
Overlapping and Distinct Features of Cardiac Pathology in Inherited Human and Murine Ether Lipid Deficiency

 , , , , , , , , ,
, , , , , , , , ,
Abstract
:1. Introduction
2. Results
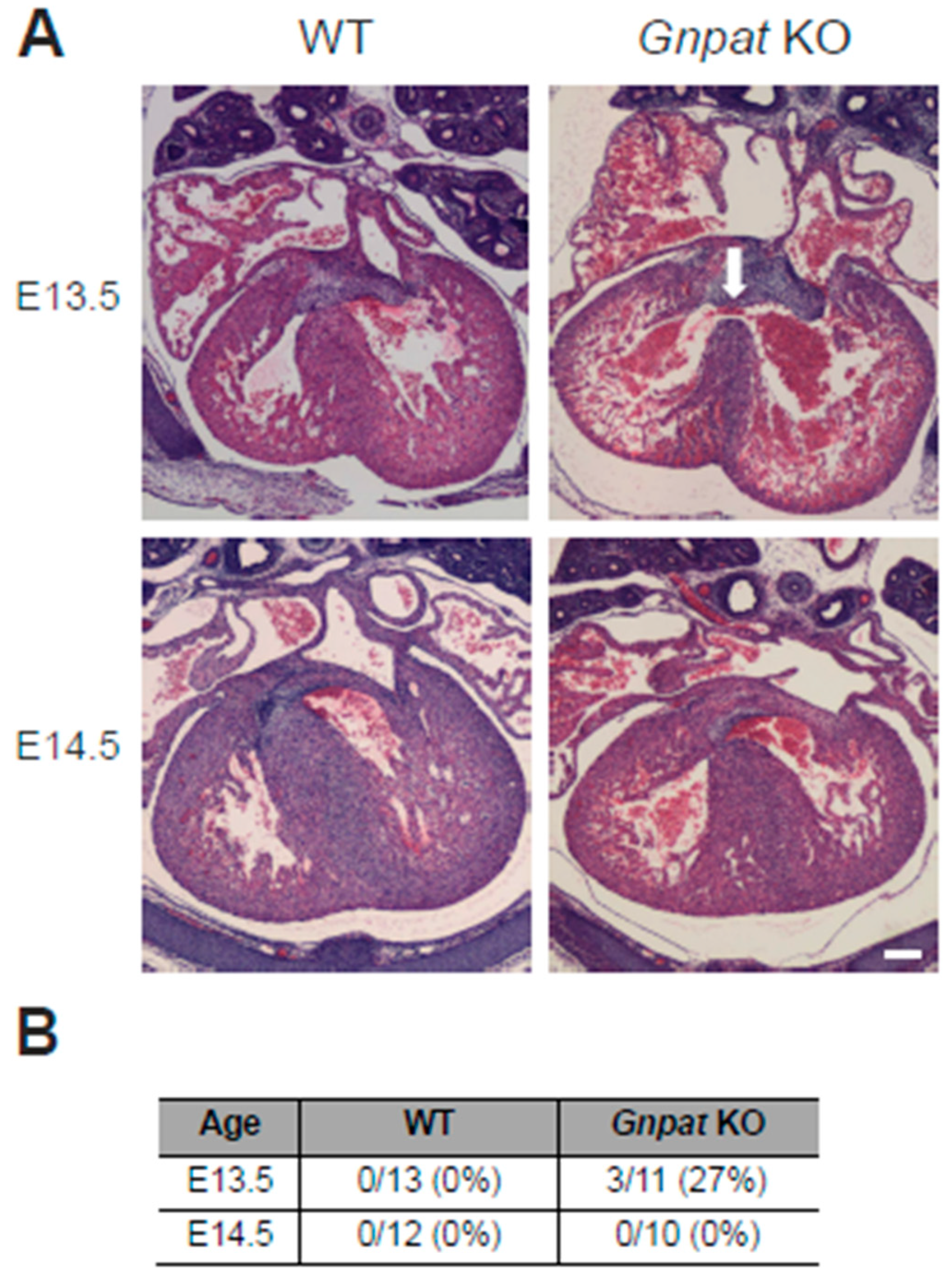
2.1. Ventricular Septal Defects Are Present in Developing Gnpat KO Mice
2.2. In Vivo Assessment of Cardiac Function Reveals Normal Ejection Fraction but Reduced Stroke Volume in Middle-Aged Ether-Lipid-Deficient Mice
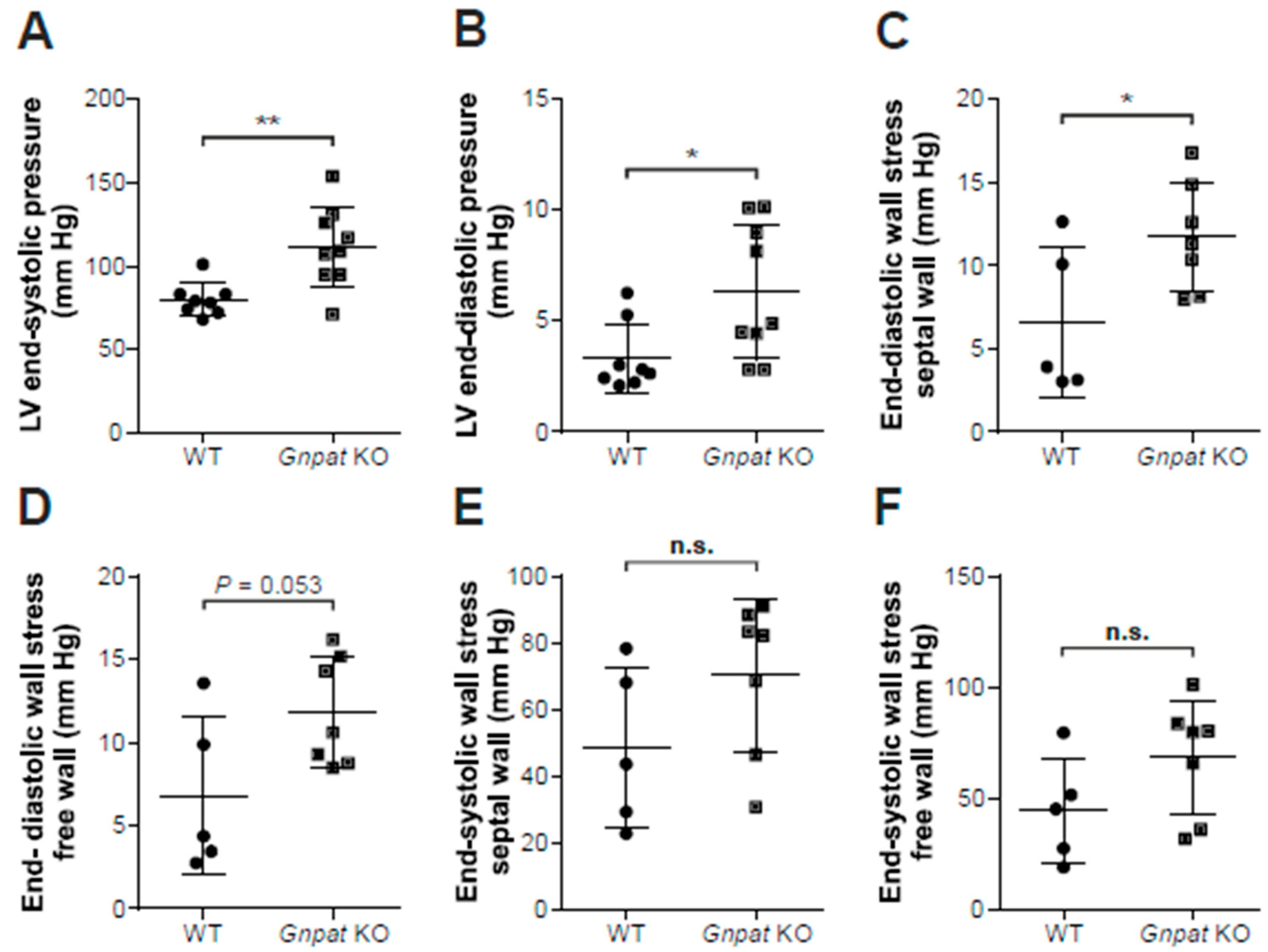
2.3. Ether-Lipid-Deficient Mice Show Increased Left Ventricular Pressure
2.4. No Overt Cardiac Fibrosis in Gnpat KO Mice
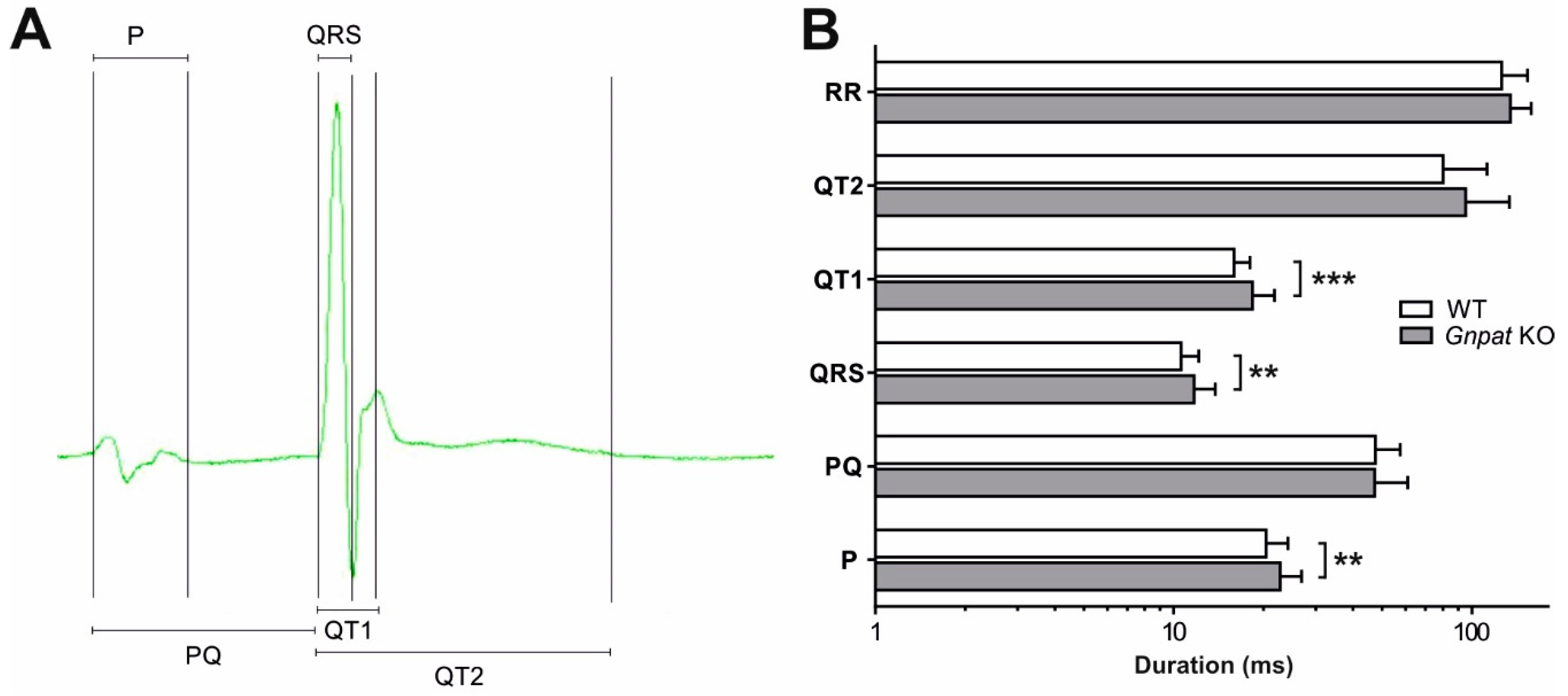
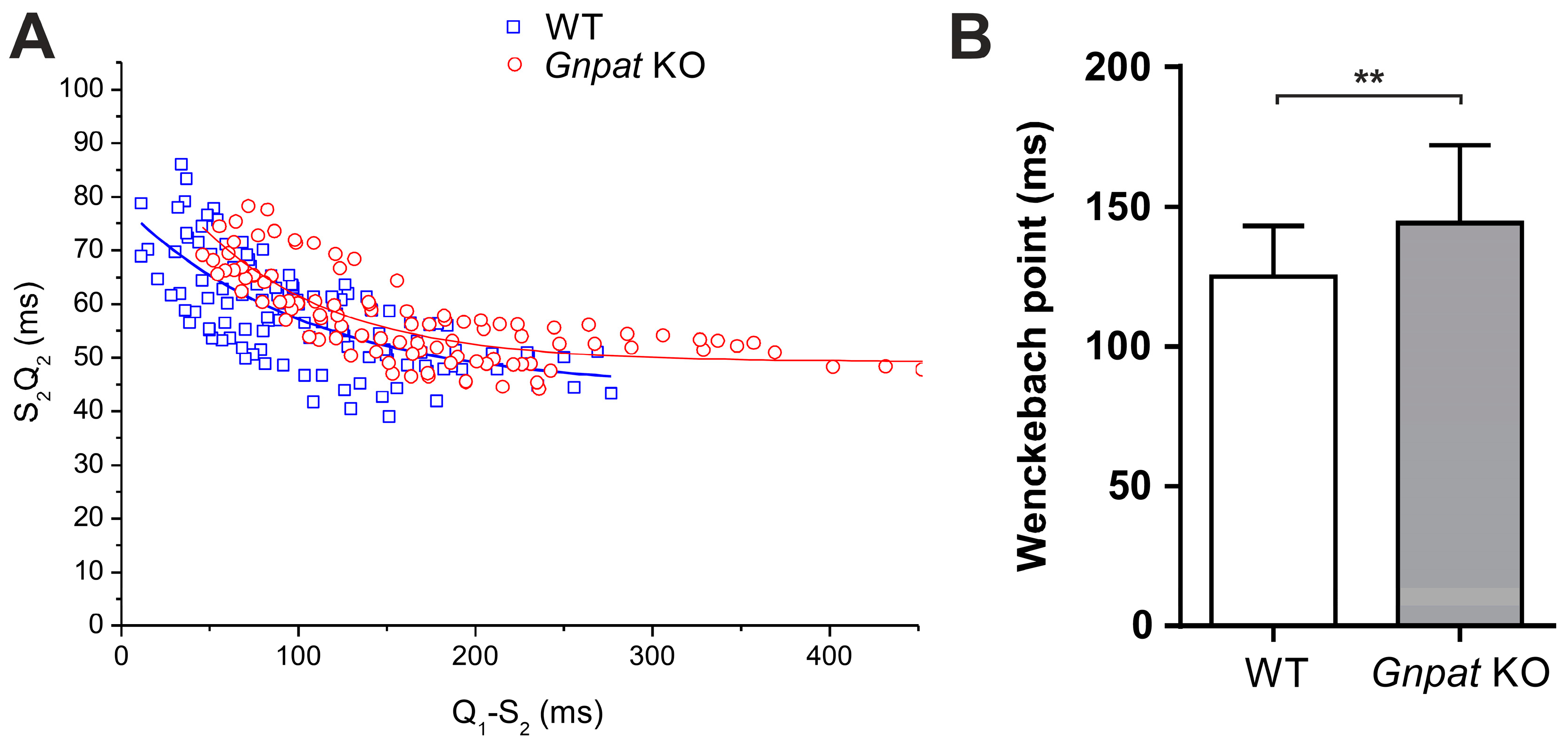
2.5. Ether Lipid Deficiency Leads to Compromised Cardiac Impulse Conduction
2.6. Human RCDP Patient Data Indicate a Complex Cardiac Pathology upon Ether Lipid Deficiency
3. Discussion
4. Material and Methods
4.1. Human Patient Data
4.2. Mice
4.3. Magnetic Resonance Imaging and Post-Processing
4.4. Echocardiography
4.5. Assessment of Left Ventricular Hemodynamic Function In Vivo
4.6. Electrocardiography
4.7. Histology and Immunohistochemistry
4.8. Real-Time Quantitative PCR
4.9. Determination of Monoamines
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [PubMed] [Green Version]
- Braverman, N.; Steel, G.; Obie, C.; Moser, A.; Moser, H.; Gould, S.J.; Valle, D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet. 1997, 15, 369–376. [Google Scholar] [CrossRef]
- Wanders, R.J.; Schumacher, H.; Heikoop, J.; Schutgens, R.B.; Tager, J.M. Human dihydroxyacetonephosphate acyltransferase deficiency: A new peroxisomal disorder. J. Inherit. Metab. Dis. 1992, 15, 389–391. [Google Scholar] [PubMed]
- Wanders, R.J.; Dekker, C.; Hovarth, V.A.; Schutgens, R.B.; Tager, J.M.; Van Laer, P.; Lecoutere, D. Human alkyldihydroxyacetonephosphate synthase deficiency: A new peroxisomal disorder. J. Inherit. Metab. Dis. 1994, 17, 315–318. [Google Scholar] [PubMed]
- Buchert, R.; Tawamie, H.; Smith, C.; Uebe, S.; Innes, A.M.; Al Hallak, B.; Ekici, A.B.; Sticht, H.; Schwarze, B.; Lamont, R.E.; et al. A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency. Am. J. Hum. Genet. 2014, 95, 602–610. [Google Scholar]
- Baroy, T.; Koster, J.; Stromme, P.; Ebberink, M.S.; Misceo, D.; Ferdinandusse, S.; Holmgren, A.; Hughes, T.; Merckoll, E.; Westvik, J.; et al. A novel type of rhizomelic chondrodysplasia punctata, RCDP5, is caused by loss of the PEX5 long isoform. Hum. Mol. Genet. 2015, 24, 5845–5854. [Google Scholar] [CrossRef] [Green Version]
- Dorninger, F.; Werner, E.R.; Berger, J.; Watschinger, K. Regulation of plasmalogen metabolism and traffic in mammals: The fog begins to lift. Front. Cell. Dev. Biol. 2022, 10, 946393. [Google Scholar] [CrossRef]
- Dorninger, F.; Forss-Petter, S.; Wimmer, I.; Berger, J. Plasmalogens, platelet-activating factor and beyond—Ether lipids in signaling and neurodegeneration. Neurobiol. Dis. 2020, 145, 105061. [Google Scholar] [CrossRef]
- Dorninger, F.; Forss-Petter, S.; Berger, J. From peroxisomal disorders to common neurodegenerative diseases—The role of ether phospholipids in the nervous system. FEBS Lett. 2017, 591, 2761–2788. [Google Scholar] [CrossRef] [Green Version]
- Lessig, J.; Fuchs, B. Plasmalogens in biological systems: Their role in oxidative processes in biological membranes, their contribution to pathological processes and aging and plasmalogen analysis. Curr. Med. Chem. 2009, 16, 2021–2041. [Google Scholar]
- Koivuniemi, A. The biophysical properties of plasmalogens originating from their unique molecular architecture. FEBS Lett. 2017, 591, 2700–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagan, N.; Zoeller, R.A. Plasmalogens: Biosynthesis and functions. Prog. Lipid Res. 2001, 40, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kimura, A.K.; Ren, M.; Berno, B.; Xu, Y.; Schlame, M.; Epand, R.M. Substantial Decrease in Plasmalogen in the Heart Associated with Tafazzin Deficiency. Biochemistry 2018, 57, 2162–2175. [Google Scholar] [CrossRef] [Green Version]
- Huffnagel, I.C.; Clur, S.A.; Bams-Mengerink, A.M.; Blom, N.A.; Wanders, R.J.; Waterham, H.R.; Poll-The, B.T. Rhizomelic chondrodysplasia punctata and cardiac pathology. J. Med. Genet. 2013, 50, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Duker, A.L.; Eldridge, G.; Braverman, N.E.; Bober, M.B. Congenital heart defects common in rhizomelic chondrodysplasia punctata. Am. J. Med. Genet. A 2016, 170, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Rodemer, C.; Thai, T.P.; Brugger, B.; Kaercher, T.; Werner, H.; Nave, K.A.; Wieland, F.; Gorgas, K.; Just, W.W. Inactivation of ether lipid biosynthesis causes male infertility, defects in eye development and optic nerve hypoplasia in mice. Hum. Mol. Genet. 2003, 12, 1881–1895. [Google Scholar] [CrossRef] [Green Version]
- Todt, H.; Dorninger, F.; Rothauer, P.J.; Fischer, C.M.; Schranz, M.; Bruegger, B.; Luchtenborg, C.; Ebner, J.; Hilber, K.; Koenig, X.; et al. Oral batyl alcohol supplementation rescues decreased cardiac conduction in ether phospholipid-deficient mice. J. Inherit. Metab. Dis. 2020, 43, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S.; Singal, T. Phospholipid-mediated signaling and heart disease. Subcell. Biochem. 2008, 49, 299–324. [Google Scholar] [PubMed]
- Lim, H.Y.; Wang, W.; Wessells, R.J.; Ocorr, K.; Bodmer, R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev. 2011, 25, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graessler, J.; Schwudke, D.; Schwarz, P.E.; Herzog, R.; Shevchenko, A.; Bornstein, S.R. Top-down lipidomics reveals ether lipid deficiency in blood plasma of hypertensive patients. PLoS ONE 2009, 4, e6261. [Google Scholar] [CrossRef] [Green Version]
- Dorninger, F.; Vaz, F.M.; Waterham, H.R.; Klinken, J.B.V.; Zeitler, G.; Forss-Petter, S.; Berger, J.; Wiesinger, C. Ether lipid transfer across the blood-brain and placental barriers does not improve by inactivation of the most abundant ABC transporters. Brain Res. Bull. 2022, 189, 69–79. [Google Scholar] [CrossRef]
- Savolainen, S.M.; Foley, J.F.; Elmore, S.A. Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol. Pathol. 2009, 37, 395–414. [Google Scholar] [CrossRef] [PubMed]
- Brites, P.; Mooyer, P.A.; El Mrabet, L.; Waterham, H.R.; Wanders, R.J. Plasmalogens participate in very-long-chain fatty acid-induced pathology. Brain 2009, 132, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorninger, F.; Konig, T.; Scholze, P.; Berger, M.L.; Zeitler, G.; Wiesinger, C.; Gundacker, A.; Pollak, D.D.; Huck, S.; Just, W.W.; et al. Disturbed neurotransmitter homeostasis in ether lipid deficiency. Hum. Mol. Genet. 2019, 28, 2046–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa-Araujo, R.; Oliveira, J.S.; Ricciardi Cruz, A. Cardiac levels of norepinephrine, dopamine, serotonin and histamine in Chagas’ disease. Int. J. Cardiol. 1991, 31, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Pyatskowit, J.W.; Prohaska, J.R. Rodent brain and heart catecholamine levels are altered by different models of copper deficiency. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 145, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallatah, W.; Schouten, M.; Yergeau, C.; Di Pietro, E.; Engelen, M.; Waterham, H.R.; Poll-The, B.T.; Braverman, N. Clinical, biochemical, and molecular characterization of mild (nonclassic) rhizomelic chondrodysplasia punctata. J. Inherit. Metab. Dis. 2021, 44, 1021–1038. [Google Scholar] [CrossRef] [PubMed]
- Rudic, B.; Schimpf, R.; Borggrefe, M. Short QT Syndrome—Review of Diagnosis and Treatment. Arrhythm. Electrophysiol. Rev. 2014, 3, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Recent Advances in Short QT Syndrome. Front. Cardiovasc. Med. 2018, 5, 149. [Google Scholar] [CrossRef] [Green Version]
- Dorninger, F.; Brodde, A.; Braverman, N.E.; Moser, A.B.; Just, W.W.; Forss-Petter, S.; Brugger, B.; Berger, J. Homeostasis of phospholipids—The level of phosphatidylethanolamine tightly adapts to changes in ethanolamine plasmalogens. Biochim. Biophys. Acta 2015, 1851, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Noyon, C.; Ruysschaert, J.M.; Van Antwerpen, P.; Govaerts, C. Phosphatidylethanolamine Is a Key Regulator of Membrane Fluidity in Eukaryotic Cells. J. Biol. Chem. 2016, 291, 3658–3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Teigler, A.; Komljenovic, D.; Draguhn, A.; Gorgas, K.; Just, W.W. Defects in myelination, paranode organization and Purkinje cell innervation in the ether lipid-deficient mouse cerebellum. Hum. Mol. Genet. 2009, 18, 1897–1908. [Google Scholar] [CrossRef] [Green Version]
- Lalowski, M.M.; Bjork, S.; Finckenberg, P.; Soliymani, R.; Tarkia, M.; Calza, G.; Blokhina, D.; Tulokas, S.; Kankainen, M.; Lakkisto, P.; et al. Characterizing the Key Metabolic Pathways of the Neonatal Mouse Heart Using a Quantitative Combinatorial Omics Approach. Front. Physiol. 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penny, D.J.; Vick, G.W., III. Ventricular septal defect. Lancet 2011, 377, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Minette, M.S.; Sahn, D.J. Ventricular septal defects. Circulation 2006, 114, 2190–2197. [Google Scholar] [CrossRef] [Green Version]
- Dorninger, F.; Herbst, R.; Kravic, B.; Camurdanoglu, B.Z.; Macinkovic, I.; Zeitler, G.; Forss-Petter, S.; Strack, S.; Khan, M.M.; Waterham, H.R.; et al. Reduced muscle strength in ether lipid-deficient mice is accompanied by altered development and function of the neuromuscular junction. J. Neurochem. 2017, 143, 569–583. [Google Scholar] [CrossRef] [Green Version]
- Phoon, C.K.; Acehan, D.; Schlame, M.; Stokes, D.L.; Edelman-Novemsky, I.; Yu, D.; Xu, Y.; Viswanathan, N.; Ren, M. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J. Am. Heart Assoc. 2012, 1, e000455. [Google Scholar] [CrossRef] [Green Version]
- Tham, Y.K.; Huynh, K.; Mellett, N.A.; Henstridge, D.C.; Kiriazis, H.; Ooi, J.Y.Y.; Matsumoto, A.; Patterson, N.L.; Sadoshima, J.; Meikle, P.J.; et al. Distinct lipidomic profiles in models of physiological and pathological cardiac remodeling, and potential therapeutic strategies. Biochim. Biophys. Acta 2018, 1863, 219–234. [Google Scholar] [CrossRef]
- Foulon, P.; De Backer, D. The hemodynamic effects of norepinephrine: Far more than an increase in blood pressure! Ann. Transl. Med. 2018, 6, S25. [Google Scholar] [CrossRef] [PubMed]
- Monnet, X.; Jabot, J.; Maizel, J.; Richard, C.; Teboul, J.-L. Norepinephrine increases cardiac preload and reduces preload dependency assessed by passive leg raising in septic shock patients*. Crit. Care Med. 2011, 39, 689–694. [Google Scholar] [CrossRef] [PubMed]
- van Rijen, H.V.; Eckardt, D.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; de Bakker, J.M. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Poelzing, S.; Rosenbaum, D.S. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1762–H1770. [Google Scholar] [CrossRef]
- Stein, M.; van Veen, T.A.; Hauer, R.N.; de Bakker, J.M.; van Rijen, H.V. A 50% reduction of excitability but not of intercellular coupling affects conduction velocity restitution and activation delay in the mouse heart. PLoS ONE 2011, 6, e20310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, D.; Figiel, S.; Felix, R.; Kouba, S.; Fromont, G.; Maheo, K.; Potier-Cartereau, M.; Chantome, A.; Vandier, C. Roles of endogenous ether lipids and associated PUFAs in the regulation of ion channels and their relevance for disease. J. Lipid Res. 2020, 61, 840–858. [Google Scholar] [CrossRef] [Green Version]
- Severs, N.J.; Rothery, S.; Dupont, E.; Coppen, S.R.; Yeh, H.I.; Ko, Y.S.; Matsushita, T.; Kaba, R.; Halliday, D. Immunocytochemical analysis of connexin expression in the healthy and diseased cardiovascular system. Microsc. Res. Tech. 2001, 52, 301–322. [Google Scholar] [CrossRef]
- Beauchamp, P.; Yamada, K.A.; Baertschi, A.J.; Green, K.; Kanter, E.M.; Saffitz, J.E.; Kleber, A.G. Relative contributions of connexins 40 and 43 to atrial impulse propagation in synthetic strands of neonatal and fetal murine cardiomyocytes. Circ. Res. 2006, 99, 1216–1224. [Google Scholar] [CrossRef] [Green Version]
- Kolcz, J.; Drukala, J.; Bzowska, M.; Rajwa, B.; Korohoda, W.; Malec, E. The expression of connexin 43 in children with Tetralogy of Fallot. Cell. Mol. Biol. Lett. 2005, 10, 287–303. [Google Scholar]
- Fontes, M.S.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys. Acta 2012, 1818, 2020–2029. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, S.K.; Johnson, W.W. Cardiac conduction abnormalities in children with Duchenne’s progressive muscular dystrophy: Electrocardiographic features and morphologic correlates. Circulation 1982, 66, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Bia, B.L.; Cassidy, P.J.; Young, M.E.; Rafael, J.A.; Leighton, B.; Davies, K.E.; Radda, G.K.; Clarke, K. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 1999, 31, 1857–1862. [Google Scholar] [CrossRef]
- Higham, P.D.; Campbell, R.W. QT dispersion. Br. Heart J. 1994, 71, 508–510. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou-Nana, M.I.; Nanas, J.N.; Karagounis, L.A.; Tsagalou, E.P.; Alexopoulos, G.E.; Toumanidis, S.; Gerali, S.; Stamatelopoulos, S.F.; Moulopoulos, S.D. Relation of dispersion of QRS and QT in patients with advanced congestive heart failure to cardiac and sudden death mortality. Am. J. Cardiol. 2000, 85, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Kearney, M.T.; Fox, K.A.; Lee, A.J.; Brooksby, W.P.; Shah, A.M.; Flapan, A.; Prescott, R.J.; Andrews, R.; Batin, P.D.; Eckberg, D.L.; et al. Predicting sudden death in patients with mild to moderate chronic heart failure. Heart 2004, 90, 1137–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Harding, S.A.; Jimenez, A.; Larsen, P.D. Standard 12-lead electrocardiography measures predictive of increased appropriate therapy in implantable cardioverter defibrillator recipients. Europace 2013, 15, 892–898. [Google Scholar] [CrossRef]
- Yotsukura, M.; Yamamoto, A.; Kajiwara, T.; Nishimura, T.; Sakata, K.; Ishihara, T.; Ishikawa, K. QT dispersion in patients with Duchenne-type progressive muscular dystrophy. Am. Heart J. 1999, 137, 672–677. [Google Scholar] [CrossRef]
- Baker, L.C.; London, B.; Choi, B.R.; Koren, G.; Salama, G. Enhanced dispersion of repolarization and refractoriness in transgenic mouse hearts promotes reentrant ventricular tachycardia. Circ. Res. 2000, 86, 396–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.A.; Zhang, Y.; Grace, A.A.; Huang, C.L. In vivo studies of Scn5a+/- mice modeling Brugada syndrome demonstrate both conduction and repolarization abnormalities. J. Electrocardiol. 2010, 43, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Saba, S.; London, B.; Ganz, L. Autonomic blockade unmasks maturational differences in rate-dependent atrioventricular nodal conduction and facilitation in the mouse. J. Cardiovasc. Electrophysiol. 2003, 14, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar]
- Villafane, J.; Atallah, J.; Gollob, M.H.; Maury, P.; Wolpert, C.; Gebauer, R.; Watanabe, H.; Horie, M.; Anttonen, O.; Kannankeril, P.; et al. Long-term follow-up of a pediatric cohort with short QT syndrome. J. Am. Coll. Cardiol. 2013, 61, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Anttonen, O.; Junttila, M.J.; Rissanen, H.; Reunanen, A.; Viitasalo, M.; Huikuri, H.V. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007, 116, 714–720. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.R.; Beeton, C. Differences in ion channel phenotype and function between humans and animal models. Front. Biosci. 2018, 23, 43–64. [Google Scholar]
- Heiberg, E.; Sjogren, J.; Ugander, M.; Carlsson, M.; Engblom, H.; Arheden, H. Design and validation of Segment—Freely available software for cardiovascular image analysis. BMC Med. Imaging 2010, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Perera-Gonzalez, M.; Kiss, A.; Kaiser, P.; Holzweber, M.; Nagel, F.; Watzinger, S.; Acar, E.; Szabo, P.L.; Goncalves, I.F.; Weber, L.; et al. The Role of Tenascin C in Cardiac Reverse Remodeling Following Banding-Debanding of the Ascending Aorta. Int. J. Mol. Sci. 2021, 22, 2023. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2018, 129, 531–545. [Google Scholar] [CrossRef] [Green Version]
- London, B. Cardiac arrhythmias: From (transgenic) mice to men. J. Cardiovasc. Electrophysiol. 2001, 12, 1089–1091. [Google Scholar] [CrossRef]
- Danik, S.; Cabo, C.; Chiello, C.; Kang, S.; Wit, A.L.; Coromilas, J. Correlation of repolarization of ventricular monophasic action potential with ECG in the murine heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H372–H381. [Google Scholar] [CrossRef] [Green Version]
- Boukens, B.J.; Hoogendijk, M.G.; Verkerk, A.O.; Linnenbank, A.; van Dam, P.; Remme, C.A.; Fiolet, J.W.; Opthof, T.; Christoffels, V.M.; Coronel, R. Early repolarization in mice causes overestimation of ventricular activation time by the QRS duration. Cardiovasc. Res. 2013, 97, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Iden, J.B.; Kovithavongs, K.; Gulamhusein, R.; Duff, H.J.; Kavanagh, K.M. In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J. Physiol. 2004, 555, 267–279. [Google Scholar] [CrossRef]
- Wung, S.-F. Twelve-Lead Electrocardiogram. In AACN Procedure Manual for High Acuity, Progressive, and Critical Care; Wiegand, D.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 494–500. [Google Scholar]
- Postema, P.G.; Wilde, A.A. The measurement of the QT interval. Curr. Cardiol. Rev. 2014, 10, 287–294. [Google Scholar] [CrossRef]
- Bauer, J.; Lassmann, H. Neuropathological Techniques to Investigate Central Nervous System Sections in Multiple Sclerosis. Methods Mol. Biol. 2016, 1304, 211–229. [Google Scholar]
- Hortnagl, H.; Berger, M.L.; Sperk, G.; Pifl, C. Regional heterogeneity in the distribution of neurotransmitter markers in the rat hippocampus. Neuroscience 1991, 45, 261–272. [Google Scholar] [CrossRef]
- Pifl, C.; Schingnitz, G.; Hornykiewicz, O. Effect of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on the regional distribution of brain monoamines in the rhesus monkey. Neuroscience 1991, 44, 591–605. [Google Scholar] [CrossRef]
- Peneder, T.M.; Scholze, P.; Berger, M.L.; Reither, H.; Heinze, G.; Bertl, J.; Bauer, J.; Richfield, E.K.; Hornykiewicz, O.; Pifl, C. Chronic exposure to manganese decreases striatal dopamine turnover in human alpha-synuclein transgenic mice. Neuroscience 2011, 180, 280–292. [Google Scholar] [CrossRef]
- Horrocks, L.A. Content, composition, and metabolism of mammalian and avian lipids that contain ether groups. In Ether Lipids: Chemistry and Biology; Snyder, F., Ed.; Academic Press: New York, NY, USA, 1972; pp. 177–272. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Change in Gnpat KO Mice |
|---|---|
| Heart weight/body weight | ↑ |
| MRI | |
| LV end-diastolic volume | ↓ |
| LV end-systolic volume | = |
| RV end-diastolic volume | = |
| LV mass | ↓ |
| LV end-diastolic diameter | ↓ |
| LV end-systolic diameter | = |
| Ejection fraction | = |
| Stroke volume | ↓ |
| Wall thickness (systolic and diastolic) | = |
| Wall stress (diastolic) | ↑ |
| Wall stress (systolic) | = |
| In vivo hemodynamic examination | |
| LV systolic pressure | ↑ |
| LV end-diastolic pressure | ↑ |
| Echocardiography | |
| Ejection fraction | = |
| Fractional shortening | = |
| Stroke volume | ↓ |
| Wall thickness (systolic and diastolic) | = |
| ECG | |
| P wave duration | ↑ |
| QRS length | ↑ |
| R/S ratio | ↓ |
| Dispersion | ↑ |
| Wenckebach point | ↑ |
| WT | Gnpat KO | |
|---|---|---|
| R/S ratio | 10.61 ± 14.33 (n = 45) | 4.96 ± 6.11 * (n = 40) |
| Dispersion (QRS) | 3.17 ± 1.38 ms (n = 28) | 4.88 ± 2.33 ms ** (n = 25) |
| Dispersion (QT1) | 4.68 ± 2.04 ms (n = 36) | 6.89 ± 4.26 ms * (n = 27) |
| Reference | Fraction of Patients with Cardiac Pathology/Total Patients | Main Pathological Findings |
|---|---|---|
| [14] | 13/18 | ASD, MVP, PDA, PS, ToF |
| [15] | 9/14 | ASD, MVP, ToF, VSD, ARD |
| [27] | 5/11 | ASD, MVP, PDA, PS |
| This study | 1/2 * | ASD |
| RCDP Patient (Age in yrs) | QTc Interval (s) | Sinus Rhythm | Comment |
|---|---|---|---|
| 1 (12) | 0.337 * | Marked sinus arrhythmia | ST elevation (early repolarization normal variant) |
| 2 (7) | 0.400 | Normal sinus rhythm | Normal ECG |
| 3 (3) | 0.315 * | Normal sinus rhythm | +T in V1 (RVH with pressure overload) |
| 4 (3) | 0.331 * | Normal sinus rhythm | RSr’ in V1 (mild intraventricular conduction delay—normal variant) |
| 5 (3) | 0.395 | Normal sinus rhythm | rSR’ in V1 (RVH with volume overload 1 or mild conduction delay 2) |
| 6 (31) | 0.392 | Normal sinus rhythm | Deep S in V5-V6 with no other signs of RVH (likely artifact of chest abnormality) |
| 7 (21) | 0.395 | Normal sinus rhythm | rSR’ in V1 (RVH with volume overload 1 or mild conduction delay 2, borderline tall p wave in lead II—possible RA enlargement) |
| 8 (5) | 0.371 | Sinus arrhythmia | Normal ECG |
| 9 (10) | 0.381 | Normal sinus rhythm | Normal ECG |
| 10 (3) | 0.388 | Normal sinus rhythm | rSR’ in V1 (RVH with volume overload or mild conduction delay) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorninger, F.; Kiss, A.; Rothauer, P.; Stiglbauer-Tscholakoff, A.; Kummer, S.; Fallatah, W.; Perera-Gonzalez, M.; Hamza, O.; König, T.; Bober, M.B.; et al. Overlapping and Distinct Features of Cardiac Pathology in Inherited Human and Murine Ether Lipid Deficiency. Int. J. Mol. Sci. 2023, 24, 1884. https://doi.org/10.3390/ijms24031884
Dorninger F, Kiss A, Rothauer P, Stiglbauer-Tscholakoff A, Kummer S, Fallatah W, Perera-Gonzalez M, Hamza O, König T, Bober MB, et al. Overlapping and Distinct Features of Cardiac Pathology in Inherited Human and Murine Ether Lipid Deficiency. International Journal of Molecular Sciences. 2023; 24(3):1884. https://doi.org/10.3390/ijms24031884
Chicago/Turabian StyleDorninger, Fabian, Attila Kiss, Peter Rothauer, Alexander Stiglbauer-Tscholakoff, Stefan Kummer, Wedad Fallatah, Mireia Perera-Gonzalez, Ouafa Hamza, Theresa König, Michael B. Bober, and et al. 2023. "Overlapping and Distinct Features of Cardiac Pathology in Inherited Human and Murine Ether Lipid Deficiency" International Journal of Molecular Sciences 24, no. 3: 1884. https://doi.org/10.3390/ijms24031884