
The COPD-Associated Polymorphism Impairs the CFTR Function to Suppress Excessive IL-8 Production upon Environmental Pathogen Exposure

Abstract
:1. Introduction
2. Results
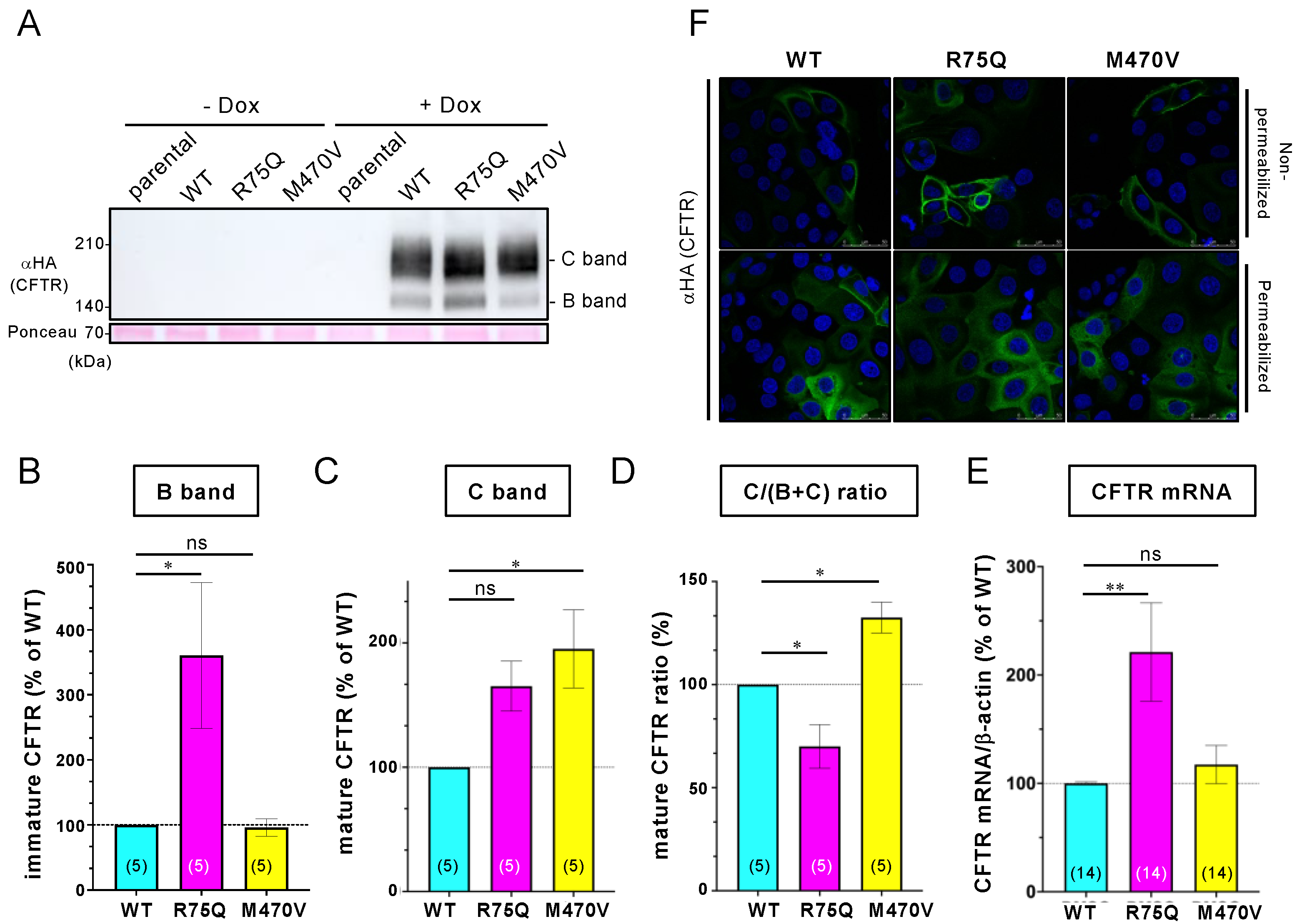
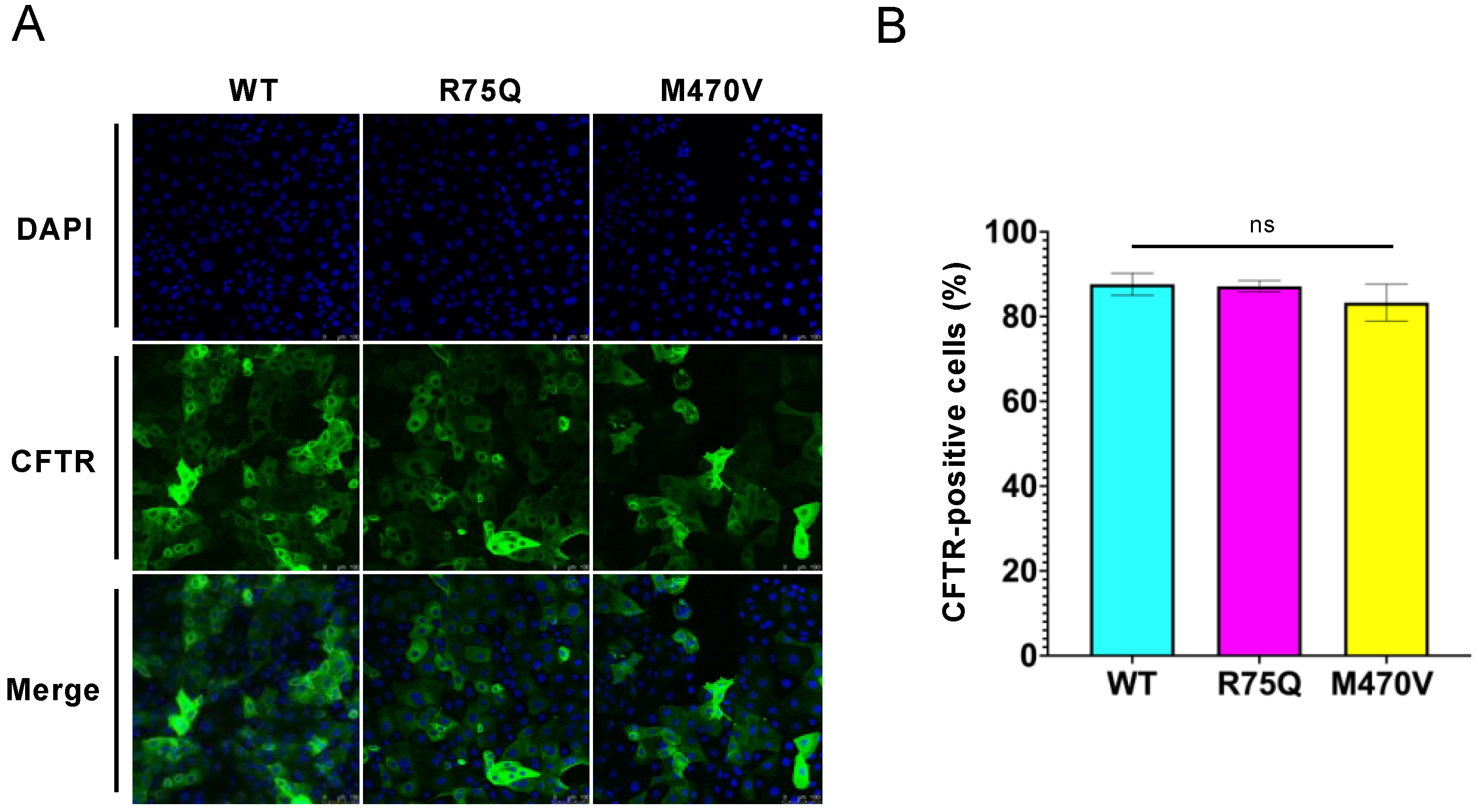
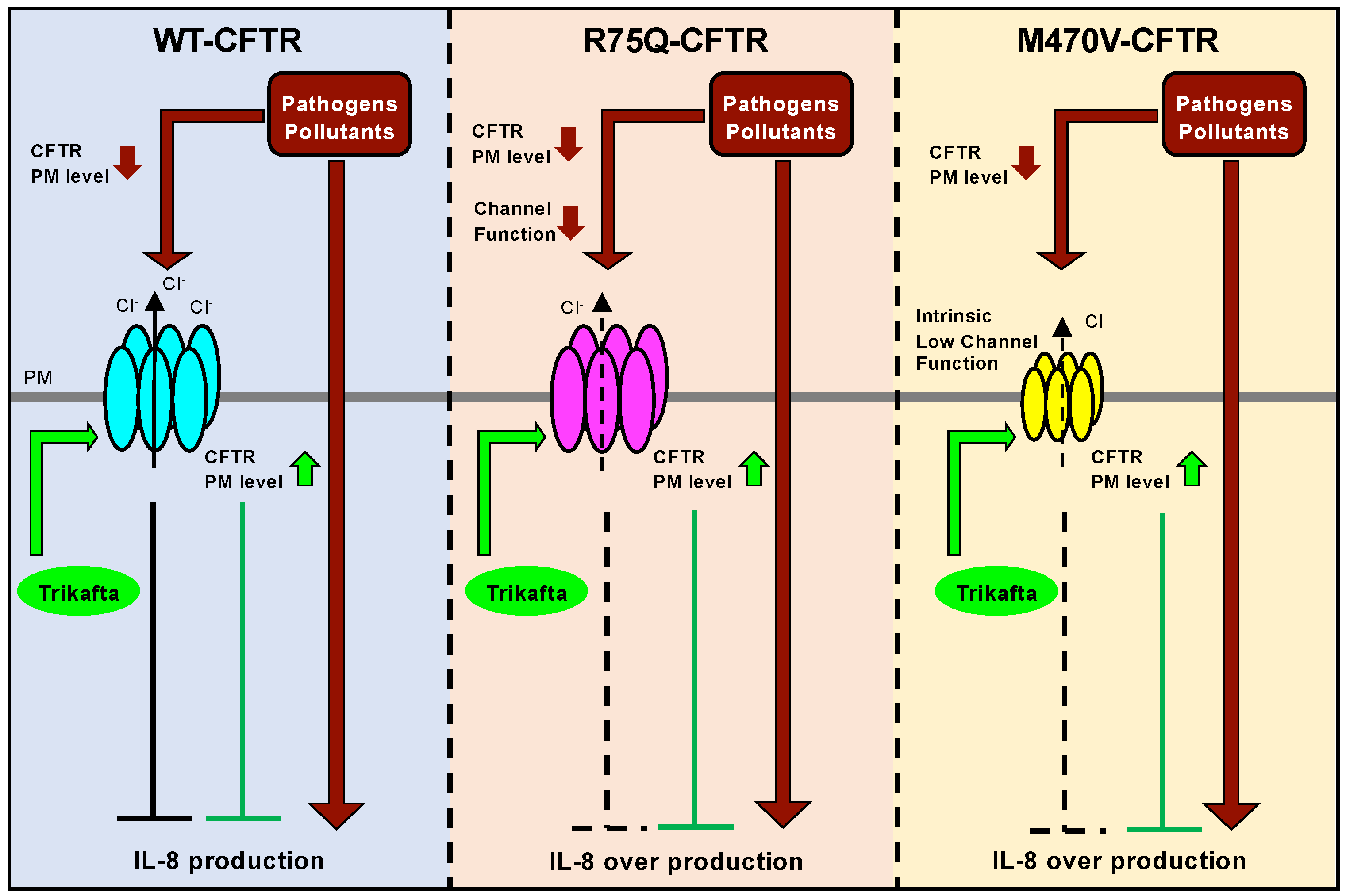
2.1. R75Q or M470V Polymorphism Loses CFTR’s Function to Suppress the Proinflammatory Cytokine Secretion
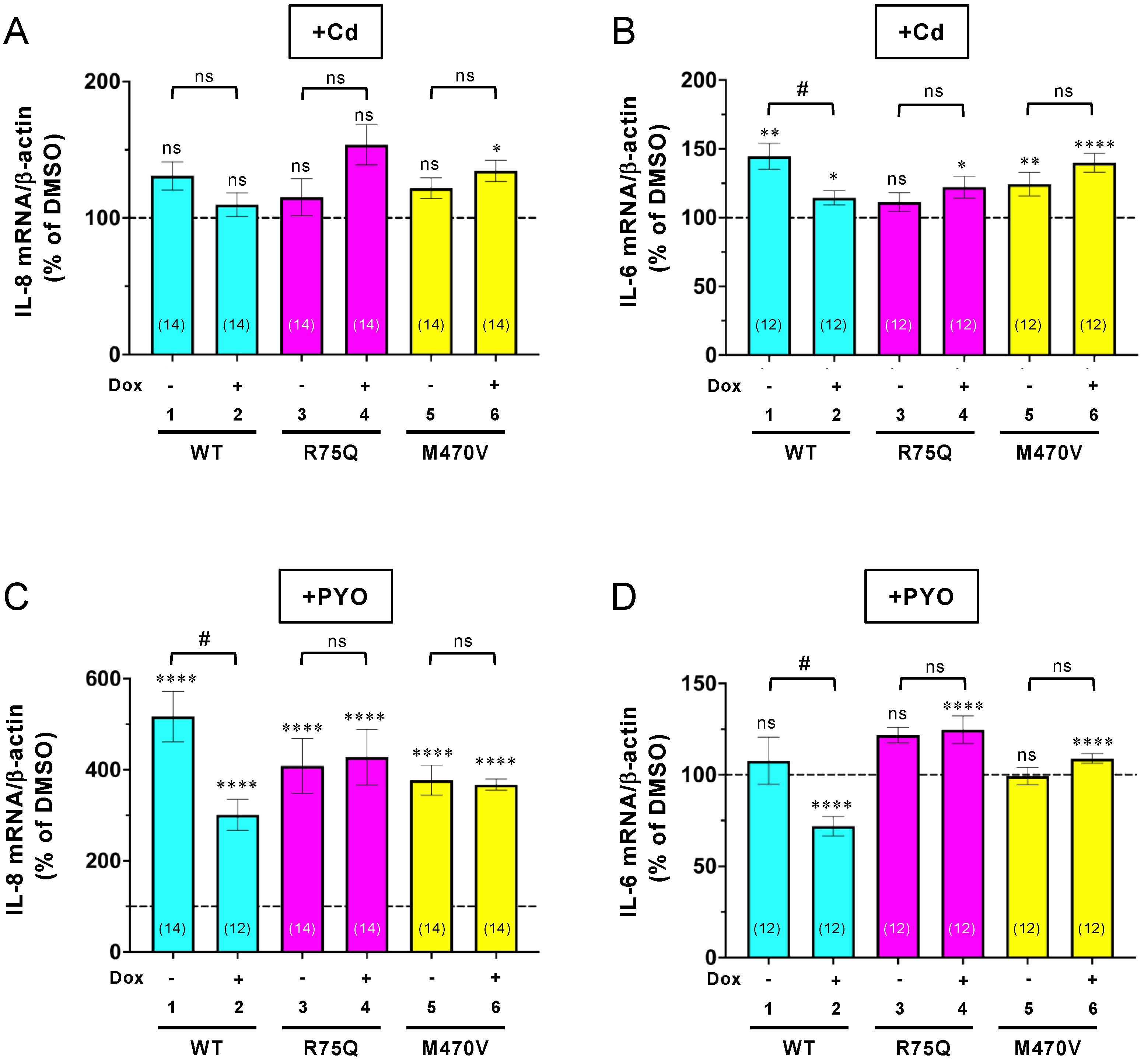
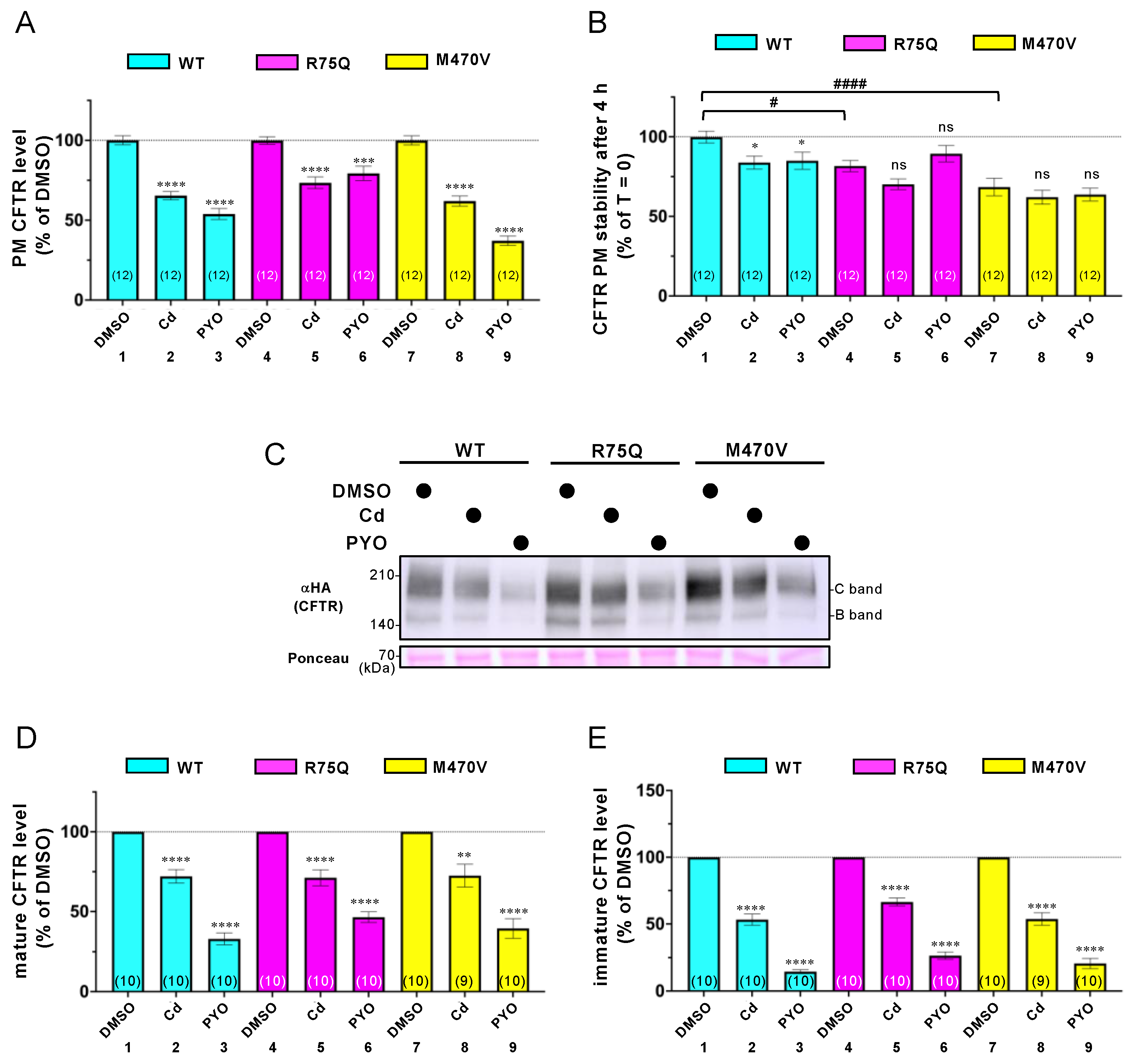
2.2. Sustained Exposure to Cd or PYO Reduces the PM Level of R75Q-CFTR and M470V-CFTR Proteins to the Same Extent as WT-CFTR
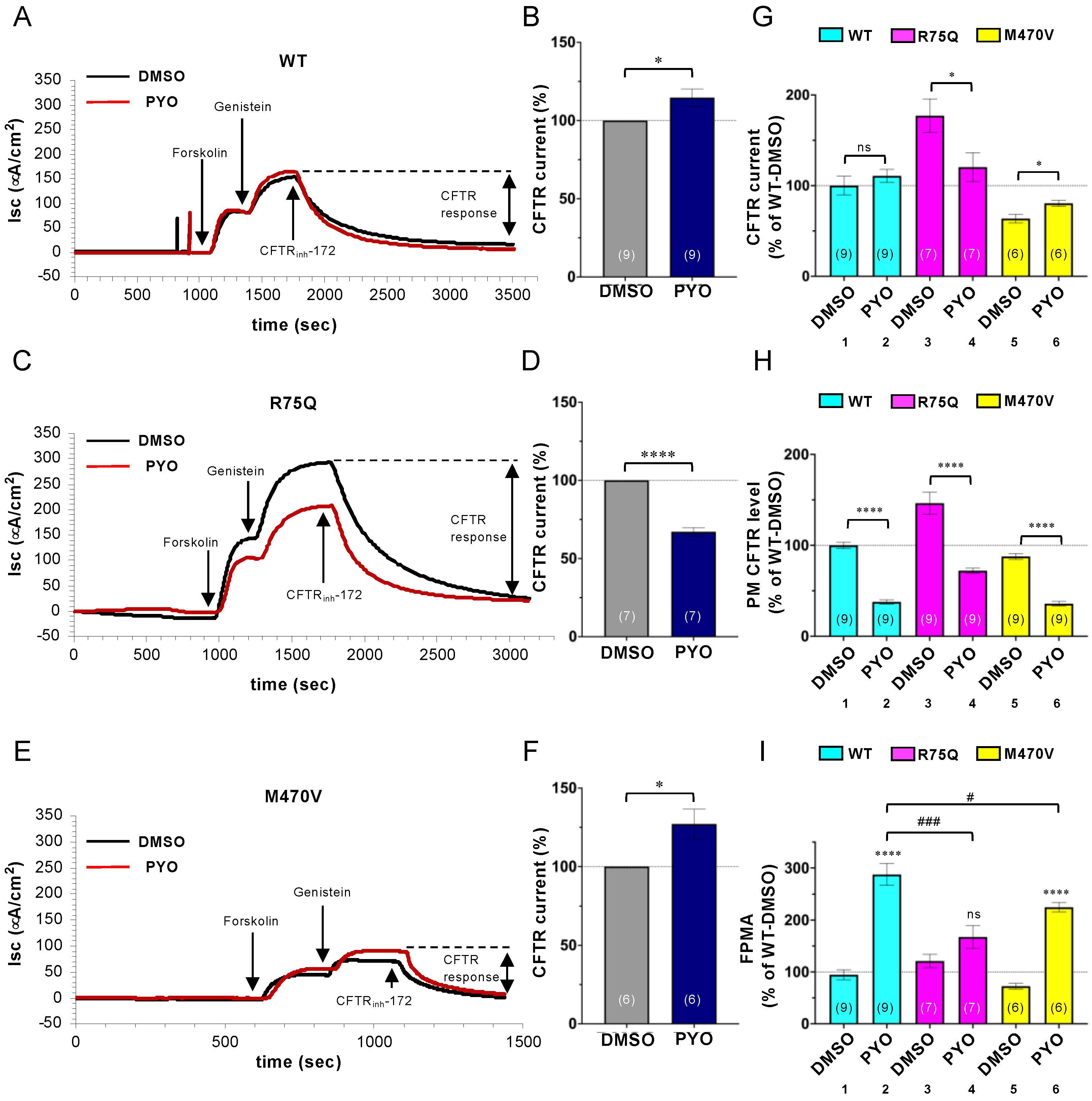
2.3. Chronic PYO Exposure Inactivates the R75Q-CFTR Channel
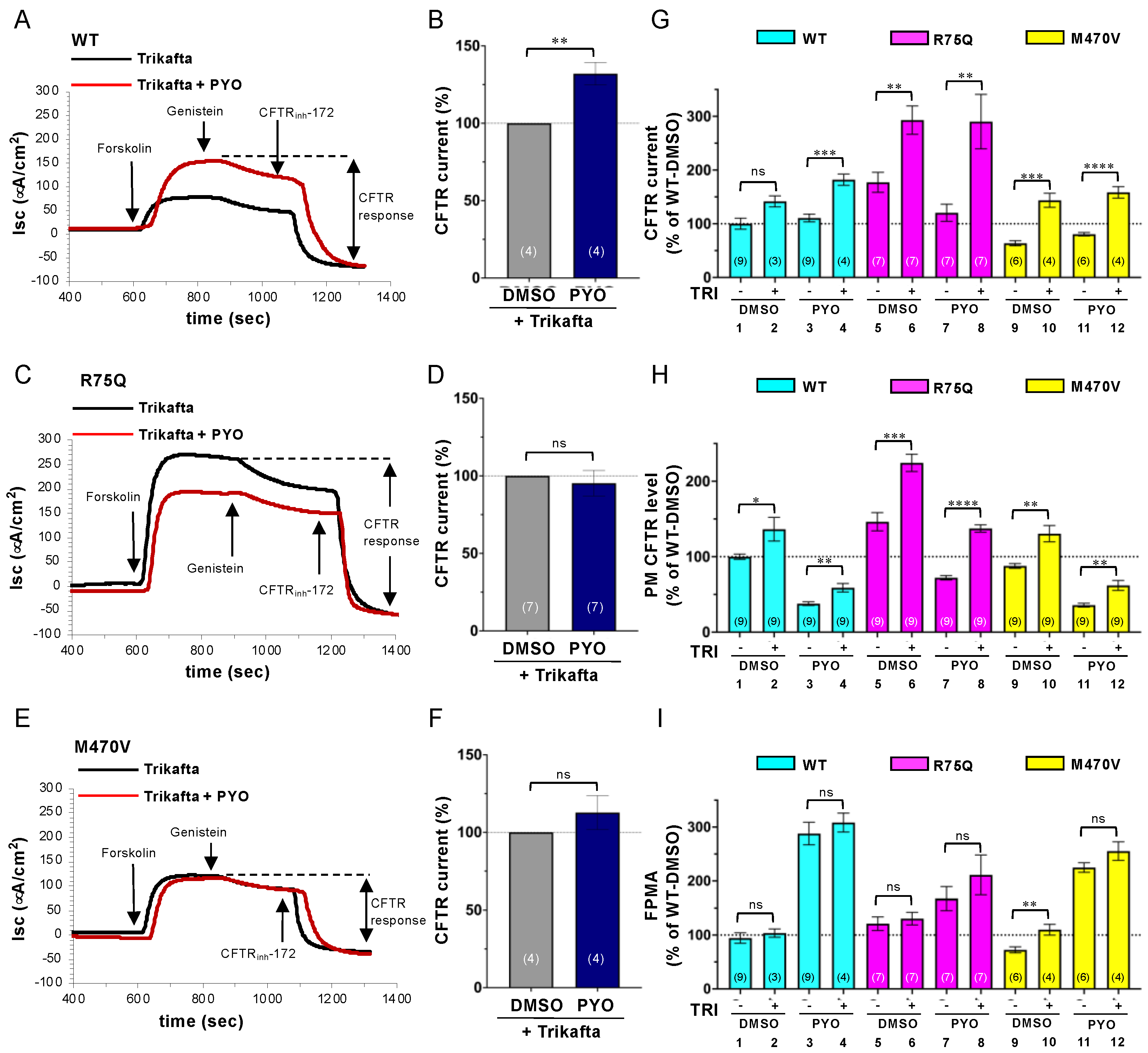
2.4. Trikafta Corrects the Cell Surface Expression of CFTR upon PYO Exposure
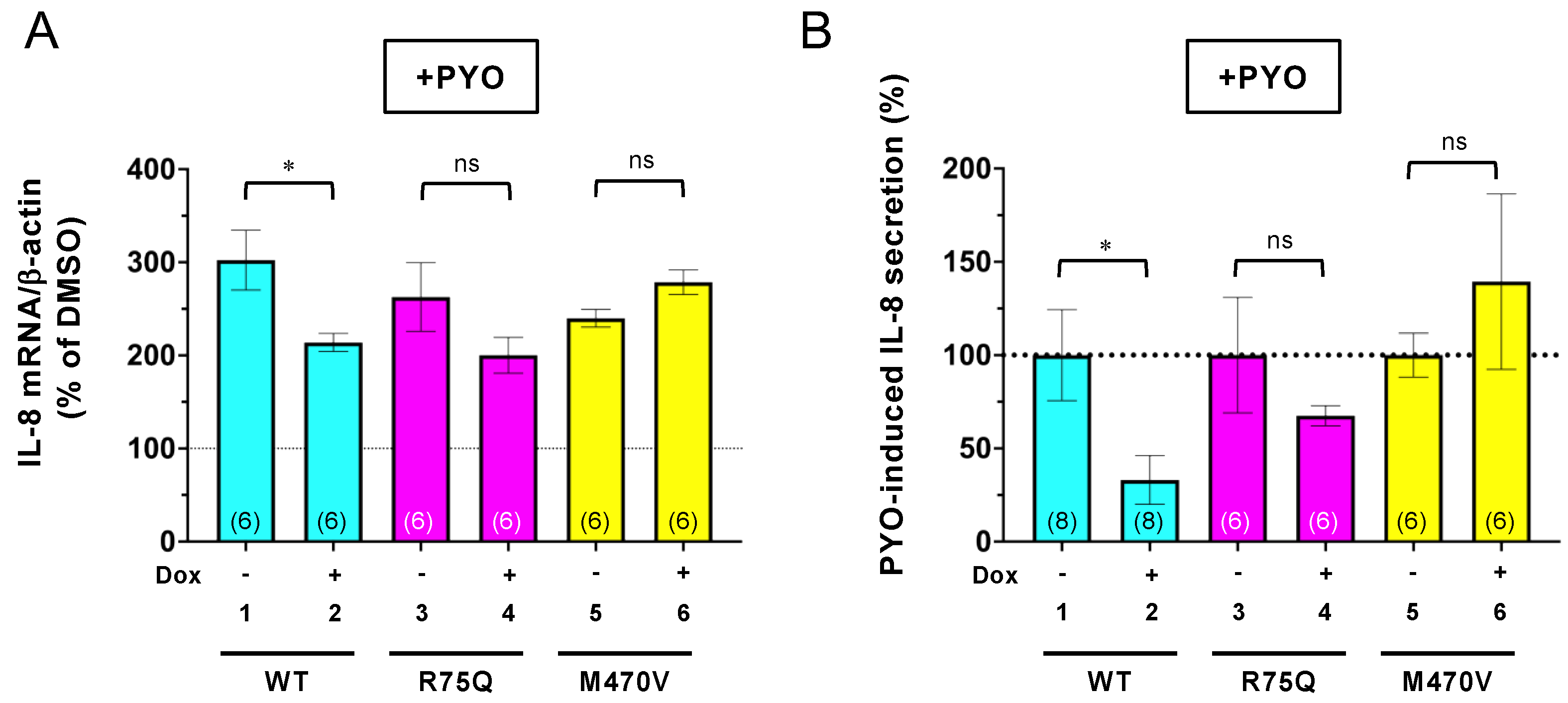
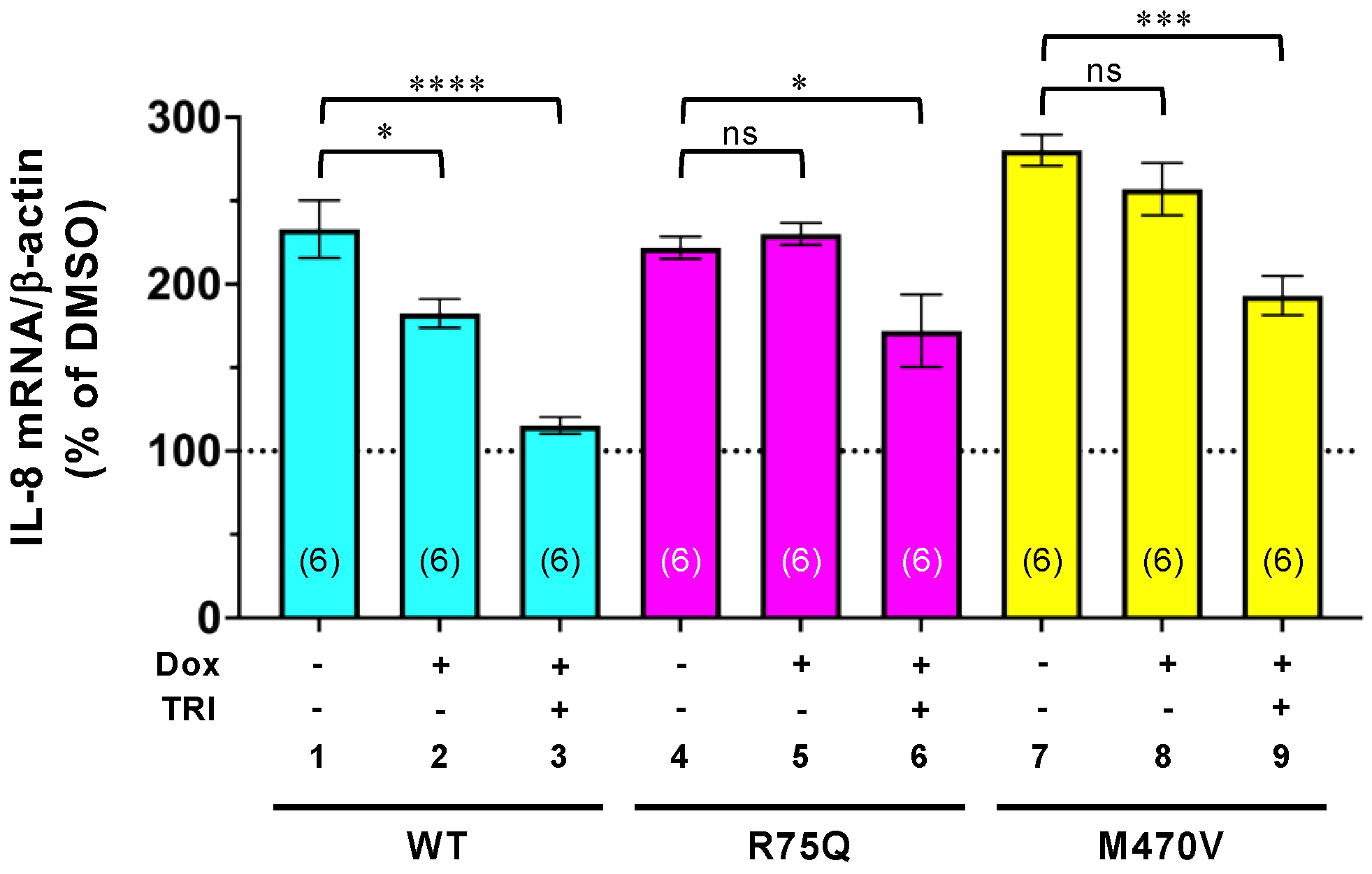
2.5. Trikafta Corrects the Function of R75Q- or M470V-CFTR to Suppress the PYO-induced IL-8 Production
3. Discussion
4. Materials and Methods
4.1. Constructs
4.2. Cell Culture and Reagents
4.3. Immunocytochemistry
4.4. Quantitative Real-Time PCR
4.5. IL-8 ELISA
4.6. Cell Surface Density and Cell Surface Stability Measurements of CFTR
4.7. Western Blotting
4.8. Short-Circuit Current Measurement
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christenson, S.; Smith, B.; Bafadhel, M.; Putcha, N. Chronic obstructive pulmonary disease. Lancet 2022, 399, 2227–2242. [Google Scholar] [CrossRef]
- WHO. Chronic Obstructive Pulmonary Disease (COPD); WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Dransfield, M.; Rowe, S.; Vogelmeier, C.; Wedzicha, J.; Criner, G.; Han, M.; Martinez, F.; Calverley, P. Cystic Fibrosis Transmembrane Conductance Regulator: Roles in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2022, 205, 631–640. [Google Scholar] [CrossRef]
- Riordan, J.; Rommens, J.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Mall, M.; Grubb, B.; Harkema, J.; O’Neal, W.; Boucher, R. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef]
- Pilewski, J.; Frizzell, R. Role of CFTR in airway disease. Physiol. Rev. 1999, 79, S215–S255. [Google Scholar] [CrossRef] [Green Version]
- Cantin, A. Cystic Fibrosis Transmembrane Conductance Regulator. Implications in Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2016, 13, S150–S155. [Google Scholar]
- Dransfield, M.; Wilhelm, A.; Flanagan, B.; Courville, C.; Tidwell, S.; Raju, S.; Gaggar, A.; Steele, C.; Tang, L.; Liu, B.; et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013, 144, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Min, T.; Mazur, S.; Vij, N. Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J. Immunol. 2011, 186, 602–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Liang, X.; Zhao, M.; Liu, S.; Huang, Y.; Idell, S.; Li, X.; Ji, H. Correlation of apical fluid-regulating channel proteins with lung function in human COPD lungs. PLoS ONE 2014, 9, e109725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreindler, J.; Jackson, A.; Kemp, P.; Bridges, R.; Danahay, H. Inhibition of chloride secretion in human bronchial epithelial cells by cigarette smoke extract. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L894–L902. [Google Scholar] [CrossRef] [Green Version]
- Raju, S.V.; Jackson, P.; Courville, C.; McNicholas, C.; Sloane, P.; Sabbatini, G.; Tidwell, S.; Tang, L.; Liu, B.; Fortenberry, J.; et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am. J. Respir. Crit. Care Med. 2013, 188, 1321–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marklew, A.; Patel, W.; Moore, P.; Tan, C.; Smith, A.; Sassano, M.; Gray, M.; Tarran, R. Cigarette Smoke Exposure Induces Retrograde Trafficking of CFTR to the Endoplasmic Reticulum. Sci. Rep. 2019, 9, 13655. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, J.; Sheridan, J.; Polk, W.; Davies, C.; Tarran, R. Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J. Biol. Chem. 2014, 289, 7671–7681. [Google Scholar] [CrossRef] [Green Version]
- Rennolds, J.; Butler, S.; Maloney, K.; Boyaka, P.; Davis, I.; Knoell, D.; Parinandi, N.; Cormet-Boyaka, E. Cadmium regulates the expression of the CFTR chloride channel in human airway epithelial cells. Toxicol. Sci. 2010, 116, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco-Hernández, L.; Quintana-Gallego, E.; Calero, C.; Reinoso-Arija, R.; Ruiz-Duque, B.; López-Campos, J. Dysfunction in the Cystic Fibrosis Transmembrane Regulator in Chronic Obstructive Pulmonary Disease as a Potential Target for Personalised Medicine. Biomedicines 2021, 9, 1437. [Google Scholar] [CrossRef]
- Hanrahan, J.; Abu-Arish, A.; Wong, F.; Turner, M.; Carlile, G.; Thomas, D.; Cantin, A. Chronic obstructive pulmonary disease and the modulation of CFTR by acute exposure to cigarette smoke. Am. J. Physiol. Cell Physiol. 2022, 323, C1374–C1392. [Google Scholar] [CrossRef] [PubMed]
- Fresquez, M.; Pappas, R.; Watson, C. Establishment of toxic metal reference range in tobacco from US cigarettes. J. Anal. Toxicol. 2013, 37, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fang, J.; Leonard, S.; Rao, K. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic. Biol. Med. 2004, 36, 1434–1443. [Google Scholar] [CrossRef]
- Jin, T.; Nordberg, M.; Frech, W.; Dumont, X.; Bernard, A.; Ye, T.; Kong, Q.; Wang, Z.; Li, P.; Lundström, N.; et al. Cadmium biomonitoring and renal dysfunction among a population environmentally exposed to cadmium from smelting in China (ChinaCad). Biometals 2002, 15, 397–410. [Google Scholar] [CrossRef]
- Nordberg, G. Lung cancer and exposure to environmental cadmium. Lancet Oncol. 2006, 7, 99–101. [Google Scholar] [CrossRef]
- Pääkkö, P.; Anttila, S.; Kokkonen, P.; Kalliomäki, P. Cadmium in lung tissue as marker for smoking. Lancet 1988, 1, 477. [Google Scholar] [CrossRef]
- Chambers, R.; Laurent, G.; Westergren-Thorsson, G. Cadmium inhibits proteoglycan and procollagen production by cultured human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 1998, 19, 498–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnúr, A.; Premchandar, A.; Bagdany, M.; Lukacs, G. Phosphorylation-dependent modulation of CFTR macromolecular signalling complex activity by cigarette smoke condensate in airway epithelia. Sci. Rep. 2019, 9, 12706. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T. The many faces of Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Clin. Infect. Dis. 2008, 47, 1534–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.; Prince, A. Cystic fibrosis: A mucosal immunodeficiency syndrome. Nat. Med. 2012, 18, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Kuang, Z.; Xu, Y.; Walling, B.; Lau, G. Pyocyanin-induced mucin production is associated with redox modification of FOXA2. Respir. Res. 2013, 14, 82. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.; Sykes, D.; Watson, D.; Rutman, A.; Taylor, G.; Cole, P. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect. Immun. 1988, 56, 2515–2517. [Google Scholar] [CrossRef] [Green Version]
- Gloyne, L.; Grant, G.; Perkins, A.; Powell, K.; McDermott, C.; Johnson, P.; Anderson, G.; Kiefel, M.; Anoopkumar-Dukie, S. Pyocyanin-induced toxicity in A549 respiratory cells is causally linked to oxidative stress. Toxicol. In Vitro 2011, 25, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Young, L.; Chen, Y.; Ran, H.; Meyers, M.; Joseph, P.; Cho, Y.; Hassett, D.; Lau, G. Pseudomonas aeruginosa pyocyanin inactivates lung epithelial vacuolar ATPase-dependent cystic fibrosis transmembrane conductance regulator expression and localization. Cell Microbiol. 2006, 8, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, C.; Fischer, H.; Kim, E.-J. Oxidative stress caused by pyocyanin impairs CFTR Cl—Transport in human bronchial epithelial cells. Free. Radic. Biol. Med. 2008, 45, 1653–1662. [Google Scholar] [CrossRef] [Green Version]
- Domenech, A.; Puig, C.; Martí, S.; Santos, S.; Fernández, A.; Calatayud, L.; Dorca, J.; Ardanuy, C.; Liñares, J. Infectious etiology of acute exacerbations in severe COPD patients. J. Infect. 2013, 67, 516–523. [Google Scholar] [CrossRef]
- Garcia-Vidal, C.; Almagro, P.; Romaní, V.; Rodríguez-Carballeira, M.; Cuchi, E.; Canales, L.; Blasco, D.; Heredia, J.; Garau, J. Pseudomonas aeruginosa in patients hospitalised for COPD exacerbation: A prospective study. Eur. Respir. J. 2009, 34, 1072–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo-Troyano, A.; Melo, V.; Marcos, P.; Laserna, E.; Peiro, M.; Suarez-Cuartin, G.; Perea, L.; Feliu, A.; Plaza, V.; Faverio, P.; et al. Pseudomonas aeruginosa in Chronic Obstructive Pulmonary Disease Patients with Frequent Hospitalized Exacerbations: A Prospective Multicentre Study. Respiration 2018, 96, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Tzetis, M.; Efthymiadou, A.; Strofalis, S.; Psychou, P.; Dimakou, A.; Pouliou, E.; Doudounakis, S.; Kanavakis, E. CFTR gene mutations—Including three novel nucleotide substitutions—And haplotype background in patients with asthma, disseminated bronchiectasis and chronic obstructive pulmonary disease. Hum. Genet. 2001, 108, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Larusch, J.; Sun, X.; Aloe, A.; Lamb, J.; Hawes, R.; Cotton, P.; Brand, R.; Anderson, M.; Money, M.; et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011, 140, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Cuppens, H.; Lin, W.; Jaspers, M.; Costes, B.; Teng, H.; Vankeerberghen, A.; Jorissen, M.; Droogmans, G.; Reynaert, I.; Goossens, M.; et al. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J. Clin. Investig. 1998, 101, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Bossard, F.; Goepp, J.; Verkman, A.S.; Galietta, L.J.; Hanrahan, J.W.; Lukacs, G.L. Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol. Biol. Cell 2012, 23, 4188–4202. [Google Scholar] [CrossRef]
- Stankovic, M.; Nikolic, A.; Divac, A.; Tomovic, A.; Petrovic-Stanojevic, N.; Andjelic, M.; Dopudja-Pantic, V.; Surlan, M.; Vujicic, I.; Ponomarev, D.; et al. The CFTR M470V gene variant as a potential modifier of COPD severity: Study of Serbian population. Genet. Test 2008, 12, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Divac, A.; Nikolic, A.; Mitic-Milikic, M.; Nagorni-Obradovic, L.; Petrovic-Stanojevic, N.; Dopudja-Pantic, V.; Nadaskic, R.; Savic, A.; Radojkovic, D. High frequency of the R75Q CFTR variation in patients with chronic obstructive pulmonary disease. J. Cyst. Fibros. 2004, 3, 189–191. [Google Scholar] [CrossRef] [Green Version]
- LaRusch, J.; Jung, J.; General, I. Mechanisms of CFTR Functional Variants That Impair Regulated Bicarbonate Permeation and Increase Risk for Pancreatitis but Not for Cystic Fibrosis. PLoS Genet. 2014, 10, e1004376. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, C.; Collnot, E.; Baldes, C.; Becker, U.; Laue, M.; Kim, K.; Lehr, C. Towards an in vitro model of cystic fibrosis small airway epithelium: Characterisation of the human bronchial epithelial cell line CFBE41o-. Cell Tissue Res. 2006, 323, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Veit, G.; Sakai, R.; Aki, M.; Fujihara, T.; Higashi, M.; Susuki-Miyata, S.; Miyata, M.; Fukuda, N.; Yoshida, A.; et al. Chaperone-Independent Peripheral Quality Control of CFTR by RFFL E3 Ligase. Dev. Cell 2018, 44, 694–708.e7. [Google Scholar] [CrossRef] [Green Version]
- Bebok, Z.; Collawn, J.; Wakefield, J.; Parker, W.; Li, Y.; Varga, K.; Sorscher, E.; Clancy, J. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J. Physiol. 2005, 569, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Monsó, E.; Soler, N.; Torres, F.; Angrill, J.; Riise, G.; Zalacaín, R.; Morera, J.; Torres, A. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern. Med. 2005, 165, 891–897. [Google Scholar] [CrossRef]
- Miravitlles, M.; Espinosa, C.; Fernández-Laso, E.; Martos, J.; Maldonado, J.; Gallego, M. Relationship between bacterial flora in sputum and functional impairment in patients with acute exacerbations of COPD. Study Group of Bacterial Infection in COPD. Chest 1999, 116, 40–46. [Google Scholar] [CrossRef]
- Eller, J.; Ede, A.; Schaberg, T.; Niederman, M.; Mauch, H.; Lode, H. Infective exacerbations of chronic bronchitis: Relation between bacteriologic etiology and lung function. Chest 1998, 113, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.; Brauer, A.; Eschberger, K.; Lobbins, P.; Grove, L.; Cai, X.; Sethi, S. Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 853–860. [Google Scholar] [CrossRef]
- Clunes, L.; Davies, C.; Coakley, R.; Aleksandrov, A.; Henderson, A.; Zeman, K.; Worthington, E.; Gentzsch, M.; Kreda, S.; Cholon, D.; et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012, 26, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Bagdany, M.; Veit, G.; Fukuda, R.; Avramescu, R.; Okiyoneda, T.; Baaklini, I.; Singh, J.; Sovak, G.; Xu, H.; Apaja, P.; et al. Chaperones rescue the energetic landscape of mutant CFTR at single molecule and in cell. Nat. Commun. 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Cowley, E.; Linsdell, P. Oxidant stress stimulates anion secretion from the human airway epithelial cell line Calu-3: Implications for cystic fibrosis lung disease. J. Physiol. 2002, 543, 201–209. [Google Scholar] [CrossRef]
- Hoy, S. Elexacaftor/Ivacaftor/Tezacaftor: First Approval. Drugs 2019, 79, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Roldan, A.; Hancock, M.; Da Fonte, D.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Boncoeur, E.; Roque, T.; Bonvin, E.; Saint-Criq, V.; Bonora, M.; Clement, A.; Tabary, O.; Henrion-Caude, A.; Jacquot, J. Cystic fibrosis transmembrane conductance regulator controls lung proteasomal degradation and nuclear factor-kappaB activity in conditions of oxidative stress. Am. J. Pathol. 2008, 172, 1184–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantin, A.; Bilodeau, G.; Ouellet, C.; Liao, J.; Hanrahan, J. Oxidant stress suppresses CFTR expression. Am. J. Physiol. Cell Physiol. 2006, 290, C262–C270. [Google Scholar] [CrossRef] [Green Version]
- Bérubé, J.; Roussel, L.; Nattagh, L.; Rousseau, S. Loss of cystic fibrosis transmembrane conductance regulator function enhances activation of p38 and ERK MAPKs, increasing interleukin-6 synthesis in airway epithelial cells exposed to Pseudomonas aeruginosa. J. Biol. Chem. 2010, 285, 22299–22307. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, A.; Tang, X.; Hoegger, M.; Alaiwa, M.A.; Ramachandran, S.; Moninger, T.; Karp, P.; Wohlford-Lenane, C.; Haagsman, H.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Salinas, D.; Nielson, D.; Verkman, A. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell Physiol. 2006, 290, C741–C749. [Google Scholar] [CrossRef]
- Cho, S.; Urata, Y.; Iida, T.; Goto, S.; Yamaguchi, M.; Sumikawa, K.; Kondo, T. Glutathione downregulates the phosphorylation of I kappa B: Autoloop regulation of the NF-kappa B-mediated expression of NF-kappa B subunits by TNF-alpha in mouse vascular endothelial cells. Biochem. Biophys. Res. Commun. 1998, 253, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Huang, X.; Warren, J. Intracellular glutathione redox status modulates MCP-1 expression in pulmonary granulomatous vasculitis. Lab Invest. 1999, 79, 837–847. [Google Scholar]
- Christman, J.; Sadikot, R.; Blackwell, T. The role of nuclear factor-kappa B in pulmonary diseases. Chest 2000, 117, 1482–1487. [Google Scholar] [CrossRef]
- Hashimoto, S.; Gon, Y.; Matsumoto, K.; Takeshita, I.; Asai, Y.; Machino, T.; Horie, T. Regulation by intracellular glutathione of TNF-alpha-induced p38 MAP kinase activation and RANTES production by human pulmonary vascular endothelial cells. Allergy 2000, 55, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Gosset, P.; Wallaert, B.; Tonnel, A.; Fourneau, C. Thiol regulation of the production of TNF-alpha, IL-6 and IL-8 by human alveolar macrophages. Eur. Respir. J. 1999, 14, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Jun, I.; Shin, D.; Yoon, J.; Piao, H.; Jung, J.; Park, H.; Cheng, M.; Bahar, I.; Whitcomb, D.; et al. Regulation of CFTR Bicarbonate Channel Activity by WNK1: Implications for Pancreatitis and CFTR-Related Disorders. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 79–103. [Google Scholar] [CrossRef] [Green Version]
- Wong, F.; AbuArish, A.; Matthes, E.; Turner, M.; Greene, L.; Cloutier, A.; Robert, R.; Thomas, D.; Cosa, G.; Cantin, A.; et al. Cigarette smoke activates CFTR through ROS-stimulated cAMP signaling in human bronchial epithelial cells. Am. J. Physiol. Cell Physiol. 2018, 314, C118–C134. [Google Scholar] [CrossRef] [PubMed]
- Harrington, M.; Gunderson, K.; Kopito, R. Redox reagents and divalent cations alter the kinetics of cystic fibrosis transmembrane conductance regulator channel gating. J. Biol. Chem. 1999, 274, 27536–27544. [Google Scholar] [CrossRef] [Green Version]
- Harrington, M.; Kopito, R. Cysteine residues in the nucleotide binding domains regulate the conductance state of CFTR channels. Biophys. J. 2002, 82, 1278–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, G.; Fu, L.; Rowe, S.; Collawn, J. The therapeutic potential of CFTR modulators for COPD and other airway diseases. Curr. Opin. Pharmacol. 2017, 34, 132–139. [Google Scholar] [CrossRef]
- Lambert, J.; Raju, S.; Tang, L.; McNicholas, C.; Li, Y.; Courville, C.; Farris, R.; Coricor, G.; Smoot, L.; Mazur, M.; et al. Cystic fibrosis transmembrane conductance regulator activation by roflumilast contributes to therapeutic benefit in chronic bronchitis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloane, P.; Shastry, S.; Wilhelm, A.; Courville, C.; Tang, L.; Backer, K.; Levin, E.; Raju, S.; Li, Y.; Mazur, M.; et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS ONE 2012, 7, e39809. [Google Scholar] [CrossRef] [Green Version]
- Raju, S.; Lin, V.; Liu, L.; McNicholas, C.; Karki, S.; Sloane, P.; Tang, L.; Jackson, P.; Wang, W.; Wilson, L.; et al. The Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor Augments Mucociliary Clearance Abrogating Cystic Fibrosis Transmembrane Conductance Regulator Inhibition by Cigarette Smoke. Am. J. Respir. Cell Mol. Biol. 2017, 56, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Barrière, H.; Bagdány, M.; Rabeh, W.; Du, K.; Höhfeld, J.; Young, J.; Lukacs, G. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Glozman, R.; Okiyoneda, T.; Mulvihill, C.; Rini, J.; Barriere, H.; Lukacs, G. N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 2009, 184, 847–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Orientation | Sequence |
|---|---|---|
| Human β-actin | Forward | 5′-ACTCTTCCAGCCTTCCTTCC-3′ |
| Reverse | 5′-GAGGAGCAATGATCTTGATCTTC-3′ | |
| Human IL-8 | Forward | 5′-TCCTGATTTCTGCAAGCTCTG-3′ |
| Reverse | 5′-GTCCACTCTCAATCACTCTCAG-3′ | |
| Human IL-6 | Forward | 5′-GCACTGGCAGAAAACAACCT-3′ |
| Reverse | 5′-CAGGGGTGGTTATTGCATCT-3′ | |
| Human CFTR | Forward | 5′-AGTGGAGGAAAGCCTTTGGAGT-3′ |
| Reverse | 5′-ACAGATCTGAGCCCAACCTCA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinata, D.; Fukuda, R.; Okiyoneda, T. The COPD-Associated Polymorphism Impairs the CFTR Function to Suppress Excessive IL-8 Production upon Environmental Pathogen Exposure. Int. J. Mol. Sci. 2023, 24, 2305. https://doi.org/10.3390/ijms24032305
Hinata D, Fukuda R, Okiyoneda T. The COPD-Associated Polymorphism Impairs the CFTR Function to Suppress Excessive IL-8 Production upon Environmental Pathogen Exposure. International Journal of Molecular Sciences. 2023; 24(3):2305. https://doi.org/10.3390/ijms24032305
Chicago/Turabian StyleHinata, Daichi, Ryosuke Fukuda, and Tsukasa Okiyoneda. 2023. "The COPD-Associated Polymorphism Impairs the CFTR Function to Suppress Excessive IL-8 Production upon Environmental Pathogen Exposure" International Journal of Molecular Sciences 24, no. 3: 2305. https://doi.org/10.3390/ijms24032305