, 724–730. [Google Scholar] [CrossRef]
- Walia, J.; Steinberg, C.; Laksman, Z. Brugada syndrome: Updated perspectives. RRCC 2019, 10, 19–32. [Google Scholar] [CrossRef]
- Li, K.H.C.; Lee, S.; Yin, C.; Liu, T.; Ngarmukos, T.; Conte, G.; Yan, G.-X.; Sy, R.W.; Letsas, K.P.; Tse, G. Brugada syndrome: A comprehensive review of pathophysiological mechanisms and risk stratification strategies. Int. J. Cardiol. Heart Vasc. 2020, 26, 100468. [Google Scholar] [CrossRef]
- Brugada, R.; Brugada, J.; Antzelevitch, C.; Kirsch, G.E.; Potenza, D.; Towbin, J.A.; Brugada, P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 2000, 101, 510–515. [Google Scholar] [CrossRef]
- Shimizu, W.; Antzelevitch, C.; Suyama, K.; Kurita, T.; Taguchi, A.; Aihara, N.; Takaki, H.; Sunagawa, K.; Kamakura, S. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2000, 11, 1320–1329. [Google Scholar] [CrossRef]
- Hong, K.; Brugada, J.; Oliva, A.; Berruezo-Sanchez, A.; Potenza, D.; Pollevick, G.D.; Guerchicoff, A.; Matsuo, K.; Burashnikov, E.; Dumaine, R.; et al. Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syndrome caused by SCN5A mutations. Circulation 2004, 110, 3023–3027. [Google Scholar] [CrossRef]
- Meregalli, P.G.; Ruijter, J.M.; Hofman, N.; Bezzina, C.R.; Wilde, A.A.M.; Tan, H.L. Diagnostic value of flecainide testing in unmasking SCN5A-related Brugada syndrome. J. Cardiovasc. Electrophysiol. 2006, 17, 857–864. [Google Scholar] [CrossRef]
- Gavrielatos, G.; Letsas, K.P.; Pappas, L.K.; Efremidis, M.; Sideris, A.; Kardaras, F. Sensitivity and specificity of sodium channel blocking test in the diagnosis of Brugada syndrome. Int. J. Cardiol. 2010, 141, e31–e33. [Google Scholar] [CrossRef]
- Wolpert, C.; Echternach, C.; Veltmann, C.; Antzelevitch, C.; Thomas, G.P.; Spehl, S.; Streitner, F.; Kuschyk, J.; Schimpf, R.; Haase, K.K.; et al. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm 2005, 2, 254–260. [Google Scholar] [CrossRef]
- Brugada, P.; Brugada, J.; Brugada, R. Arrhythmia induction by antiarrhythmic drugs. Pacing Clin. Electrophysiol. 2000, 23, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Brugada, R. Fever and Brugada syndrome. Pacing Clin. Electrophysiol. 2002, 25, 1537–1539. [Google Scholar] [CrossRef]
- Junttila, M.J.; Gonzalez, M.; Lizotte, E.; Benito, B.; Vernooy, K.; Sarkozy, A.; Huikuri, H.V.; Brugada, P.; Brugada, J.; Brugada, R. Induced Brugada-type electrocardiogram, a sign for imminent malignant arrhythmias. Circulation 2008, 117, 1890–1893. [Google Scholar] [CrossRef]
- Adler, A.; Topaz, G.; Heller, K.; Zeltser, D.; Ohayon, T.; Rozovski, U.; Halkin, A.; Rosso, R.; Ben-Shachar, S.; Antzelevitch, C.; et al. Fever-induced Brugada pattern: How common is it and what does it mean? Heart Rhythm 2013, 10, 1375–1382. [Google Scholar] [CrossRef]
- Sangwatanaroj, S.; Prechawat, S.; Sunsaneewitayakul, B.; Sitthisook, S.; Tosukhowong, P.; Tungsanga, K. New electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur. Heart J. 2001, 22, 2290–2296. [Google Scholar] [CrossRef]
- Teijeiro, R.; Garro, H.A.; Acunzo, R.S.; Albino, E.; Chiale, P.A. Recording of high V1-V3 precordial leads may be essential to the diagnosis of Brugada syndrome during the ajmaline test. J. Cardiovasc. Pharmacol. Ther. 2006, 11, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Márquez, M.F.; Allende, R.; Morales, J.L. Unmasking the Brugada syndrome with high parasternal leads. Europace 2007, 9, 1216. [Google Scholar] [CrossRef] [PubMed]
- Baranchuk, A.; Nguyen, T.; Ryu, M.H.; Femenía, F.; Zareba, W.; Wilde, A.A.M.; Shimizu, W.; Brugada, P.; Pérez-Riera, A.R. Brugada phenocopy: New terminology and proposed classification. Ann. Noninvasive Electrocardiol. 2012, 17, 299–314. [Google Scholar] [CrossRef]
- Arce, M.; Riera, A.R.P.; Femenía, F.; Baranchuk, A. Brugada electrocardiographic phenocopy in a patient with chronic Chagasic cardiomyopathy. Cardiol. J. 2010, 17, 525–527. [Google Scholar]
- Gourraud, J.-B.; Barc, J.; Thollet, A.; Le Scouarnec, S.; Le Marec, H.; Schott, J.-J.; Redon, R.; Probst, V. The Brugada Syndrome: A Rare Arrhythmia Disorder with Complex Inheritance. Front. Cardiovasc. Med. 2016, 3, 9. [Google Scholar] [CrossRef]
- Tse, G.; Yeo, J.M. Conduction abnormalities and ventricular arrhythmogenesis: The roles of sodium channels and gap junctions. Int. J. Cardiol. Heart Vasc. 2015, 9, 75–82. [Google Scholar] [CrossRef]
- Kyndt, F.; Probst, V.; Potet, F.; Demolombe, S.; Chevallier, J.C.; Baro, I.; Moisan, J.P.; Boisseau, P.; Schott, J.J.; Escande, D.; et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 2001, 104, 3081–3086. [Google Scholar] [CrossRef]
- Martini, B.; Nava, A.; Thiene, G.; Buja, G.F.; Canciani, B.; Scognamiglio, R.; Daliento, L.; Dalla Volta, S. Ventricular fibrillation without apparent heart disease: Description of six cases. Am. Heart J. 1989, 118, 1203–1209. [Google Scholar] [CrossRef]
- Leoni, A.-L.; Gavillet, B.; Rougier, J.-S.; Marionneau, C.; Probst, V.; Le Scouarnec, S.; Schott, J.-J.; Demolombe, S.; Bruneval, P.; Huang, C.L.H.; et al. Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/-) mouse model. PLoS ONE 2010, 5, e9298. [Google Scholar] [CrossRef]
- Schweizer, P.A.; Fink, T.; Yampolsky, P.; Seyler, C.; Fabritz, L.; Kirchhof, P.; Becker, R.; Koenen, M.; Katus, H.A.; Thomas, D. Generation and characterization of SCN5A loss-of-function mutant mice modeling human brugada syndrome. Eur. Heart J. 2013, 34, 4556. [Google Scholar] [CrossRef]
- Boukens, B.J.; Sylva, M.; de Gier-de Vries, C.; Remme, C.A.; Bezzina, C.R.; Christoffels, V.M.; Coronel, R. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ. Res. 2013, 113, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Nagase, S.; Kusano, K.F.; Morita, H.; Fujimoto, Y.; Kakishita, M.; Nakamura, K.; Emori, T.; Matsubara, H.; Ohe, T. Epicardial electrogram of the right ventricular outflow tract in patients with the brugada syndrome. J. Am. Coll. Cardiol. 2002, 39, 1992–1995. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H. Electrocardiographic patterns of the Brugada syndrome in right ventricular infarction/ischemia. Am. J. Cardiol. 2000, 86, 1056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sacher, F.; Hoffmayer, K.; O’Hara, T.; Strom, M.; Cuculich, P.; Silva, J.; Cooper, D.; Faddis, M.; Hocini, M.; et al. Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 2015, 131, 1950–1959. [Google Scholar] [CrossRef] [Green Version]
- Veldkamp, M.W.; Viswanathan, P.C.; Bezzina, C.; Baartscheer, A.; Wilde, A.A.; Balser, J.R. Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ. Res. 2000, 86, E91–E97. [Google Scholar] [CrossRef]
- Maoz, A.; Christini, D.J.; Krogh-Madsen, T. Dependence of phase-2 reentry and repolarization dispersion on epicardial and transmural ionic heterogeneity: A simulation study. Europace 2014, 16, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.X.; Antzelevitch, C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 1999, 100, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Maury, P.; Sacher, F.; Gourraud, J.-B.; Pasquié, J.-L.; Raczka, F.; Bongard, V.; Duparc, A.; Mondoly, P.; Sadron, M.; Chatel, S.; et al. Increased Tpeak-Tend interval is highly and independently related to arrhythmic events in Brugada syndrome. Heart Rhythm 2015, 12, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Pollevick, G.D.; Cordeiro, J.M.; Casis, O.; Sanguinetti, M.C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A.; Wollnik, B.; et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007, 115, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Pastore, J.M.; Girouard, S.D.; Laurita, K.R.; Akar, F.G.; Rosenbaum, D.S. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 1999, 99, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, M.G.; Potse, M.; Linnenbank, A.C.; Verkerk, A.O.; den Ruijter, H.M.; van Amersfoorth, S.C.M.; Klaver, E.C.; Beekman, L.; Bezzina, C.R.; Postema, P.G.; et al. Mechanism of right precordial ST-segment elevation in structural heart disease: Excitation failure by current-to-load mismatch. Heart Rhythm 2010, 7, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, M.G.; Potse, M.; Vinet, A.; de Bakker, J.M.T.; Coronel, R. ST segment elevation by current-to-load mismatch: An experimental and computational study. Heart Rhythm 2011, 8, 111–118. [Google Scholar] [CrossRef]
- Hoogendijk, M.G.; Opthof, T.; Postema, P.G.; Wilde, A.A.M.; de Bakker, J.M.T.; Coronel, R. The Brugada ECG pattern: A marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2010, 3, 283–290. [Google Scholar] [CrossRef]
- Ten Sande, J.N.; Coronel, R.; Conrath, C.E.; Driessen, A.H.G.; de Groot, J.R.; Tan, H.L.; Nademanee, K.; Wilde, A.A.M.; de Bakker, J.M.T.; van Dessel, P.F.H.M. ST-Segment Elevation and Fractionated Electrograms in Brugada Syndrome Patients Arise From the Same Structurally Abnormal Subepicardial RVOT Area but Have a Different Mechanism. Circ. Arrhythm. Electrophysiol. 2015, 8, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.G.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef]
- Postema, P.G.; van Dessel, P.F.H.M.; de Bakker, J.M.T.; Dekker, L.R.C.; Linnenbank, A.C.; Hoogendijk, M.G.; Coronel, R.; Tijssen, J.G.P.; Wilde, A.A.M.; Tan, H.L. Slow and discontinuous conduction conspire in Brugada syndrome: A right ventricular mapping and stimulation study. Circ. Arrhythm. Electrophysiol. 2008, 1, 379–386. [Google Scholar] [CrossRef]
- Robyns, T.; Lu, H.R.; Gallacher, D.J.; Garweg, C.; Ector, J.; Willems, R.; Janssens, S.; Nuyens, D. Evaluation of Index of Cardio-Electrophysiological Balance (iCEB) as a New Biomarker for the Identification of Patients at Increased Arrhythmic Risk. Ann. Noninvasive Electrocardiol. 2016, 21, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Tse, G.; Yan, B.P. Traditional and novel electrocardiographic conduction and repolarization markers of sudden cardiac death. Europace 2017, 19, 712–721. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Postema, P.G.; Di Diego, J.M.; Viskin, S.; Morita, H.; Fish, J.M.; Antzelevitch, C. The pathophysiological mechanism underlying Brugada syndrome: Depolarization versus repolarization. J. Mol. Cell. Cardiol. 2010, 49, 543–553. [Google Scholar] [CrossRef]
- Kobayashi, T.; Shintani, U.; Yamamoto, T.; Shida, S.; Isshiki, N.; Tanaka, T.; Ohmoto, Y.; Kitamura, M.; Kato, S.; Misaki, M. Familial occurrence of electrocardiographic abnormalities of the Brugada-type. Intern. Med. 1996, 35, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Finneran, W.; Feld, G.K. Familial sudden cardiac death associated with a terminal QRS abnormality on surface 12-lead electrocardiogram in the index case. J. Cardiovasc. Electrophysiol. 1998, 9, 642–647. [Google Scholar] [CrossRef]
- Brugada, P.; Brugada, R.; Brugada, J. Sudden death in patients and relatives with the syndrome of right bundle branch block, ST segment elevation in the precordial leads V(1)to V(3)and sudden death. Eur. Heart J. 2000, 21, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Wilde, A.A.M.; Barc, J.; Sacher, F.; Babuty, D.; Mabo, P.; Mansourati, J.; Le Scouarnec, S.; Kyndt, F.; Le Caignec, C.; et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet. 2009, 2, 552–557. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Pappone, C. Brugada syndrome: Oligogenic or mendelian disease? Int. J. Mol. Sci. 2020, 21, 1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micaglio, E.; Monasky, M.M.; Ciconte, G.; Vicedomini, G.; Conti, M.; Mecarocci, V.; Giannelli, L.; Giordano, F.; Pollina, A.; Saviano, M.; et al. Novel SCN5A frameshift mutation in brugada syndrome associated with complex arrhythmic phenotype. Front. Genet. 2019, 10, 547. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, S.; Di Resta, C.; Rocchetti, M.; Barile, L.; Rizzetto, R.; Summa, A.; Severi, S.; Sommariva, E.; Pappone, C.; Ferrari, M.; et al. A Brugada syndrome mutation (p.S216L) and its modulation by p.H558R polymorphism: Standard and dynamic characterization. Cardiovasc. Res. 2011, 91, 606–616. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.-B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Giachino, D.; Ciconte, G.; Giannelli, L.; Locati, E.T.; Ramondini, E.; Cotugno, R.; Vicedomini, G.; Borrelli, V.; et al. Genotype-Phenotype Correlation in a Family with Brugada Syndrome Harboring the Novel p.Gln371* Nonsense Variant in the SCN5A Gene. Int. J. Mol. Sci. 2019, 20, 5522. [Google Scholar] [CrossRef]
- Zaklyazminskaya, E.; Dzemeshkevich, S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim. Biophys. Acta 2016, 1863, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yin, L.; Shen, C.; Hu, K.; Ge, J.; Sun, A. SCN5A variants: Association with cardiac disorders. Front. Physiol. 2018, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Perez-Serra, A.; Del Olmo, B.; Jordá, P.; et al. Genetic interpretation and clinical translation of minor genes related to Brugada syndrome. Hum. Mutat. 2019, 40, 749–764. [Google Scholar] [CrossRef]
- Coppola, G.; Corrado, E.; Curnis, A.; Maglia, G.; Oriente, D.; Mignano, A.; Brugada, P. Update on brugada syndrome 2019. Curr. Probl. Cardiol. 2021, 46, 100454. [Google Scholar] [CrossRef]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Eastaugh, L.J.; James, P.A.; Phelan, D.G.; Davis, A.M. Brugada syndrome caused by a large deletion in SCN5A only detected by multiplex ligation-dependent probe amplification. J. Cardiovasc. Electrophysiol. 2011, 22, 1073–1076. [Google Scholar] [CrossRef]
- Mademont-Soler, I.; Pinsach-Abuin, M.L.; Riuró, H.; Mates, J.; Pérez-Serra, A.; Coll, M.; Porres, J.M.; Del Olmo, B.; Iglesias, A.; Selga, E.; et al. Large genomic imbalances in brugada syndrome. PLoS ONE 2016, 11, e0163514. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, K.; Ohno, S.; Ozawa, J.; Hayano, M.; Hattori, T.; Kobori, A.; Yahata, M.; Aburadani, I.; Watanabe, S.; Matsumoto, Y.; et al. Copy number variations of SCN5A in Brugada syndrome. Heart Rhythm 2018, 15, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Lizotte, E.; Junttila, M.J.; Dube, M.P.; Hong, K.; Benito, B.; DE Zutter, M.; Henkens, S.; Sarkozy, A.; Huikuri, H.V.; Towbin, J.; et al. Genetic modulation of brugada syndrome by a common polymorphism. J. Cardiovasc. Electrophysiol. 2009, 20, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Tadros, R.; Tan, H.L.; ESCAPE-NET Investigators; El Mathari, S.; Kors, J.A.; Postema, P.G.; Lahrouchi, N.; Beekman, L.; Radivojkov-Blagojevic, M.; Amin, A.S.; et al. Predicting cardiac electrical response to sodium-channel blockade and Brugada syndrome using polygenic risk scores. Eur. Heart J. 2019, 40, 3097–3107. [Google Scholar] [CrossRef] [PubMed]
- Barc, J.; Tadros, R.; Glinge, C.; Chiang, D.Y.; Jouni, M.; Simonet, F.; Jurgens, S.J.; Baudic, M.; Nicastro, M.; Potet, F.; et al. Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat. Genet. 2022, 54, 232–239. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Benedetti, S.; Di Resta, C.; Vicedomini, G.; Borrelli, V.; Ghiroldi, A.; Piccoli, M.; Anastasia, L.; et al. Genotype/phenotype relationship in a consanguineal family with brugada syndrome harboring the R1632C missense variant in the SCN5A gene. Front. Physiol. 2019, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Micaglio, E.; Monasky, M.M.; Ciconte, G.; Vicedomini, G.; Conti, M.; Mecarocci, V.; Giannelli, L.; Giordano, F.; Pollina, A.; Saviano, M.; et al. SCN5A nonsense mutation and NF1 frameshift mutation in a family with brugada syndrome and neurofibromatosis. Front. Genet. 2019, 10, 50. [Google Scholar] [CrossRef]
- Micaglio, E.; Monasky, M.M.; Resta, N.; Bagnulo, R.; Ciconte, G.; Giannelli, L.; Locati, E.T.; Vicedomini, G.; Borrelli, V.; Ghiroldi, A.; et al. Novel SCN5A p.W697X Nonsense Mutation Segregation in a Family with Brugada Syndrome. Int. J. Mol. Sci. 2019, 20, 4920. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Vicedomini, G.; Locati, E.T.; Ciconte, G.; Giannelli, L.; Giordano, F.; Crisà, S.; Vecchi, M.; Borrelli, V.; et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace 2019, 21, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Yeates, L.; Ingles, J.; Gray, B.; Singarayar, S.; Sy, R.W.; Semsarian, C.; Bagnall, R.D. A balanced translocation disrupting SCN5A in a family with Brugada syndrome and sudden cardiac death. Heart Rhythm 2019, 16, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Yagihara, N.; Watanabe, H.; Barnett, P.; Duboscq-Bidot, L.; Thomas, A.C.; Yang, P.; Ohno, S.; Hasegawa, K.; Kuwano, R.; Chatel, S.; et al. Variants in the SCN5A promoter associated with various arrhythmia phenotypes. J. Am. Heart Assoc. 2016, 5, 3644. [Google Scholar] [CrossRef] [PubMed]
- Pappone, C.; Monasky, M.M.; Ciconte, G. Epicardial ablation in genetic cardiomyopathies: A new frontier. Eur. Heart J. Suppl. 2019, 21, B61–B66. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Albers, S.; Cyganek, L.; Zhao, Z.; Lan, H.; Li, X.; Xu, Q.; Kleinsorge, M.; Huang, M.; Liao, Z.; et al. A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. Europace 2019, 21, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Behr, E.R.; Savio-Galimberti, E.; Barc, J.; Holst, A.G.; Petropoulou, E.; Prins, B.P.; Jabbari, J.; Torchio, M.; Berthet, M.; Mizusawa, Y.; et al. Role of common and rare variants in SCN10A: Results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 2015, 106, 520–529. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhao, J.; Terracciano, C.M.N.; Bezzina, C.R.; Zhang, W.; Kaba, R.; Navaratnarajah, M.; Lotlikar, A.; Sehmi, J.S.; Kooner, M.K.; et al. Genetic variation in SCN10A influences cardiac conduction. Nat. Genet. 2010, 42, 149–152. [Google Scholar] [CrossRef]
- Qi, B.; Wei, Y.; Chen, S.; Zhou, G.; Li, H.; Xu, J.; Ding, Y.; Lu, X.; Zhao, L.; Zhang, F.; et al. Nav1.8 channels in ganglionated plexi modulate atrial fibrillation inducibility. Cardiovasc. Res. 2014, 102, 480–486. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Sakabe, N.J.; Savic, D.; Nobrega, M.A. Transcriptional enhancers in development and disease. Genome Biol. 2012, 13, 238. [Google Scholar] [CrossRef]
- GWAS Catalog. Available online: https://www.ebi.ac.uk/gwas/home (accessed on 12 January 2023).
- Sotoodehnia, N.; Isaacs, A.; de Bakker, P.I.W.; Dörr, M.; Newton-Cheh, C.; Nolte, I.M.; van der Harst, P.; Müller, M.; Eijgelsheim, M.; Alonso, A.; et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 2010, 42, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akopian, A.N.; Sivilotti, L.; Wood, J.N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996, 379, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Benn, S.C.; Costigan, M.; Tate, S.; Fitzgerald, M.; Woolf, C.J. Developmental expression of the TTX-resistant voltage-gated sodium channels Nav1.8 (SNS) and Nav1.9 (SNS2) in primary sensory neurons. J. Neurosci. 2001, 21, 6077–6085. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, T.; Noguchi, K. Comparative study of voltage-gated sodium channel α-subunits in non-overlapping four neuronal populations in the rat dorsal root ganglion. Neurosci. Res. 2011, 70, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Remme, C.A.; Schumacher, C.A.; Scicluna, B.P.; Wolswinkel, R.; de Jonge, B.; Bezzina, C.R.; Veldkamp, M.W. Functional Nav1.8 channels in intracardiac neurons: The link between SCN10A and cardiac electrophysiology. Circ. Res. 2012, 111, 333–343. [Google Scholar] [CrossRef]
- Stroud, D.M.; Yang, T.; Bersell, K.; Kryshtal, D.O.; Nagao, S.; Shaffer, C.; Short, L.; Hall, L.; Atack, T.C.; Zhang, W.; et al. Contrasting Nav1.8 Activity in Scn10a-/- Ventricular Myocytes and the Intact Heart. J. Am. Heart Assoc. 2016, 5, 2946. [Google Scholar] [CrossRef]
- Yang, T.; Atack, T.C.; Stroud, D.M.; Zhang, W.; Hall, L.; Roden, D.M. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 2012, 111, 322–332. [Google Scholar] [CrossRef]
- Dybkova, N.; Ahmad, S.; Pabel, S.; Tirilomis, P.; Hartmann, N.; Fischer, T.H.; Bengel, P.; Tirilomis, T.; Ljubojevic, S.; Renner, A.; et al. Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc. Res. 2018, 114, 1728–1737. [Google Scholar] [CrossRef]
- Casini, S.; Marchal, G.A.; Kawasaki, M.; Nariswari, F.A.; Portero, V.; van den Berg, N.W.E.; Guan, K.; Driessen, A.H.G.; Veldkamp, M.W.; Mengarelli, I.; et al. Absence of Functional Nav1.8 Channels in Non-diseased Atrial and Ventricular Cardiomyocytes. Cardiovasc. Drugs Ther. 2019, 33, 649–660. [Google Scholar] [CrossRef]
- van den Boogaard, M.; Smemo, S.; Burnicka-Turek, O.; Arnolds, D.E.; van de Werken, H.J.G.; Klous, P.; McKean, D.; Muehlschlegel, J.D.; Moosmann, J.; Toka, O.; et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Investig. 2014, 124, 1844–1852. [Google Scholar] [CrossRef]
- van den Boogaard, M.; Wong, L.Y.E.; Tessadori, F.; Bakker, M.L.; Dreizehnter, L.K.; Wakker, V.; Bezzina, C.R.; ’t Hoen, P.A.C.; Bakkers, J.; Barnett, P.; et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Investig. 2012, 122, 2519–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, J.C.K.; Mohan, R.A.; van den Boogaard, M.; Hilvering, C.R.E.; Jenkins, C.; Wakker, V.; Bianchi, V.; de Laat, W.; Barnett, P.; Boukens, B.J.; et al. An enhancer cluster controls gene activity and topology of the SCN5A-SCN10A locus in vivo. Nat. Commun. 2019, 10, 4943. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Agustín, A.; Pinsach-Abuin, M.L.; Pagans, S. Role of Non-Coding Variants in Brugada Syndrome. Int. J. Mol. Sci. 2020, 21, 8556. [Google Scholar] [CrossRef] [PubMed]
- Portero, V.; Wilders, R.; Casini, S.; Charpentier, F.; Verkerk, A.O.; Remme, C.A. KV4.3 expression modulates nav1.5 sodium current. Front. Physiol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Gütter, C.; Benndorf, K.; Zimmer, T. Characterization of N-terminally mutated cardiac Na(+) channels associated with long QT syndrome 3 and Brugada syndrome. Front. Physiol. 2013, 4, 153. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Keller, D.I.; Huang, H.; Fressart, V.; Schmied, C.; Timour, Q.; Chahine, M. Mexiletine differentially restores the trafficking defects caused by two brugada syndrome mutations. Front. Pharmacol. 2012, 3, 62. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, C.; Abriel, H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 2015, 82, 36–47. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Wu, X.; Yang, Z.; Wu, L.; Yong, S.L.; Zhang, C.; Hu, K.; Wang, Q.K.; Chen, Q. MOG1 rescues defective trafficking of Na(v)1.5 mutations in Brugada syndrome and sick sinus syndrome. Circ. Arrhythm. Electrophysiol. 2013, 6, 392–401. [Google Scholar] [CrossRef]
- Valdivia, C.R.; Ueda, K.; Ackerman, M.J.; Makielski, J.C. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1446–H1452. [Google Scholar] [CrossRef]
- Huang, L.; Tang, S.; Chen, Y.; Zhang, L.; Yin, K.; Wu, Y.; Zheng, J.; Wu, Q.; Makielski, J.C.; Cheng, J. Molecular pathological study on LRRC10 in sudden unexplained nocturnal death syndrome in the Chinese Han population. Int. J. Legal Med. 2017, 131, 621–628. [Google Scholar] [CrossRef]
- Monasky, M.M.; Pappone, C.; Piccoli, M.; Ghiroldi, A.; Micaglio, E.; Anastasia, L. Calcium in brugada syndrome: Questions for future research. Front. Physiol. 2018, 9, 1088. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.W.; Holst, A.G.; Olesen, S.-P.; Olesen, M.S. The genetic component of Brugada syndrome. Front. Physiol. 2013, 4, 179. [Google Scholar] [CrossRef]
- Juang, J.-M.J.; Horie, M. Genetics of Brugada syndrome. J. Arrhythm. 2016, 32, 418–425. [Google Scholar] [CrossRef]
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef]
- Janin, A.; Bessière, F.; Georgescu, T.; Chanavat, V.; Chevalier, P.; Millat, G. TRPM4 mutations to cause autosomal recessive and not autosomal dominant Brugada type 1 syndrome. Eur. J. Med. Genet. 2019, 62, 103527. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Parisi, P.; Oliva, A.; Coll Vidal, M.; Partemi, S.; Campuzano, O.; Iglesias, A.; Pisani, D.; Pascali, V.L.; Paolino, M.C.; Villa, M.P.; et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 2013, 105, 415–418. [Google Scholar] [CrossRef]
- Sandorfi, G.; Clemens, B.; Csanadi, Z. Electrical storm in the brain and in the heart: Epilepsy and Brugada syndrome. Mayo Clin. Proc. 2013, 88, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Veltmann, C.; Barajas-Martinez, H.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Pfeiffer, R.; Cáceres, G.; Burashnikov, E.; Antzelevitch, C.; Hu, D. Further insights in the most common SCN5A mutation causing overlapping phenotype of long QT syndrome, brugada syndrome, and conduction defect. J. Am. Heart Assoc. 2016, 5, 3379. [Google Scholar] [CrossRef]
- Camacho Velásquez, J.L.; Rivero Sanz, E.; Velazquez Benito, A.; Mauri Llerda, J.A. Epilepsia y síndrome de Brugada. Neurología 2017, 32, 58–60. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Denecke, S.; Koehler, B. Results of ajmaline testing in patients with arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int. J. Cardiol. 2004, 95, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haim, Y.; Asimaki, A.; Behr, E.R. Brugada syndrome and arrhythmogenic cardiomyopathy: Overlapping disorders of the connexome? Europace 2021, 23, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Agullo-Pascual, E.; Cerrone, M.; Delmar, M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. FEBS Lett. 2014, 588, 1322–1330. [Google Scholar] [CrossRef]
- Corrado, D.; Zorzi, A.; Cerrone, M.; Rigato, I.; Mongillo, M.; Bauce, B.; Delmar, M. Relationship between arrhythmogenic right ventricular cardiomyopathy and brugada syndrome: New insights from molecular biology and clinical implications. Circ. Arrhythm. Electrophysiol. 2016, 9, e003631. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Delmar, M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 2014, 24, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Scheirlynck, E.; Chivulescu, M.; Lie, Ø.H.; Motoc, A.; Koulalis, J.; de Asmundis, C.; Sieira, J.; Chierchia, G.-B.; Brugada, P.; Cosyns, B.; et al. Worse prognosis in brugada syndrome patients with arrhythmogenic cardiomyopathy features. JACC Clin. Electrophysiol. 2020, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Micaglio, E.; Locati, E.T.; Pappone, C. Evaluating the use of genetics in brugada syndrome risk stratification. Front. Cardiovasc. Med. 2021, 8, 652027. [Google Scholar] [CrossRef]
- Zhu, K.; Bao, X.; Wang, Y.; Lu, T.; Zhang, L. Human induced pluripotent stem cell (hiPSC)-derived cardiomyocyte modelling of cardiovascular diseases for natural compound discovery. Biomed. Pharmacother. 2023, 157, 113970. [Google Scholar] [CrossRef]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Vermglinchan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Müller, J.; Zhao, Z.; Cyganek, L.; Zhong, R.; Zhang, F.; Kleinsorge, M.; Lan, H.; Li, X.; Xu, Q.; et al. Studying Brugada Syndrome With an SCN1B Variants in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2019, 7, 261. [Google Scholar] [CrossRef]
- Blok, M.; Boukens, B.J. Mechanisms of arrhythmias in the brugada syndrome. Int. J. Mol. Sci. 2020, 21, 7051. [Google Scholar] [CrossRef] [PubMed]
- Belbachir, N.; Portero, V.; Al Sayed, Z.R.; Gourraud, J.-B.; Dilasser, F.; Jesel, L.; Guo, H.; Wu, H.; Gaborit, N.; Guilluy, C.; et al. RRAD mutation causes electrical and cytoskeletal defects in cardiomyocytes derived from a familial case of Brugada syndrome. Eur. Heart J. 2019, 40, 3081–3094. [Google Scholar] [CrossRef] [PubMed]
- Mizusawa, Y.; Wilde, A.A.M. Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2012, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Veltmann, C.; Eckardt, L.; Meregalli, P.G.; Gaita, F.; Tan, H.L.; Babuty, D.; Sacher, F.; Giustetto, C.; Schulze-Bahr, E.; et al. Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation 2010, 121, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Gasparini, M.; Napolitano, C.; Della Bella, P.; Ottonelli, A.G.; Sassone, B.; Giordano, U.; Pappone, C.; Mascioli, G.; Rossetti, G.; et al. Risk stratification in Brugada syndrome: Results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J. Am. Coll. Cardiol. 2012, 59, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Conte, G.; Sieira, J.; Ciconte, G.; de Asmundis, C.; Chierchia, G.-B.; Baltogiannis, G.; Di Giovanni, G.; La Meir, M.; Wellens, F.; Czapla, J.; et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: A 20-year single-center experience. J. Am. Coll. Cardiol. 2015, 65, 879–888. [Google Scholar] [CrossRef]
- Viskin, S.; Rogowski, O. Asymptomatic Brugada syndrome: A cardiac ticking time-bomb? Europace 2007, 9, 707–710. [Google Scholar] [CrossRef]
- Letsas, K.P.; Georgopoulos, S.; Vlachos, K.; Karamichalakis, N.; Liatakis, I.; Korantzopoulos, P.; Liu, T.; Efremidis, M.; Sideris, A. Brugada syndrome:risk stratification and management. J. Atr. Fibrillation 2016, 9, 1413. [Google Scholar] [CrossRef]
- Hamilton, R.M. Implantable devices in young patients: Hitting the reset button on risk versus benefit. Heart Rhythm 2016, 13, 455–456. [Google Scholar] [CrossRef]
- Honarbakhsh, S.; Providencia, R.; Lambiase, P.D. Risk stratification in brugada syndrome: Current status and emerging approaches. Arrhythm. Electrophysiol. Rev. 2018, 7, 79–83. [Google Scholar] [CrossRef]
- Viskin, S.; Rosso, R.; Friedensohn, L.; Havakuk, O.; Wilde, A.A.M. Everybody has Brugada syndrome until proven otherwise? Heart Rhythm 2015, 12, 1595–1598. [Google Scholar] [CrossRef]
- Anselm, D.D.; Evans, J.M.; Baranchuk, A. Brugada phenocopy: A new electrocardiogram phenomenon. World J. Cardiol. 2014, 6, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Delise, P.; Allocca, G.; Marras, E.; Giustetto, C.; Gaita, F.; Sciarra, L.; Calo, L.; Proclemer, A.; Marziali, M.; Rebellato, L.; et al. Risk stratification in individuals with the Brugada type 1 ECG pattern without previous cardiac arrest: Usefulness of a combined clinical and electrophysiologic approach. Eur. Heart J. 2011, 32, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Kamakura, T.; Morita, H.; Tokioka, K.; Nakajima, I.; Wada, M.; Ishibashi, K.; Miyamoto, K.; Noda, T.; Aiba, T.; et al. Risk stratification in patients with Brugada syndrome without previous cardiac arrest–prognostic value of combined risk factors. Circ. J. 2015, 79, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Fauchier, L.; Isorni, M.A.; Clementy, N.; Pierre, B.; Simeon, E.; Babuty, D. Prognostic value of programmed ventricular stimulation in Brugada syndrome according to clinical presentation: An updated meta-analysis of worldwide published data. Int. J. Cardiol. 2013, 168, 3027–3029. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, S.; Ohe, T.; Nakazawa, K.; Aizawa, Y.; Shimizu, A.; Horie, M.; Ogawa, S.; Okumura, K.; Tsuchihashi, K.; Sugi, K.; et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1-V3. Circ. Arrhythm. Electrophysiol. 2009, 2, 495–503. [Google Scholar] [CrossRef]
- Brugada, J.; Brugada, R.; Antzelevitch, C.; Towbin, J.; Nademanee, K.; Brugada, P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation 2002, 105, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Gehi, A.K.; Duong, T.D.; Metz, L.D.; Gomes, J.A.; Mehta, D. Risk stratification of individuals with the Brugada electrocardiogram: A meta-analysis. J. Cardiovasc. Electrophysiol. 2006, 17, 577–583. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Giordano, U.; Bloise, R.; Giustetto, C.; De Nardis, R.; Grillo, M.; et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation 2002, 105, 1342–1347. [Google Scholar] [CrossRef]
- Sacher, F.; Arsac, F.; Wilton, S.B.; Derval, N.; Denis, A.; de Guillebon, M.; Ramoul, K.; Bordachar, P.; Ritter, P.; Hocini, M.; et al. Syncope in Brugada syndrome patients: Prevalence, characteristics, and outcome. Heart Rhythm 2012, 9, 1272–1279. [Google Scholar] [CrossRef]
- Letsas, K.P.; Efremidis, M.; Gavrielatos, G.; Filippatos, G.S.; Sideris, A.; Kardaras, F. Neurally mediated susceptibility in individuals with Brugada-type ECG pattern. Pacing Clin. Electrophysiol. 2008, 31, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Giustetto, C.; Drago, S.; Demarchi, P.G.; Dalmasso, P.; Bianchi, F.; Masi, A.S.; Carvalho, P.; Occhetta, E.; Rossetti, G.; Riccardi, R.; et al. Risk stratification of the patients with Brugada type electrocardiogram: A community-based prospective study. Europace 2009, 11, 507–513. [Google Scholar] [CrossRef]
- Sieira, J.; Conte, G.; Ciconte, G.; de Asmundis, C.; Chierchia, G.-B.; Baltogiannis, G.; Di Giovanni, G.; Saitoh, Y.; Irfan, G.; Casado-Arroyo, R.; et al. Prognostic value of programmed electrical stimulation in Brugada syndrome: 20 years experience. Circ. Arrhythm. Electrophysiol. 2015, 8, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Letsas, K.P.; Liu, T.; Shao, Q.; Korantzopoulos, P.; Giannopoulos, G.; Vlachos, K.; Georgopoulos, S.; Trikas, A.; Efremidis, M.; Deftereos, S.; et al. Meta-Analysis on Risk Stratification of Asymptomatic Individuals With the Brugada Phenotype. Am. J. Cardiol. 2015, 116, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Letsas, K.P.; Baranchuk, A.; Shao, Q.; Tse, G.; Zhang, N.; Zhang, Z.; Hu, D.; Li, G.; Liu, T. Meta-analysis of Fragmented QRS as an Electrocardiographic Predictor for Arrhythmic Events in Patients with Brugada Syndrome. Front. Physiol. 2017, 8, 678. [Google Scholar] [CrossRef]
- Rattanawong, P.; Riangwiwat, T.; Prasitlumkum, N.; Limpruttidham, N.; Kanjanahattakij, N.; Chongsathidkiet, P.; Vutthikraivit, W.; Chung, E.H. Baseline fragmented QRS increases the risk of major arrhythmic events in Brugada syndrome: Systematic review and meta-analysis. Ann. Noninvasive Electrocardiol. 2018, 23, e12507. [Google Scholar] [CrossRef]
- Morita, H.; Watanabe, A.; Morimoto, Y.; Kawada, S.; Tachibana, M.; Nakagawa, K.; Nishii, N.; Ito, H. Distribution and prognostic significance of fragmented QRS in patients with brugada syndrome. Circ. Arrhythm. Electrophysiol. 2017, 10, 4765. [Google Scholar] [CrossRef]
- Morita, H.; Watanabe, A.; Kawada, S.; Miyamoto, M.; Morimoto, Y.; Nakagawa, K.; Nishii, N.; Nakamura, K.; Ito, H. Identification of electrocardiographic risk markers for the initial and recurrent episodes of ventricular fibrillation in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2018, 29, 107–114. [Google Scholar] [CrossRef]
- Morita, H.; Kusano, K.F.; Miura, D.; Nagase, S.; Nakamura, K.; Morita, S.T.; Ohe, T.; Zipes, D.P.; Wu, J. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation 2008, 118, 1697–1704. [Google Scholar] [CrossRef]
- Tokioka, K.; Kusano, K.F.; Morita, H.; Miura, D.; Nishii, N.; Nagase, S.; Nakamura, K.; Kohno, K.; Ito, H.; Ohe, T. Electrocardiographic parameters and fatal arrhythmic events in patients with Brugada syndrome: Combination of depolarization and repolarization abnormalities. J. Am. Coll. Cardiol. 2014, 63, 2131–2138. [Google Scholar] [CrossRef]
- Maury, P.; Rollin, A.; Sacher, F.; Gourraud, J.-B.; Raczka, F.; Pasquié, J.-L.; Duparc, A.; Mondoly, P.; Cardin, C.; Delay, M.; et al. Prevalence and prognostic role of various conduction disturbances in patients with the Brugada syndrome. Am. J. Cardiol. 2013, 112, 1384–1389. [Google Scholar] [CrossRef]
- Junttila, M.J.; Brugada, P.; Hong, K.; Lizotte, E.; DE Zutter, M.; Sarkozy, A.; Brugada, J.; Benito, B.; Perkiomaki, J.S.; Mäkikallio, T.H.; et al. Differences in 12-lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J. Cardiovasc. Electrophysiol. 2008, 19, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Yokoyama, Y.; Aonuma, K.; Aihara, N.; Hiraoka, M.; Japan Idiopathic Ventricular Fibrillation Study (J-IVFS) Investigators. Clinical characteristics and risk stratification in symptomatic and asymptomatic patients with brugada syndrome: Multicenter study in Japan. J. Cardiovasc. Electrophysiol. 2007, 18, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Castro Hevia, J.; Antzelevitch, C.; Tornés Bárzaga, F.; Dorantes Sánchez, M.; Dorticós Balea, F.; Zayas Molina, R.; Quiñones Pérez, M.A.; Fayad Rodríguez, Y. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J. Am. Coll. Cardiol. 2006, 47, 1828–1834. [Google Scholar] [CrossRef]
- Tse, G. Novel conduction-repolarization indices for the stratification of arrhythmic risk. J. Geriatr. Cardiol. 2016, 13, 811–812. [Google Scholar] [CrossRef] [PubMed]
- Tse, G. (Tpeak-Tend)/QRS and (Tpeak-Tend)/(QT × QRS): Novel markers for predicting arrhythmic risk in Brugada syndrome. EP Eur. 2017, 19, 696. [Google Scholar] [CrossRef]
- Lu, H.R.; Yan, G.-X.; Gallacher, D.J. A new biomarker--index of cardiac electrophysiological balance (iCEB)--plays an important role in drug-induced cardiac arrhythmias: Beyond QT-prolongation and Torsades de Pointes (TdPs). J. Pharmacol. Toxicol. Methods 2013, 68, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Tse, G. Both transmural dispersion of repolarization and of refractoriness are poor predictors of arrhythmogenicity: A role for iCEB (QT/QRS)? J. Geriatr. Cardiol. 2016, 13, 813–814. [Google Scholar] [CrossRef]
- Asvestas, D.; Tse, G.; Baranchuk, A.; Bazoukis, G.; Liu, T.; Saplaouras, A.; Korantzopoulos, P.; Goga, C.; Efremidis, M.; Sideris, A.; et al. High risk electrocardiographic markers in Brugada syndrome. IJC Heart Vasc. 2018, 18, 58–64. [Google Scholar] [CrossRef]
- Xia, Y.; Liang, Y.; Kongstad, O.; Liao, Q.; Holm, M.; Olsson, B.; Yuan, S. In vivo validation of the coincidence of the peak and end of the T wave with full repolarization of the epicardium and endocardium in swine. Heart Rhythm 2005, 2, 162–169. [Google Scholar] [CrossRef]
- Miyamoto, A.; Hayashi, H.; Makiyama, T.; Yoshino, T.; Mizusawa, Y.; Sugimoto, Y.; Ito, M.; Xue, J.Q.; Murakami, Y.; Horie, M. Risk determinants in individuals with a spontaneous type 1 Brugada ECG. Circ. J. 2011, 75, 844–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, T.; Kusano, K.F.; Nagase, S.; Banba, K.; Miura, D.; Nishii, N.; Watanabe, A.; Nakamura, K.; Morita, H.; Ohe, T. Clinical significance of macroscopic T-wave alternans after sodium channel blocker administration in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2008, 19, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Masrur, S.; Memon, S.; Thompson, P.D. Brugada syndrome, exercise, and exercise testing. Clin. Cardiol. 2015, 38, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Makimoto, H.; Nakagawa, E.; Takaki, H.; Yamada, Y.; Okamura, H.; Noda, T.; Satomi, K.; Suyama, K.; Aihara, N.; Kurita, T.; et al. Augmented ST-segment elevation during recovery from exercise predicts cardiac events in patients with Brugada syndrome. J. Am. Coll. Cardiol. 2010, 56, 1576–1584. [Google Scholar] [CrossRef]
- Amin, A.S.; de Groot, E.A.A.; Ruijter, J.M.; Wilde, A.A.M.; Tan, H.L. Exercise-induced ECG changes in Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2009, 2, 531–539. [Google Scholar] [CrossRef]
- Kawata, H.; Morita, H.; Yamada, Y.; Noda, T.; Satomi, K.; Aiba, T.; Isobe, M.; Nagase, S.; Nakamura, K.; Fukushima Kusano, K.; et al. Prognostic significance of early repolarization in inferolateral leads in Brugada patients with documented ventricular fibrillation: A novel risk factor for Brugada syndrome with ventricular fibrillation. Heart Rhythm 2013, 10, 1161–1168. [Google Scholar] [CrossRef]
- Sarkozy, A.; Chierchia, G.-B.; Paparella, G.; Boussy, T.; De Asmundis, C.; Roos, M.; Henkens, S.; Kaufman, L.; Buyl, R.; Brugada, R.; et al. Inferior and lateral electrocardiographic repolarization abnormalities in Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2009, 2, 154–161. [Google Scholar] [CrossRef]
- Letsas, K.P.; Sacher, F.; Probst, V.; Weber, R.; Knecht, S.; Kalusche, D.; Haïssaguerre, M.; Arentz, T. Prevalence of early repolarization pattern in inferolateral leads in patients with Brugada syndrome. Heart Rhythm 2008, 5, 1685–1689. [Google Scholar] [CrossRef]
- Babai Bigi, M.A.; Aslani, A.; Shahrzad, S. aVR sign as a risk factor for life-threatening arrhythmic events in patients with Brugada syndrome. Heart Rhythm 2007, 4, 1009–1012. [Google Scholar] [CrossRef]
- Steinberg, J.S.; Berbari, E.J. The signal-averaged electrocardiogram: Update on clinical applications. J. Cardiovasc. Electrophysiol. 1996, 7, 972–988. [Google Scholar] [CrossRef]
- Huang, Z.; Patel, C.; Li, W.; Xie, Q.; Wu, R.; Zhang, L.; Tang, R.; Wan, X.; Ma, Y.; Zhen, W.; et al. Role of signal-averaged electrocardiograms in arrhythmic risk stratification of patients with Brugada syndrome: A prospective study. Heart Rhythm 2009, 6, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, Y.; Hagiwara, N.; Kasanuki, H. Assessment of markers for identifying patients at risk for life-threatening arrhythmic events in Brugada syndrome. J. Cardiovasc. Electrophysiol. 2005, 16, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, H.; Takagi, M.; Nakagawa, E.; Yamashita, H.; Yoshiyama, M. Risk stratification in patients with Brugada syndrome: Analysis of daily fluctuations in 12-lead electrocardiogram (ECG) and signal-averaged electrocardiogram (SAECG). J. Cardiovasc. Electrophysiol. 2006, 17, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.M.W.; Ng, F.S.; Yao, C.; Roney, C.; Taraborrelli, P.; Linton, N.W.F.; Whinnett, Z.I.; Lefroy, D.C.; Davies, D.W.; Boon Lim, P.; et al. ST-Elevation Magnitude Correlates With Right Ventricular Outflow Tract Conduction Delay in Type I Brugada ECG. Circ. Arrhythm. Electrophysiol. 2017, 10, 5107. [Google Scholar] [CrossRef]
- Rudic, B.; Schimpf, R.; Veltmann, C.; Doesch, C.; Tülümen, E.; Schoenberg, S.O.; Borggrefe, M.; Papavassiliu, T. Brugada syndrome: Clinical presentation and genotype-correlation with magnetic resonance imaging parameters. Europace 2016, 18, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Van Malderen, S.C.H.; Kerkhove, D.; Theuns, D.A.M.J.; Weytjens, C.; Droogmans, S.; Tanaka, K.; Daneels, D.; Van Dooren, S.; Meuwissen, M.; Bonduelle, M.; et al. Prolonged right ventricular ejection delay identifies high risk patients and gender differences in Brugada syndrome. Int. J. Cardiol. 2015, 191, 90–96. [Google Scholar] [CrossRef]
- Ciconte, G.; Monasky, M.M.; Santinelli, V.; Micaglio, E.; Vicedomini, G.; Anastasia, L.; Negro, G.; Borrelli, V.; Giannelli, L.; Santini, F.; et al. Brugada syndrome genetics is associated with phenotype severity. Eur. Heart J. 2021, 42, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Pappone, C.; Ciconte, G.; Micaglio, E.; Monasky, M.M. Common modulators of Brugada syndrome phenotype do not affect SCN5A prognostic value. Eur. Heart J. 2021, 42, 1273–1274. [Google Scholar] [CrossRef]
- Kossaify, A.; Refaat, M. Programmed ventricular stimulation--indications and limitations: A comprehensive update and review. Hellenic J. Cardiol. 2013, 54, 39–46. [Google Scholar] [PubMed]
- Milman, A.; Hochstadt, A.; Andorin, A.; Gourraud, J.-B.; Sacher, F.; Mabo, P.; Kim, S.-H.; Conte, G.; Arbelo, E.; Kamakura, T.; et al. Time-to-first appropriate shock in patients implanted prophylactically with an implantable cardioverter-defibrillator: Data from the Survey on Arrhythmic Events in BRUgada Syndrome (SABRUS). Europace 2019, 21, 796–802. [Google Scholar] [CrossRef]
- Brugada, P.; Brugada, R.; Mont, L.; Rivero, M.; Geelen, P.; Brugada, J. Natural history of Brugada syndrome: The prognostic value of programmed electrical stimulation of the heart. J. Cardiovasc. Electrophysiol. 2003, 14, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Sroubek, J.; Probst, V.; Mazzanti, A.; Delise, P.; Hevia, J.C.; Ohkubo, K.; Zorzi, A.; Champagne, J.; Kostopoulou, A.; Yin, X.; et al. Programmed ventricular stimulation for risk stratification in the brugada syndrome: A pooled analysis. Circulation 2016, 133, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Sieira, J.; Conte, G.; Ciconte, G.; Chierchia, G.-B.; Casado-Arroyo, R.; Baltogiannis, G.; Di Giovanni, G.; Saitoh, Y.; Juliá, J.; Mugnai, G.; et al. A score model to predict risk of events in patients with Brugada Syndrome. Eur. Heart J. 2017, 38, 1756–1763. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Goronflot, T.; Anys, S.; Tixier, R.; Briand, J.; Berthome, P.; Geoffroy, O.; Clementy, N.; Mansourati, J.; Jesel, L.; et al. Robustness and relevance of predictive score in sudden cardiac death for patients with Brugada syndrome. Eur. Heart J. 2021, 42, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Honarbakhsh, S.; Providencia, R.; Garcia-Hernandez, J.; Martin, C.A.; Hunter, R.J.; Lim, W.Y.; Kirkby, C.; Graham, A.J.; Sharifzadehgan, A.; Waldmann, V.; et al. A Primary Prevention Clinical Risk Score Model for Patients With Brugada Syndrome (BRUGADA-RISK). JACC Clin. Electrophysiol. 2021, 7, 210–222. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| INHERITED CARDIAC DISEASES | GENES ASSOCIATED |
|---|---|
| LQT | SCN5A SCN1B CACNA1C CACNB2b CACNA2D1 |
| SQT | CACNA1C CACNB2b CACNA2D1 |
| SSS/PCCD | SCN5A SCN1B SCN2B SCN3B SCN10A RANGRF GPD1-L SLMAP |
| ERS | CACNA1C CACNB2b CACNA2D1 ABCC9 KCND3 KCNE3 KCNE5 KCNJ8 KCNH2 HCN4 |
| ARVC | PKP2 TRPM4 HEY2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa, I.P.; Șerban, D.N.; Mărănducă, M.A.; Șerban, I.L.; Tamba, B.I.; Tudorancea, I. Brugada Syndrome: From Molecular Mechanisms and Genetics to Risk Stratification. Int. J. Mol. Sci. 2023, 24, 3328. https://doi.org/10.3390/ijms24043328
Popa IP, Șerban DN, Mărănducă MA, Șerban IL, Tamba BI, Tudorancea I. Brugada Syndrome: From Molecular Mechanisms and Genetics to Risk Stratification. International Journal of Molecular Sciences. 2023; 24(4):3328. https://doi.org/10.3390/ijms24043328
Chicago/Turabian StylePopa, Irene Paula, Dragomir N. Șerban, Minela Aida Mărănducă, Ionela Lăcrămioara Șerban, Bogdan Ionel Tamba, and Ionuț Tudorancea. 2023. "Brugada Syndrome: From Molecular Mechanisms and Genetics to Risk Stratification" International Journal of Molecular Sciences 24, no. 4: 3328. https://doi.org/10.3390/ijms24043328