
Insight into the Structural Basis for Dual Nucleic Acid—Recognition by the Scaffold Attachment Factor B2 Protein
 , , and
, , and
Abstract
:1. Introduction
2. Results
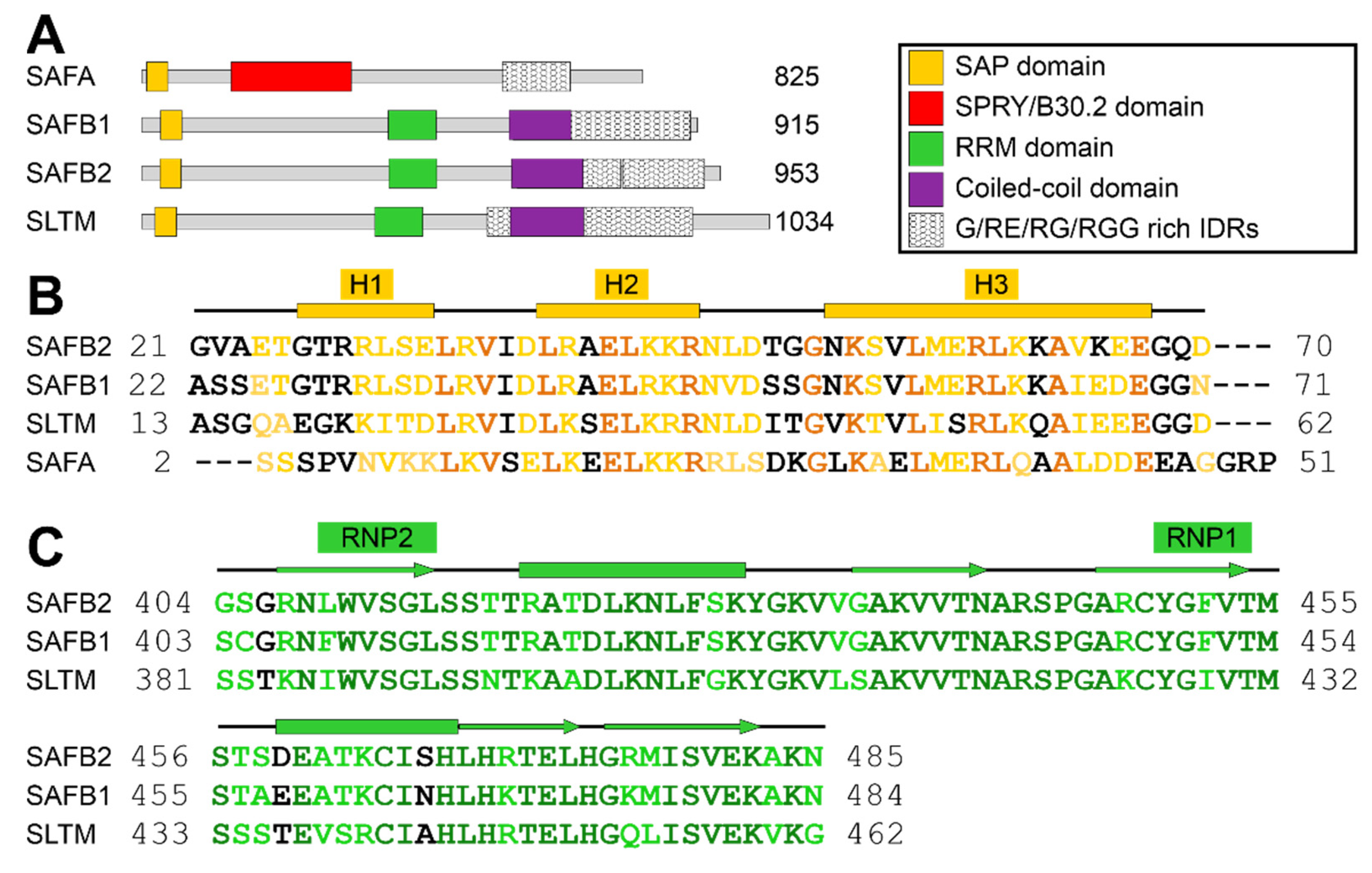
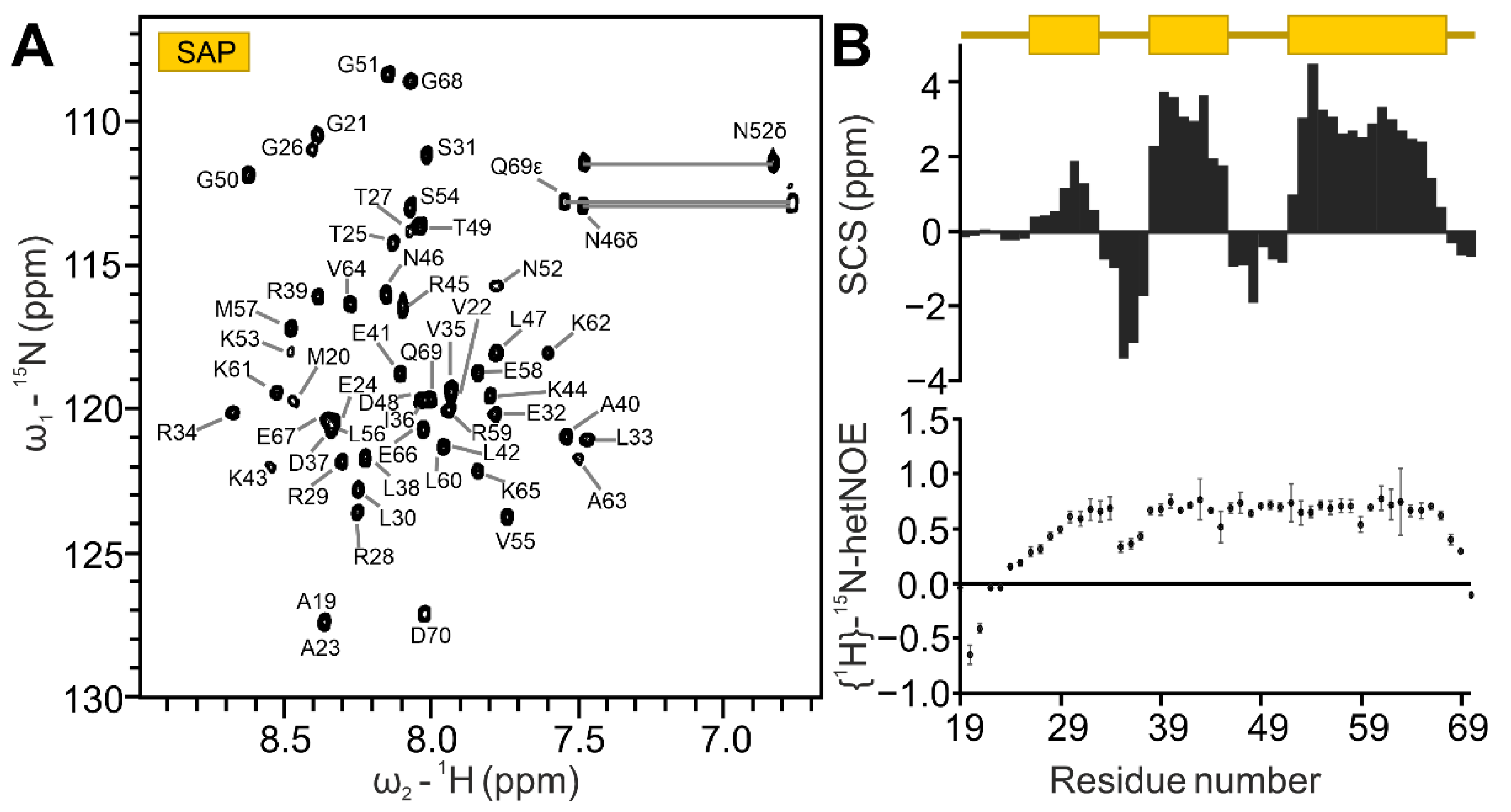
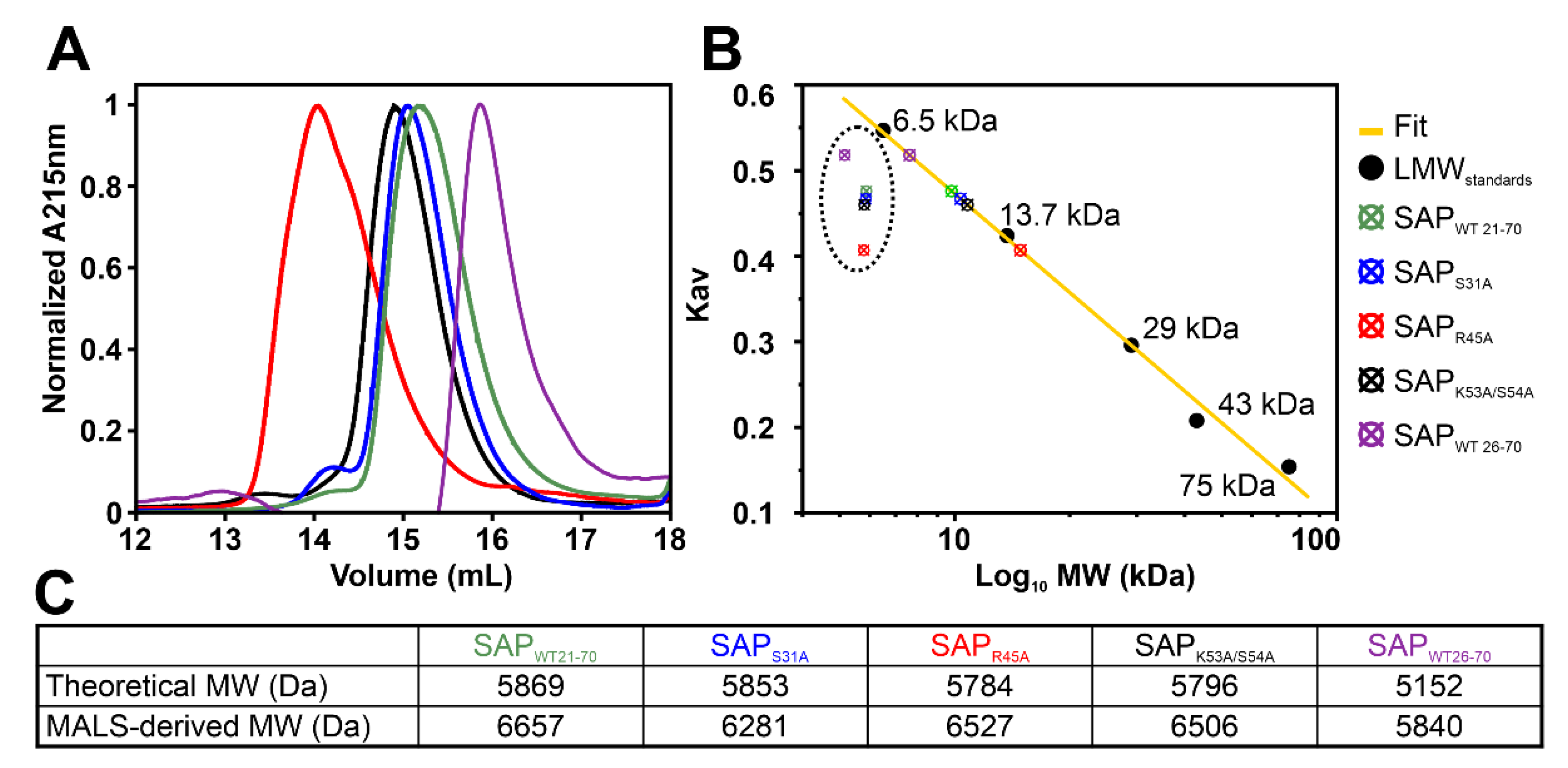
2.1. Definition of SAFB2 SAP Domain
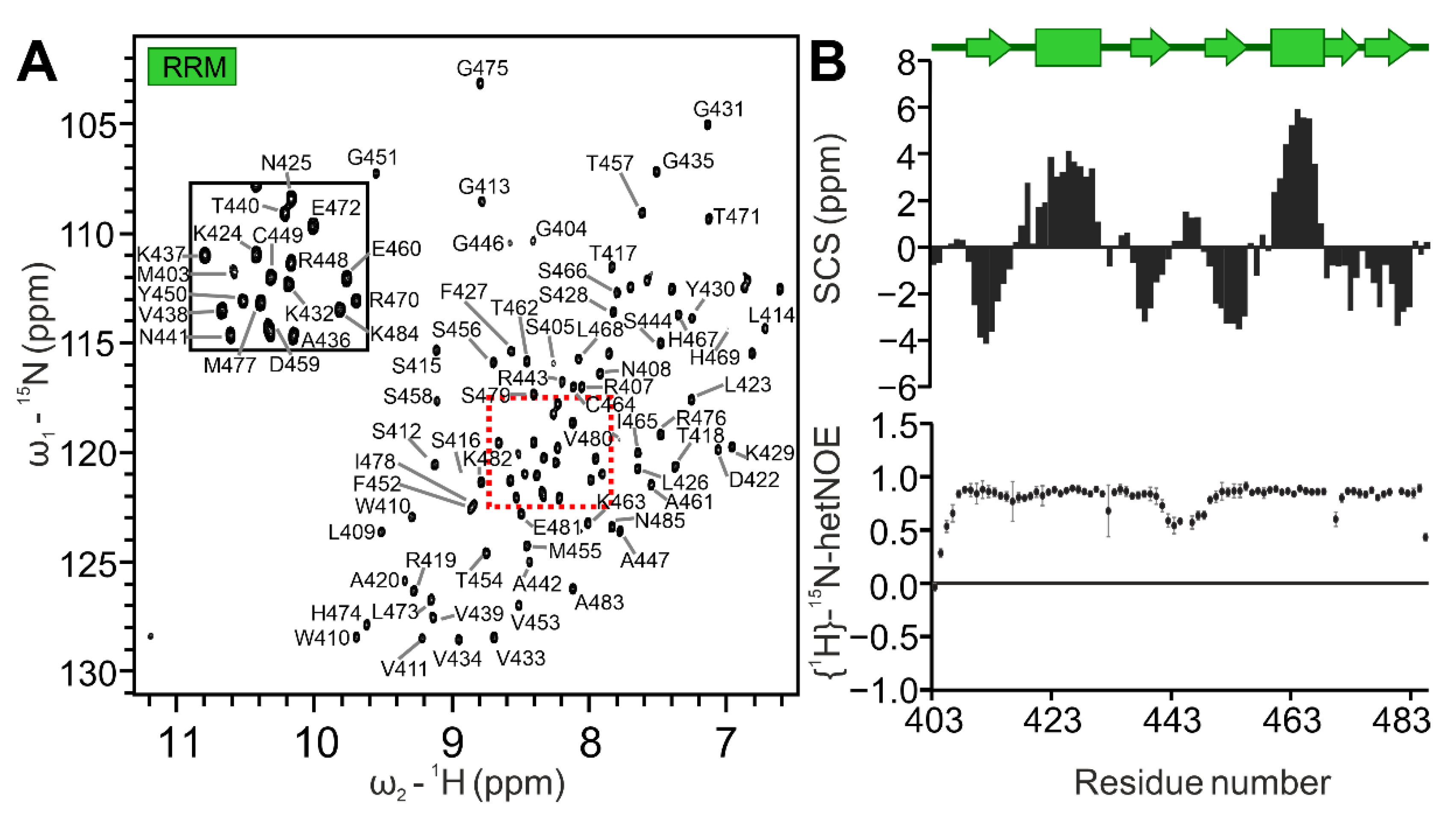
2.2. Definition of the SAFB2 RRM Domain Boundaries
2.3. Sparse NMR Data-Derived Structural Models of SAP and RRM Domains
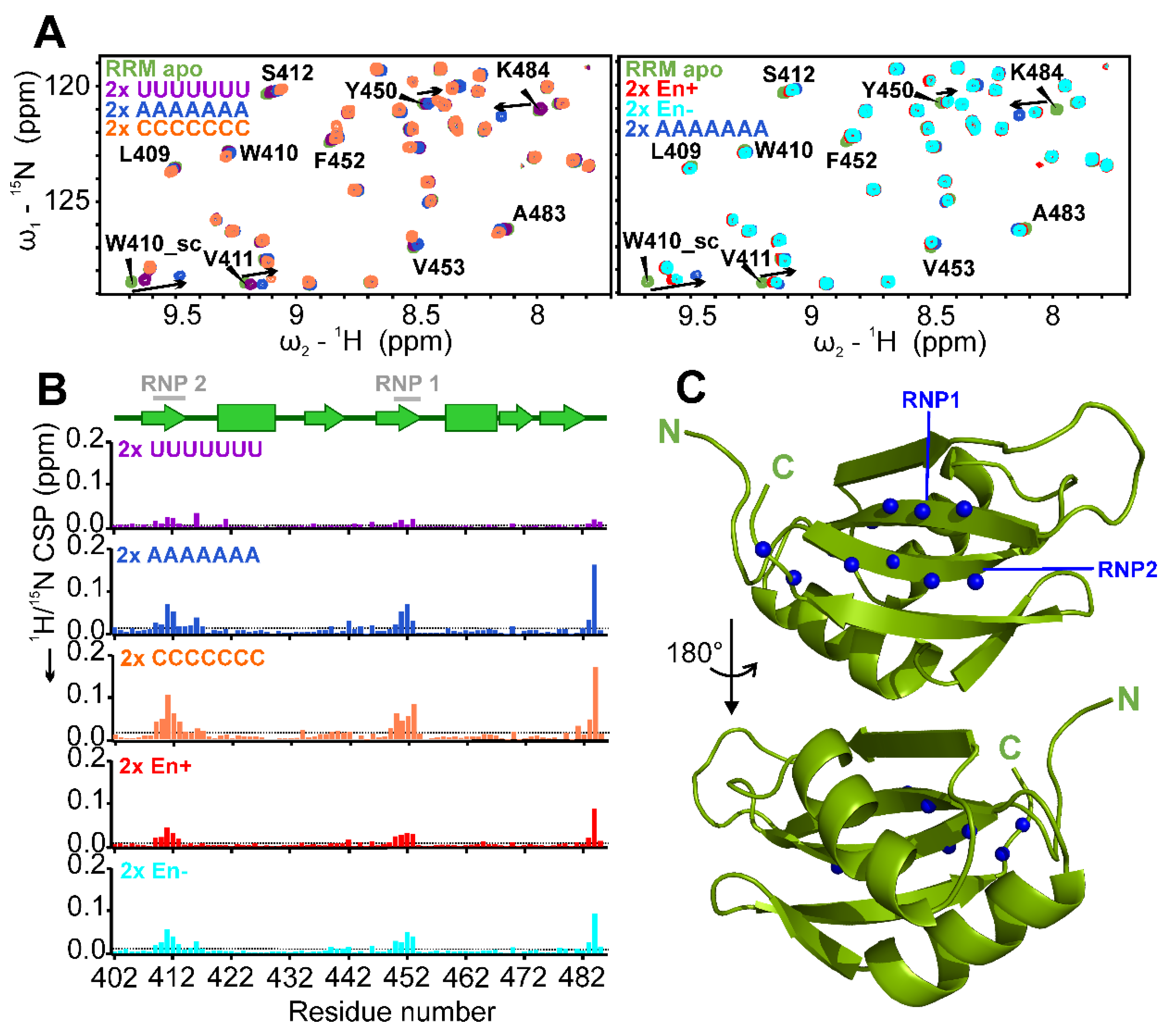
2.4. Target RNA Preferences of RRM and Mapping of Binding Site
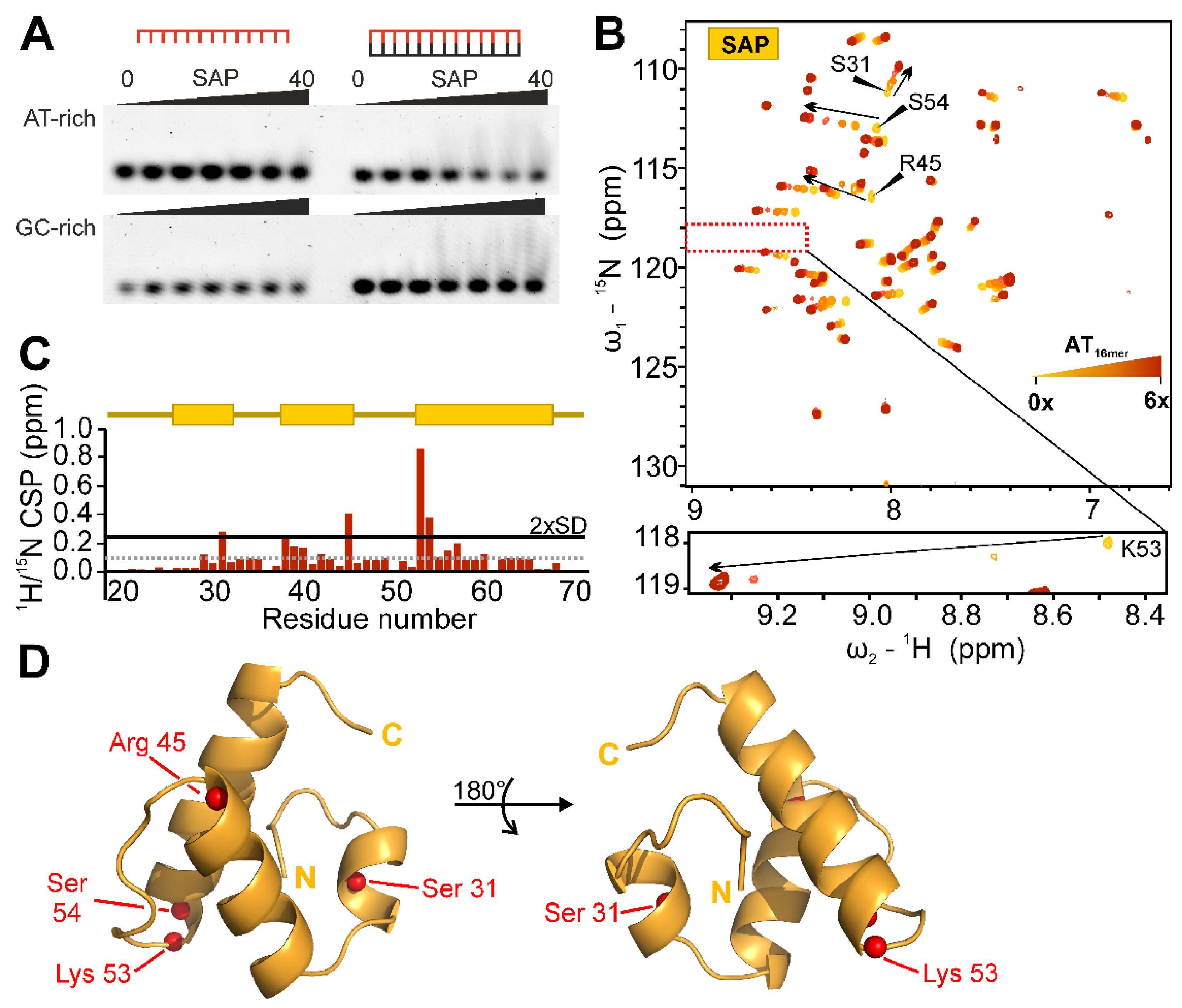
2.5. Target DNA-Preferences of SAP
2.6. The SAFB2 RRM Domain Binds Both NA Types
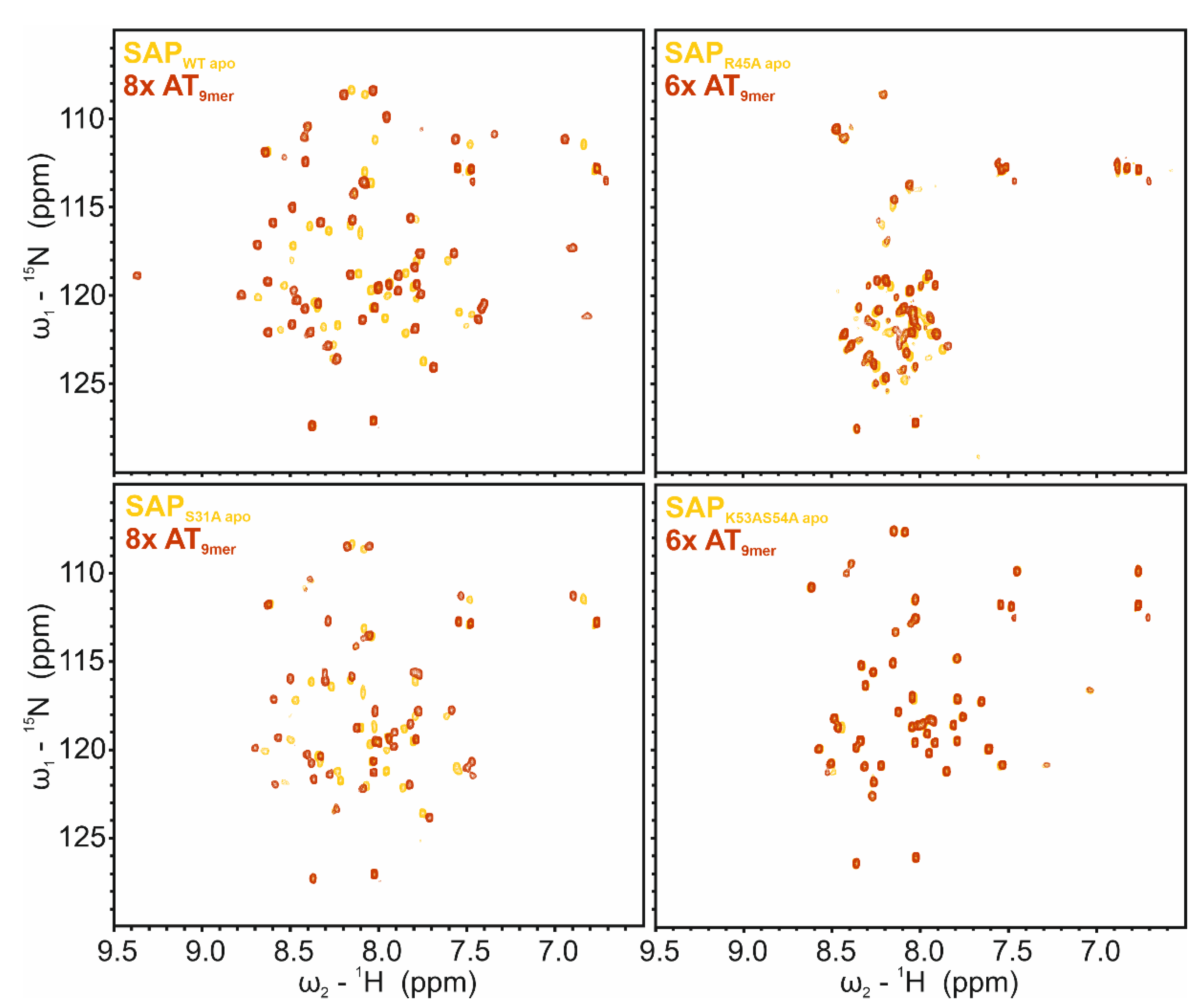
2.7. SAP Domain Mutational Study
2.8. SAP Domain Intra- and Inter-Domain Dynamics
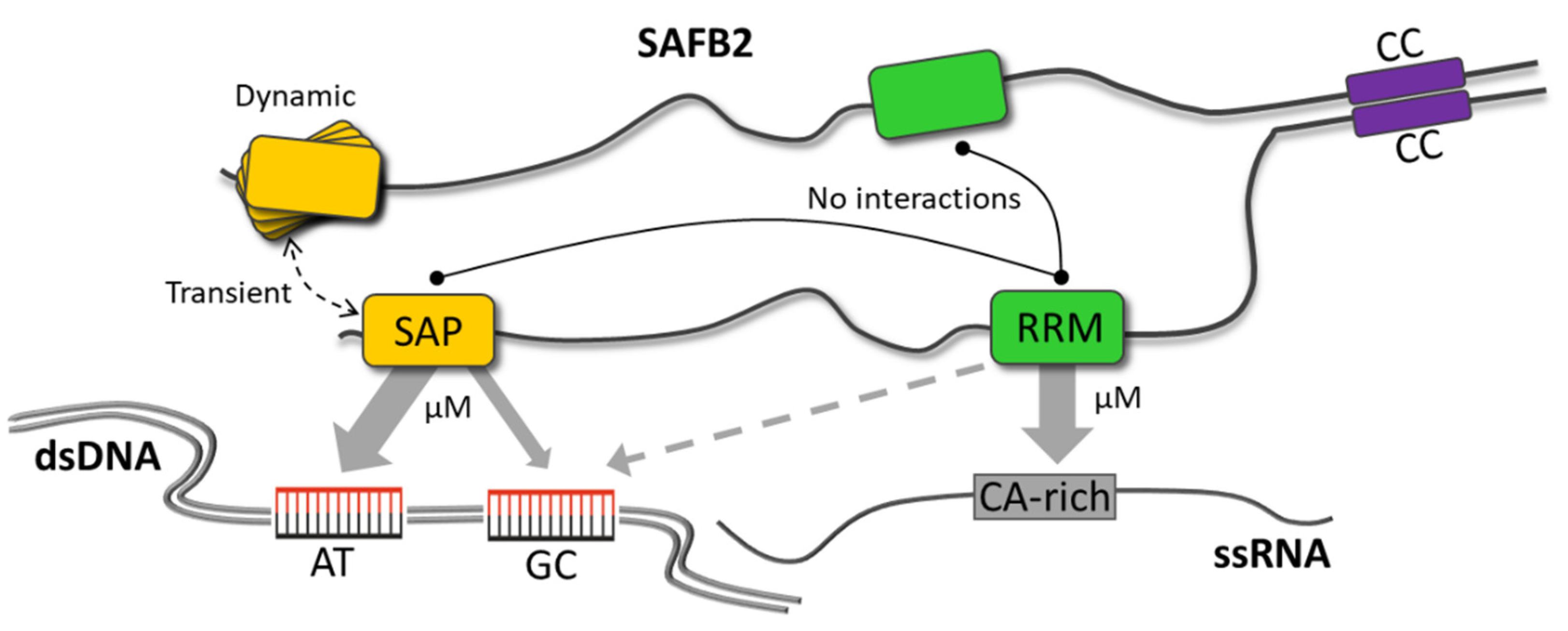
3. Discussion
4. Materials and Methods
4.1. Construct Design
4.2. Protein Production
4.3. RNA Preparation
4.4. NMR Data Acquisition and Analysis
4.5. Analytical Size-Exclusion Chromatography (aSEC) and aSEC-Based MW Determination
4.6. Size Exclusion Chromatography Coupled with Multi-Angle Light Scattering (SEC-MALS)
4.7. Electromobility Shifts Assays (EMSAs)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, R.; Chen, J.Y.; Liang, Z.; Luo, D.; Chen, G.; Lu, Z.J.; Chen, Y.; Zhou, B.; Li, H.; Du, X.; et al. Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 2019, 178, 107–121.e118. [Google Scholar] [CrossRef] [PubMed]
- Hudson, W.H.; Ortlund, E.A. The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 2014, 15, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. SMAD proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 2010, 39, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Godfrey, W.R.; Lin, J.; Zhao, G.; Kao, P.N. NF90 regulates inducible IL-2 gene expression in T cells. J. Exp. Med. 2007, 204, 971–977. [Google Scholar] [CrossRef]
- Shim, J.; Lim, H.; Yates Iii, J.; Karin, M. Nuclear export of NF90 is required for interleukin-2 mRNA stabilization. Mol. Cell 2002, 10, 1331–1344. [Google Scholar] [CrossRef]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.J.; Yuan, H.S. Structural insights into TDP43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef]
- Lukavsky, P.J. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP43. Nature Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef]
- Huang, D.B. Crystal structure of NF-κB (p50)2 complexed to a high-affinity RNA aptamer. Proc. Natl. Acad. Sci. USA 2003, 100, 9268–9273. [Google Scholar] [CrossRef]
- Müller, C.W.; Rey, F.A.; Sodeoka, M.; Verdine, G.L.; Harrison, S.C. Structure of the NF-κB p50 homodimer bound to DNA. Nature 1995, 373, 311–317. [Google Scholar] [CrossRef]
- Kiledjian, M.; Dreyfuss, G. Primary structure and binding activity of the hnRNP U protein: Binding RNA through RGG box. EMBO J. 1992, 11, 2655–2664. [Google Scholar] [CrossRef]
- Romig, H.; Fackelmayer, F.O.; Renz, A.; Ramsperger, U.; Richter, A. Characterization of SAF-A, a novel nuclear DNA binding protein from HeLa cells with high affinity for nuclear matrix/scaffold attachment DNA elements. EMBO J. 1992, 11, 3431–3440. [Google Scholar] [CrossRef]
- Renz, A.; Fackelmayer, F.O. Purification and molecular cloning of the scaffold attachment factor B (SAF-B), a novel human nuclear protein that specifically binds to S/MAR-DNA. Nucleic Acids Res. 1996, 24, 843–849. [Google Scholar] [CrossRef]
- Weighardt, F.; Cobianchi, F.; Cartegni, L.; Chiodi, I.; Villa, A.; Riva, S.; Biamonti, G. A novel hnRNP protein (HAP/SAF-B) enters a subset of hnRNP complexes and relocates in nuclear granules in response to heat shock. J. Cell Sci. 1999, 112 Pt 10, 1465–1476. [Google Scholar] [CrossRef]
- Watanabe, K.; Ohtsuki, T. Inhibition of HSF1 and SAFB Granule Formation Enhances Apoptosis Induced by Heat Stress. Int. J. Mol. Sci. 2021, 22, 4982. [Google Scholar] [CrossRef]
- Drakouli, S.; Lyberopoulou, A.; Papathanassiou, M.; Mylonis, I.; Georgatsou, E. Enhancer of rudimentary homologue interacts with scaffold attachment factor B at the nuclear matrix to regulate SR protein phosphorylation. FEBS J. 2017, 284, 2482–2500. [Google Scholar] [CrossRef]
- Norman, M.; Rivers, C.; Lee, Y.B.; Idris, J.; Uney, J. The increasing diversity of functions attributed to the SAFB family of RNA-/DNA-binding proteins. Biochem. J. 2016, 473, 4271–4288. [Google Scholar] [CrossRef]
- Huo, X.; Ji, L.; Zhang, Y.; Lv, P.; Cao, X.; Wang, Q.; Yan, Z.; Dong, S.; Du, D.; Zhang, F.; et al. The Nuclear Matrix Protein SAFB Cooperates with Major Satellite RNAs to Stabilize Heterochromatin Architecture Partially through Phase Separation. Mol. Cell 2020, 77, 368–383.e7. [Google Scholar] [CrossRef]
- Hernandez-Hernandez, J.M.; Mallappa, C.; Nasipak, B.T.; Oesterreich, S.; Imbalzano, A.N. The Scaffold attachment factor b1 (Safb1) regulates myogenic differentiation by facilitating the transition of myogenic gene chromatin from a repressed to an activated state. Nucleic Acids Res. 2013, 41, 5704–5716. [Google Scholar] [CrossRef]
- Hong, E.A.; Gautrey, H.L.; Elliott, D.J.; Tyson-Capper, A.J. SAFB1- and SAFB2-mediated transcriptional repression: Relevance to cancer. Biochem. Soc. Trans. 2012, 40, 826–830. [Google Scholar] [CrossRef]
- Luo, S.; Zhang, M.; Wu, H.; Ding, X.; Li, D.; Dong, X.; Hu, X.; Su, S.; Shang, W.; Wu, J.; et al. SAIL: A new conserved anti-fibrotic lncRNA in the heart. Basic Res. Cardiol. 2021, 116, 15. [Google Scholar] [CrossRef]
- Zhou, M.; Kuruvilla, L.; Shi, X.; Viviano, S.; Ahearn, I.M.; Amendola, C.R.; Su, W.; Badri, S.; Mahaffey, J.; Fehrenbacher, N.; et al. Scaffold association factor B (SAFB) is required for expression of prenyltransferases and RAS membrane association. Proc. Natl. Acad. Sci. USA 2020, 117, 31914–31922. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Qian, C.; You, S.; Rotinen, M.; Posadas, E.M.; Freedland, S.J.; Di Vizio, D.; Kim, J.; Freeman, M.R. Scaffold attachment factor B1 regulates androgen degradation pathways in prostate cancer. Am. J. Clin. Exp. Urol. 2021, 9, 337–349. [Google Scholar] [PubMed]
- Hutter, K.; Lohmuller, M.; Jukic, A.; Eichin, F.; Avci, S.; Labi, V.; Szabo, T.G.; Hoser, S.M.; Huttenhofer, A.; Villunger, A.; et al. SAFB2 Enables the Processing of Suboptimal Stem-Loop Structures in Clustered Primary miRNA Transcripts. Mol. Cell 2020, 78, 876–889.e876. [Google Scholar] [CrossRef]
- Xiong, F.; Wang, R.; Lee, J.H.; Li, S.; Chen, S.F.; Liao, Z.; Hasani, L.A.; Nguyen, P.T.; Zhu, X.; Krakowiak, J.; et al. RNA m(6)A modification orchestrates a LINE-1-host interaction that facilitates retrotransposition and contributes to long gene vulnerability. Cell Res. 2021, 31, 861–885. [Google Scholar] [CrossRef] [PubMed]
- Oesterreich, S. Scaffold attachment factors SAFB1 and SAFB2: Innocent bystanders or critical players in breast tumorigenesis? J. Cell. Biochem. 2003, 90, 653–661. [Google Scholar] [CrossRef]
- Hashimoto, T.; Matsuda, K.; Kawata, M. Scaffold attachment factor B (SAFB)1 and SAFB2 cooperatively inhibit the intranuclear mobility and function of ERalpha. J. Cell. Biochem. 2012, 113, 3039–3050. [Google Scholar] [CrossRef]
- Peidis, P.; Voukkalis, N.; Aggelidou, E.; Georgatsou, E.; Hadzopoulou-Cladaras, M.; Scott, R.E.; Nikolakaki, E.; Giannakouros, T. SAFB1 interacts with and suppresses the transcriptional activity of p53. FEBS Lett. 2011, 585, 78–84. [Google Scholar] [CrossRef]
- Fang, W.; Bartel, D.P. MicroRNA Clustering Assists Processing of Suboptimal MicroRNA Hairpins through the Action of the ERH Protein. Mol. Cell 2020, 78, 289–302.e6. [Google Scholar] [CrossRef]
- Erhardt, S.; Stoecklin, G. The heat’s on: Nuclear stress bodies signal intron retention. EMBO J. 2020, 39, e104154. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Metzler, W.J.; Constantine, K.L.; Friedrichs, M.S.; Bell, A.J.; Ernst, E.G.; Lavoie, T.B.; Mueller, L. Characterization of the three-dimensional solution structure of human profilin: 1H, 13C, and 15N NMR assignments and global folding pattern. Biochemistry 1993, 32, 13818–13829. [Google Scholar] [CrossRef]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K.; et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2019, 48, D882–D889. [Google Scholar] [CrossRef]
- Lambert, N.; Robertson, A.; Jangi, M.; McGeary, S.; Sharp, P.A.; Burge, C.B. RNA Bind-n-Seq: Quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol. Cell 2014, 54, 887–900. [Google Scholar] [CrossRef]
- Clery, A.; Blatter, M.; Allain, F.H. RNA recognition motifs: Boring? Not quite. Curr. Opin. Struct. Biol. 2008, 18, 290–298. [Google Scholar] [CrossRef]
- Maris, C.; Dominguez, C.; Allain, F.H.T. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005, 272, 2118–2131. [Google Scholar] [CrossRef]
- Furey, T.S. ChIP–seq and beyond: New and improved methodologies to detect and characterize protein–DNA interactions. Nat. Rev. Genet. 2012, 13, 840–852. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. SAP—A putative DNA-binding motif involved in chromosomal organization. Trends Biochem. Sci. 2000, 25, 112–114. [Google Scholar] [CrossRef]
- Weinheimer, A.S.; Paung, Y.; Rageul, J.; Khan, A.; Lo, N.; Ho, B.; Tong, M.; Alphonse, S.; Seeliger, M.A.; Kim, H. Extended DNA-binding interfaces beyond the canonical SAP domain contribute to the function of replication stress regulator SDE2 at DNA replication forks. J. Biol. Chem. 2022, 298, 102268. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Korn, S.M.; Dhamotharan, K.; Furtig, B.; Hengesbach, M.; Lohr, F.; Qureshi, N.S.; Richter, C.; Saxena, K.; Schwalbe, H.; Tants, J.N.; et al. (1)H, (13)C, and (15)N backbone chemical shift assignments of the nucleic acid-binding domain of SARS-CoV-2 non-structural protein 3e. Biomol. NMR Assign 2020, 14, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Korn, S.M.; Lambertz, R.; Furtig, B.; Hengesbach, M.; Lohr, F.; Richter, C.; Schwalbe, H.; Weigand, J.E.; Wohnert, J.; Schlundt, A. (1)H, (13)C, and (15)N backbone chemical shift assignments of the C-terminal dimerization domain of SARS-CoV-2 nucleocapsid protein. Biomol. NMR Assign. 2021, 15, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Wu, P.; Ji, P.; Zhang, J.; Gong, Q.; Wu, J.; Shi, Y. Solution structure of the second RRM domain of RBM5 and its unusual binding characters for different RNA targets. Biochemistry 2012, 51, 6667–6678. [Google Scholar] [CrossRef] [PubMed]
- Nerli, S.; McShan, A.C.; Sgourakis, N.G. Chemical shift-based methods in NMR structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 106–107, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lange, O.F.; Rossi, P.; Sgourakis, N.G.; Song, Y.; Lee, H.W.; Aramini, J.M.; Ertekin, A.; Xiao, R.; Acton, T.B.; Montelione, G.T.; et al. Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc. Natl. Acad. Sci. USA 2012, 109, 10873–10878. [Google Scholar] [CrossRef]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- Ge, H.; Zhou, D.; Tong, S.; Gao, Y.; Teng, M.; Niu, L. Crystal structure and possible dimerization of the single RRM of human PABPN1. Proteins 2008, 71, 1539–1545. [Google Scholar] [CrossRef]
- Thomsen, N.D.; Lawson, M.R.; Witkowsky, L.B.; Qu, S.; Berger, J.M. Molecular mechanisms of substrate-controlled ring dynamics and substepping in a nucleic acid-dependent hexameric motor. Proc. Natl. Acad. Sci. USA 2016, 113, E7691–E7700. [Google Scholar] [CrossRef]
- Raaijmakers, H.; Toro, I.; Birkenbihl, R.; Kemper, B.; Suck, D. Conformational flexibility in T4 endonuclease VII revealed by crystallography: Implications for substrate binding and cleavage. J. Mol. Biol. 2001, 308, 311–323. [Google Scholar] [CrossRef]
- Teplova, M.; Farazi, T.A.; Tuschl, T.; Patel, D.J. Structural basis underlying CAC RNA recognition by the RRM domain of dimeric RNA-binding protein RBPMS. Q. Rev. Biophys. 2016, 49, e1. [Google Scholar] [CrossRef]
- Schneider, T.; Hung, L.H.; Aziz, M.; Wilmen, A.; Thaum, S.; Wagner, J.; Janowski, R.; Muller, S.; Schreiner, S.; Friedhoff, P.; et al. Combinatorial recognition of clustered RNA elements by the multidomain RNA-binding protein IMP3. Nat. Commun. 2019, 10, 2266. [Google Scholar] [CrossRef]
- Tants, J.N.; Becker, L.M.; McNicoll, F.; Muller-McNicoll, M.; Schlundt, A. NMR-derived secondary structure of the full-length Ox40 mRNA 3′UTR and its multivalent binding to the immunoregulatory RBP Roquin. Nucleic Acids Res. 2022, 50, 4083–4099. [Google Scholar] [CrossRef]
- Notenboom, V.; Hibbert, R.G.; van Rossum-Fikkert, S.E.; Olsen, J.V.; Mann, M.; Sixma, T.K. Functional characterization of Rad18 domains for Rad6, ubiquitin, DNA binding and PCNA modification. Nucleic Acids Res. 2007, 35, 5819–5830. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, L.; Lin, D.; Chen, F.; Chen, D.J.; Chen, Y. The three-dimensional structure of the C-terminal DNA-binding domain of human Ku70. J. Biol. Chem. 2001, 276, 38231–38236. [Google Scholar] [CrossRef]
- Townson, S.M.; Dobrzycka, K.M.; Lee, A.V.; Air, M.; Deng, W.; Kang, K.; Jiang, S.; Kioka, N.; Michaelis, K.; Oesterreich, S. SAFB2, a new scaffold attachment factor homolog and estrogen receptor corepressor. J. Biol. Chem. 2003, 278, 20059–20068. [Google Scholar] [CrossRef]
- Sergeant, K.A.; Bourgeois, C.F.; Dalgliesh, C.; Venables, J.P.; Stevenin, J.; Elliott, D.J. Alternative RNA splicing complexes containing the scaffold attachment factor SAFB2. J. Cell Sci. 2007, 120, 309–319. [Google Scholar] [CrossRef]
- Taze, C.; Drakouli, S.; Samiotaki, M.; Panayotou, G.; Simos, G.; Georgatsou, E.; Mylonis, I. Short-term hypoxia triggers ROS and SAFB mediated nuclear matrix and mRNA splicing remodeling. Redox Biol. 2022, 58, 102545. [Google Scholar] [CrossRef]
- Daubner, G.M.; Clery, A.; Allain, F.H. RRM-RNA recognition: NMR or crystallography...and new findings. Curr. Opin. Struct. Biol. 2013, 23, 100–108. [Google Scholar] [CrossRef]
- Gobl, C.; Madl, T.; Simon, B.; Sattler, M. NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 80, 26–63. [Google Scholar] [CrossRef]
- Martinez-Lumbreras, S.; Taverniti, V.; Zorrilla, S.; Seraphin, B.; Perez-Canadillas, J.M. Gbp2 interacts with THO/TREX through a novel type of RRM domain. Nucleic Acids Res. 2016, 44, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Duszczyk, M.M.; Wischnewski, H.; Kazeeva, T.; Arora, R.; Loughlin, F.E.; von Schroetter, C.; Pradere, U.; Hall, J.; Ciaudo, C.; Allain, F.H. The solution structure of Dead End bound to AU-rich RNA reveals an unusual mode of tandem RRM-RNA recognition required for mRNA regulation. Nat. Commun. 2022, 13, 5892. [Google Scholar] [CrossRef] [PubMed]
- Clery, A.; Sinha, R.; Anczukow, O.; Corrionero, A.; Moursy, A.; Daubner, G.M.; Valcarcel, J.; Krainer, A.R.; Allain, F.H. Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc. Natl. Acad. Sci. USA 2013, 110, E2802–E2811. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Gut, H.; Chao, J.A. Structural basis of IMP3 RRM12 recognition of RNA. RNA 2018, 24, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Alvelos, M.I.; Bruggemann, M.; Sutandy, F.R.; Juan-Mateu, J.; Colli, M.L.; Busch, A.; Lopes, M.; Castela, A.; Aartsma-Rus, A.; Konig, J.; et al. The RNA-binding profile of the splicing factor SRSF6 in immortalized human pancreatic beta-cells. Life Sci. Alliance 2021, 4, e202000825. [Google Scholar] [CrossRef]
- Allain, F.H.; Gubser, C.C.; Howe, P.W.; Nagai, K.; Neuhaus, D.; Varani, G. Specificity of ribonucleoprotein interaction determined by RNA folding during complex formulation. Nature 1996, 380, 646–650. [Google Scholar] [CrossRef]
- Teplova, M.; Malinina, L.; Darnell, J.C.; Song, J.; Lu, M.; Abagyan, R.; Musunuru, K.; Teplov, A.; Burley, S.K.; Darnell, R.B.; et al. Protein-RNA and protein-protein recognition by dual KH1/2 domains of the neuronal splicing factor Nova-1. Structure 2011, 19, 930–944. [Google Scholar] [CrossRef]
- Korn, S.M.; Ulshofer, C.J.; Schneider, T.; Schlundt, A. Structures and target RNA preferences of the RNA-binding protein family of IGF2BPs: An overview. Structure 2021, 29, 787–803. [Google Scholar] [CrossRef]
- Yadav, M.; Singh, R.S.; Hogan, D.; Vidhyasagar, V.; Yang, S.; Chung, I.Y.W.; Kusalik, A.; Dmitriev, O.Y.; Cygler, M.; Wu, Y. The KH domain facilitates the substrate specificity and unwinding processivity of DDX43 helicase. J. Biol. Chem. 2021, 296, 100085. [Google Scholar] [CrossRef]
- Biertumpfel, C.; Yang, W.; Suck, D. Crystal structure of T4 endonuclease VII resolving a Holliday junction. Nature 2007, 449, 616–620. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Korn, S.M.; Schlundt, A. Structures and nucleic acid-binding preferences of the eukaryotic ARID domain. Biol. Chem. 2022, 403, 731–747. [Google Scholar] [CrossRef]
- Muto, Y.; Yokoyama, S. Structural insight into RNA recognition motifs: Versatile molecular Lego building blocks for biological systems. Wiley Interdiscip. Rev. RNA 2012, 3, 229–246. [Google Scholar] [CrossRef]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef]
- Carey, J. Affinity, Specificity, and Cooperativity of DNA Binding by Bacterial Gene Regulatory Proteins. Int. J. Mol. Sci. 2022, 23, 562. [Google Scholar] [CrossRef]
- Huang, Y.; Qiao, Y.; Zhao, Y.; Li, Y.; Yuan, J.; Zhou, J.; Sun, H.; Wang, H. Large scale RNA-binding proteins/LncRNAs interaction analysis to uncover lncRNA nuclear localization mechanisms. Brief. Bioinform. 2021, 22, bbab195. [Google Scholar] [CrossRef]
- Ron, M.; Ulitsky, I. Context-specific effects of sequence elements on subcellular localization of linear and circular RNAs. Nat. Commun. 2022, 13, 2481. [Google Scholar] [CrossRef]
- Edelheit, O.; Hanukoglu, A.; Hanukoglu, I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 2009, 9, 61. [Google Scholar] [CrossRef]
- Wacker, A.; Weigand, J.E.; Akabayov, S.R.; Altincekic, N.; Bains, J.K.; Banijamali, E.; Binas, O.; Castillo-Martinez, J.; Cetiner, E.; Ceylan, B.; et al. Secondary structure determination of conserved SARS-CoV-2 RNA elements by NMR spectroscopy. Nucleic Acids Res. 2020, 48, 12415–12435. [Google Scholar] [CrossRef]
- Morin, A.; Eisenbraun, B.; Key, J.; Sanschagrin, P.C.; Timony, M.A.; Ottaviano, M.; Sliz, P. Collaboration gets the most out of software. eLife 2013, 2, e01456. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. Chemical-Shifts as a Tool for Structure Determination. Methods Enzymol. 1994, 239, 363–392. [Google Scholar] [PubMed]
- Sklenar, V.; Bax, A. Spin-echo water suppression for the generation of pure-phase two-dimensional NMR spectra. J. Magn. Reson. 1987, 74, 469–479. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, P.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins-Struct. Funct. Bioinform. 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef]
- Shen, Y.; Lange, O.; Delaglio, F.; Rossi, P.; Aramini, J.M.; Liu, G.; Eletsky, A.; Wu, Y.; Singarapu, K.K.; Lemak, A.; et al. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA 2008, 105, 4685–4690. [Google Scholar] [CrossRef]
- Zhao, H.; Casillas, E., Jr.; Shroff, H.; Patterson, G.H.; Schuck, P. Tools for the quantitative analysis of sedimentation boundaries detected by fluorescence optical analytical ultracentrifugation. PLoS ONE 2013, 8, e77245. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence 5′ → 3′ | Usage |
|---|---|---|
| DNA Oligos used for binding studies | ||
| AT9mer_fw | CAATATAAC | Hybridized to AT9mer, for NMR titration. |
| AT9mer_rev | GTTATATTG | |
| AT16mer_fw | GCGCACAATATAACGC | Hybridized to AT16mer, for NMR titration. |
| AT16mer_rev | GCGTTATATTGTGCGC | |
| GC16mer_fw | CGCCCCCGCGCCCGCG | Hybridized to GC16mer, for NMR titration. |
| GC16mer_rev | CGCGGGCGCGGGGGCG | |
| AT13mer_fw | [FAM]-GCAATAAATACG | Hybridized to AT13mer, for fluorescent EMSAs. |
| AT13mer_rev | CGTATTTATTGC | |
| GC13mer_fw | [FAM]-GCCCCCGCGCCCG | Hybridized to GC13mer, for fluorescent EMSAs. |
| GC13mer_rev | CGGGCGCGGGGGC | |
| RNAs used for binding studies | ||
| En+ | UACACC | ENCODE (a) consensus |
| En− | CGGACU | ENCODE (a) control |
| U7 | UUUUUUU | Poly-N U-rich |
| A7 | AAAAAAA | Poly-N A-rich |
| C7 | CCCCCCC | Poly-N C-rich |
| G7 | GGGGGGG | Poly-N G-rich |
| Bulge | GGAUUUUAUGGGGCACGGACAACCCAUAUCCUGAU | Multi-characteristic model RNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korn, S.M.; Von Ehr, J.; Dhamotharan, K.; Tants, J.-N.; Abele, R.; Schlundt, A. Insight into the Structural Basis for Dual Nucleic Acid—Recognition by the Scaffold Attachment Factor B2 Protein. Int. J. Mol. Sci. 2023, 24, 3286. https://doi.org/10.3390/ijms24043286
Korn SM, Von Ehr J, Dhamotharan K, Tants J-N, Abele R, Schlundt A. Insight into the Structural Basis for Dual Nucleic Acid—Recognition by the Scaffold Attachment Factor B2 Protein. International Journal of Molecular Sciences. 2023; 24(4):3286. https://doi.org/10.3390/ijms24043286
Chicago/Turabian StyleKorn, Sophie M., Julian Von Ehr, Karthikeyan Dhamotharan, Jan-Niklas Tants, Rupert Abele, and Andreas Schlundt. 2023. "Insight into the Structural Basis for Dual Nucleic Acid—Recognition by the Scaffold Attachment Factor B2 Protein" International Journal of Molecular Sciences 24, no. 4: 3286. https://doi.org/10.3390/ijms24043286