
Integrative Analysis of Metabolome and Transcriptome Reveals the Mechanism of Color Formation in Yellow-Fleshed Kiwifruit
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
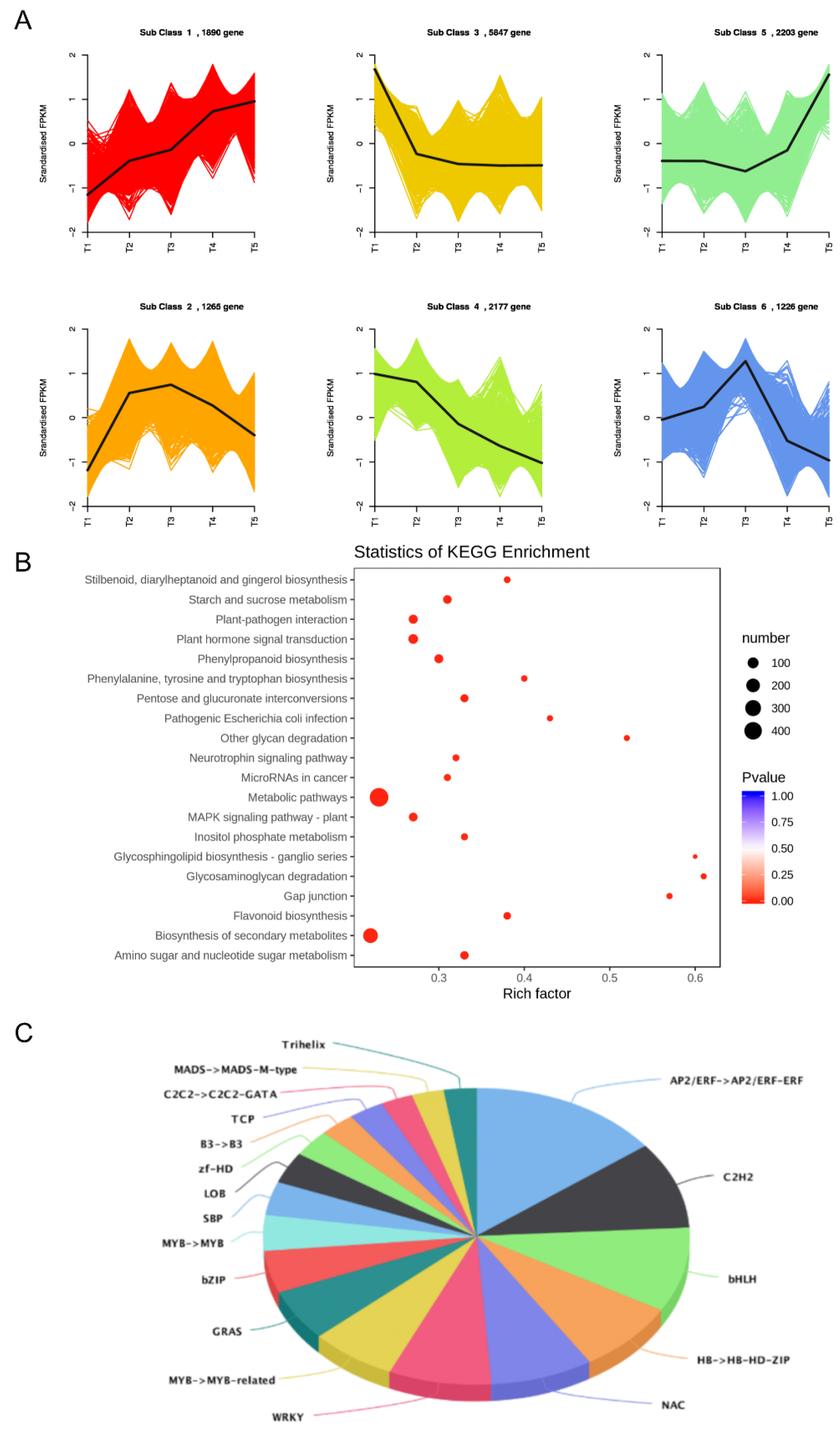
2.1. Transcriptome Profiling
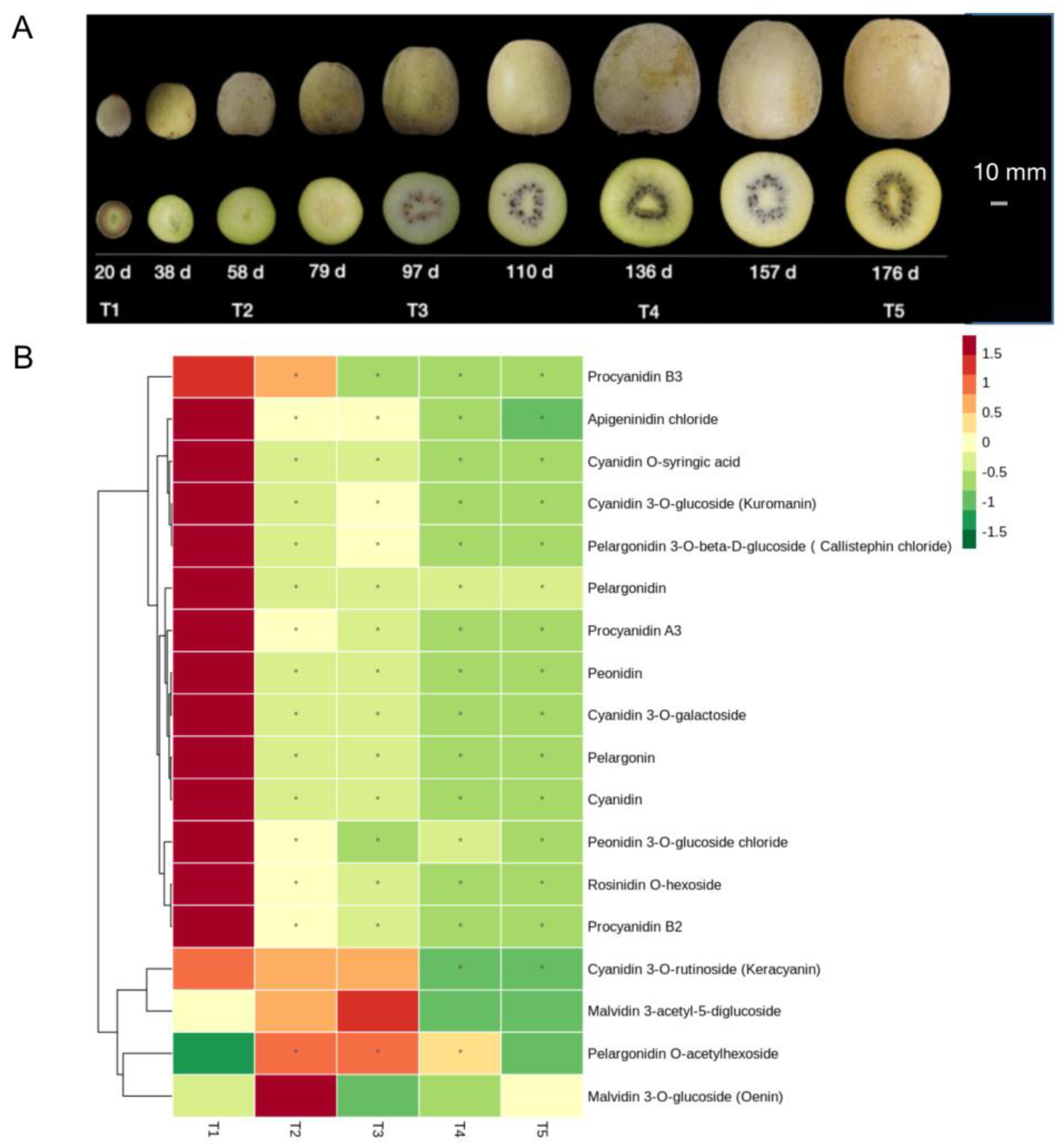
2.2. Identification of Flavonoids in the Fruits of Five Development Stages
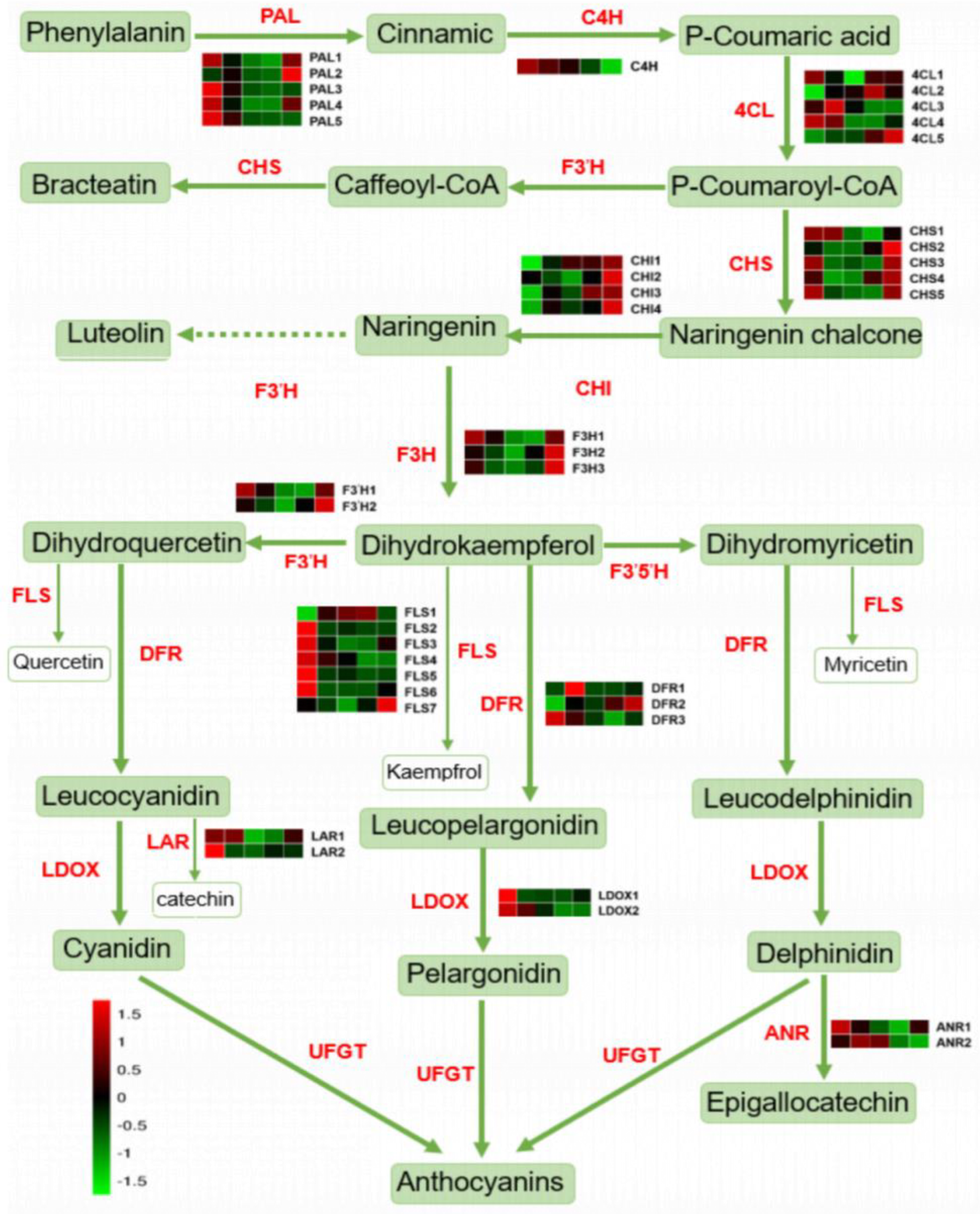
2.3. Expression of Flavonoid Synthesis-Related Genes in Five Development Stages
2.4. Candidate Transcription Factor Genes Related to Flavonoid Synthesis
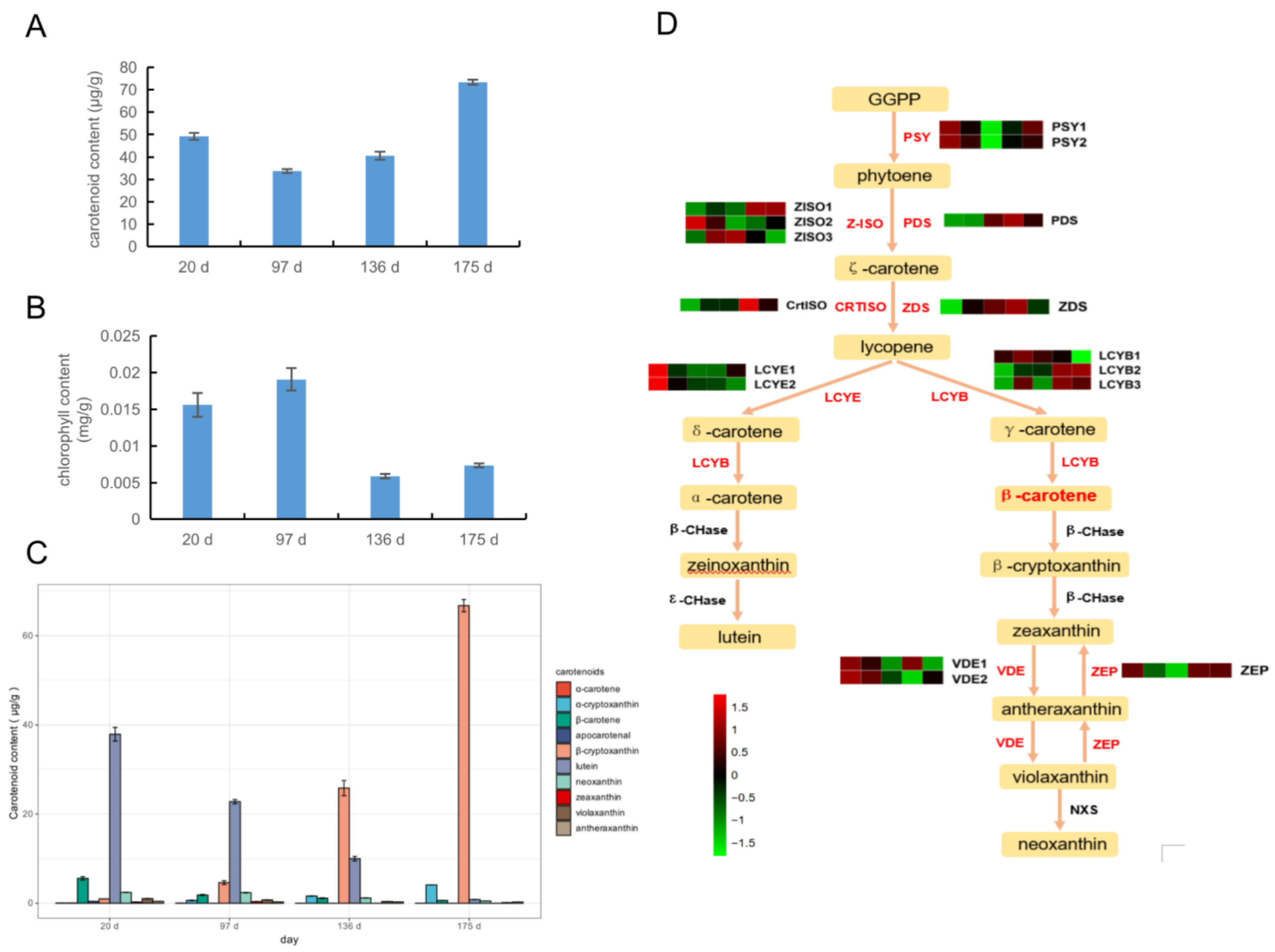
2.5. Expression of Carotenoid Synthesis Genes in Four Development Stages
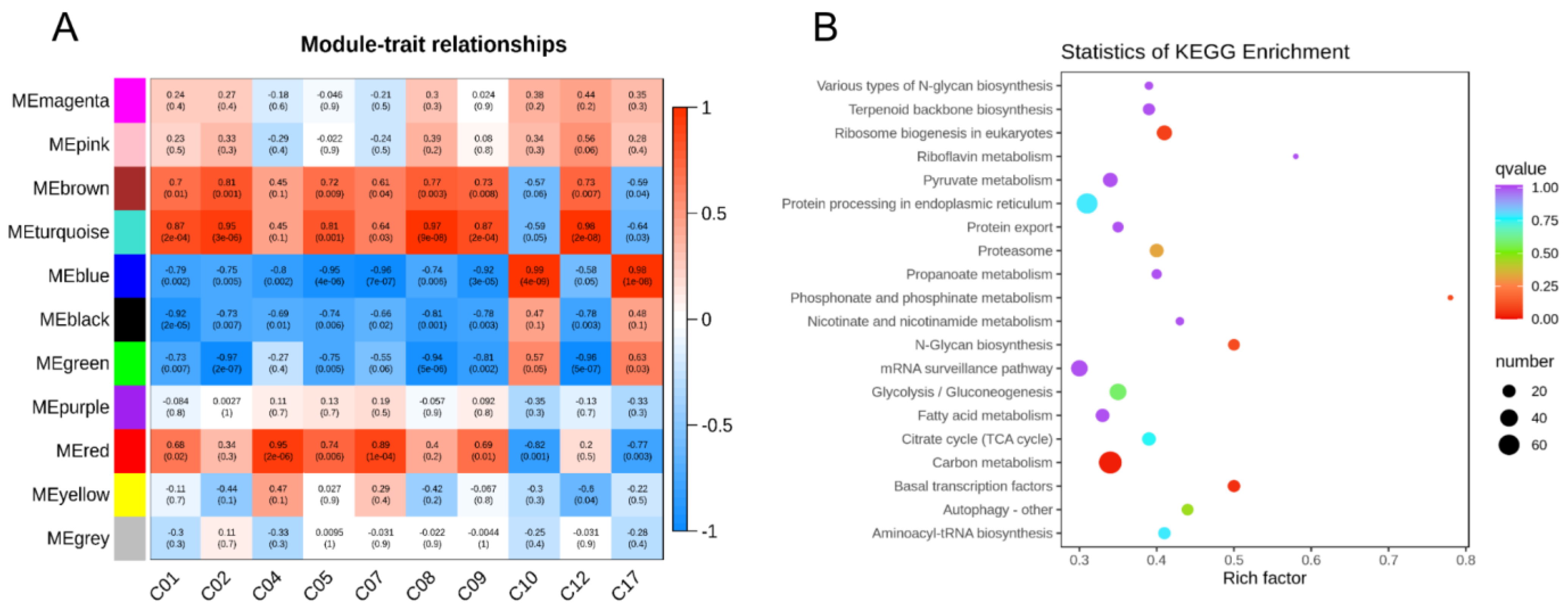
2.6. Co-Expression Network Analysis Identified Carotenoid-Related DEGs
3. Discussion
3.1. Effects of Anthocyanins and Flavonoids on Flesh Color at the Young Fruit Stage
3.2. Effects of Carotenoids and Chlorophyll on Flesh Color at Ripening
4. Materials and Methods
4.1. Plant Material and Sample Preparation
4.2. Metabolite Profiling Analysis, and Representative Flavonoid Compound Measurement
4.3. Carotenoid Identification and Quantification
4.4. RNA Isolation and Transcriptome Sequencing
4.5. Statistical Analysis
4.6. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Zhang, J.; Nageswaran, D.; Li, L. Carotenoid metabolism and regulation in horticultural crops. Hortic. Res. 2015, 2, 15036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, J.T.; Tieman, D.M.; Sims, C.A.; Odabasi, A.Z.; Clark, D.G.; Klee, H.J. Carotenoid content impacts flavor acceptability in tomato (Solanum lycopersicum). J. Sci. Food Agric. 2010, 90, 2233–2240. [Google Scholar] [CrossRef]
- He, B.; Hu, Z.; Ma, L.; Li, H.; Ai, M.; Han, J.; Zeng, B. Transcriptome analysis of different growth stages of Aspergillus oryzae reveals dynamic changes of distinct classes of genes during growth. BMC Microbiol. 2018, 18, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhou, B.; Qi, Y.; Chen, X.; Liu, C.; Liu, Z.; Ren, X. Expression Differences of Pigment Structural Genes and Transcription Factors Explain Flesh Coloration in Three Contrasting Kiwifruit Cultivars. Front. Plant Sci. 2017, 8, 1507. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.P.; Wang, Y.C.; Li, Z.Z.; Jiang, Z.W.; Yang, H.L.; Gong, J.J.; He, S.S.; Wu, S.Q.; Yang, Z.Q.; Zheng, J.; et al. High-temperature inhibition of biosynthesis and transportation of anthocyanins results in the poor red coloration in red-fleshed Actinidia chinensis. Physiol. Plant. 2015, 153, 565–583. [Google Scholar] [CrossRef]
- Montefiori, M.; McGhie, T.K.; Costa, G.; Ferguson, A.R. Pigments in the fruit of red-fleshed kiwifruit (Actinidia chinensis and Actinidia deliciosa). J. Agric. Food Chem. 2005, 53, 9526–9530. [Google Scholar] [CrossRef]
- Comeskey, D.J.; Montefiori, M.; Edwards, P.J.; McGhie, T.K. Isolation and structural identification of the anthocyanin components of red kiwifruit. J. Agric. Food Chem. 2009, 57, 2035–2039. [Google Scholar] [CrossRef]
- Morita, Y.; Hoshino, A. Recent advances in flower color variation and patterning of Japanese morning glory and petunia. Breed Sci. 2018, 68, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.; Brockman, A.; Aguirre, L.; Campbell, A.; Bean, A.; Cantero, A.; Gonzalez, A. Advances in the MYB-bHLH-WD Repeat (MBW) Pigment Regulatory Model: Addition of a WRKY Factor and Co-option of an Anthocyanin MYB for Betalain Regulation. Plant Cell Physiol. 2017, 58, 1431–1441. [Google Scholar] [CrossRef]
- Montefiori, M.; McGhie, T.K.; Hallett, I.C.; Costa, G. Changes in pigments and plastid ultrastructure during ripening of green-fleshed and yellow-fleshed kiwifruit. Sci. Hortic. 2009, 119, 377–387. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Montefiori, M.; Jameson, P.E.; Allan, A.C. The control of chlorophyll levels in maturing kiwifruit. Planta 2012, 236, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Ampomah-Dwamena, C.; McGhie, T.; Wibisono, R.; Montefiori, M.; Hellens, R.P.; Allan, A.C. The kiwifruit lycopene beta-cyclase plays a significant role in carotenoid accumulation in fruit. J. Exp. Bot. 2009, 60, 3765–3779. [Google Scholar] [CrossRef] [Green Version]
- Beck, G.; Coman, D.; Herren, E.; Ruiz-Sola, M.A.; Rodríguez-Concepción, M.; Gruissem, W.V.; Ranová, E. Characterization of the GGPP synthase gene family in Arabidopsis thaliana. Plant Mol. Biol. 2013, 82, 393–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, L.E.; Strickler, S.R.; Mueller, L.A.; Mazourek, M. An acorn squash (Cucurbita pepo ssp. ovifera) fruit and seed transcriptome as a resource for the study of fruit traits in Cucurbita. Hortic. Res. 2015, 2, 14070. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Yan, P.; Du, K.; Li, M.; Xie, Y.; Gao, P. Nutritional component analyses of kiwifruit in different development stages by metabolomic and transcriptomic approaches. J. Sci. Food Agric. 2020, 100, 2399–2409. [Google Scholar] [CrossRef]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [Green Version]
- Montefiori, M.; Comeskey, D.J.; Wohlers, M.; McGhie, T.K. Characterization and quantification of anthocyanins in red kiwifruit (Actinidia spp.). J. Agric. Food Chem. 2009, 57, 6856–6861. [Google Scholar] [CrossRef]
- Katsumoto, Y.; Fukuchi-Mizutani, M.; Fukui, Y.; Brugliera, F.; Holton, T.A.; Karan, M.; Nakamura, N.; Yonekura-Sakakibara, K.; Togami, J.; Pigeaire, A.; et al. Engineering of the rose flavonoid biosynthetic pathway successfully generated blue-hued flowers accumulating delphinidin. Plant Cell Physiol. 2007, 48, 1589–1600. [Google Scholar] [CrossRef]
- Sasaki, N.; Matsuba, Y.; Abe, Y.; Okamura, M.; Momose, M.; Umemoto, N.; Nakayama, M.; Itoh, Y.; Ozeki, Y. Recent advances in understanding the anthocyanin modification steps in carnation flowers. Sci. Hortic. 2013, 163, 37–45. [Google Scholar] [CrossRef]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef] [Green Version]
- Wang, X. Structure, function, and engineering of enzymes in isoflavonoid biosynthesis. Funct. Integr. Genom. 2011, 11, 13–22. [Google Scholar] [CrossRef]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef]
- Falcone Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef] [Green Version]
- Saengnil, K.; Lueangprasert, K.; Uthaibutra, J. Sunlight-stimulated phenylalanine ammonia-lyase (PAL) activity and anthocyanin accumulation in exocarp of ‘Mahajanaka’ mango. Maejo Int. J. Sci. Technol. 2011, 5, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Moreau, C.; Ambrose, M.J.; Turner, L.; Hill, L.; Ellis, T.H.; Hofer, J.M. The B gene of pea encodes a defective flavonoid 3’,5’-hydroxylase, and confers pink flower color. Plant Physiol. 2012, 159, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Brugliera, F. Flower colour and cytochromes P450. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120432. [Google Scholar] [CrossRef] [Green Version]
- Crowhurst, R.; Liu, Y.; Scaglione, D. The Kiwifruit Genome; Testolin, R., Huang, H.-W., Ferguson, A.R., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 8, pp. 101–114. [Google Scholar] [CrossRef]
- Tian, J.; Han, Z.Y.; Zhang, J.; Hu, Y.; Song, T.; Yao, Y. The Balance of Expression of Dihydroflavonol 4-reductase and Flavonol Synthase Regulates Flavonoid Biosynthesis and Red Foliage Coloration in Crabapples. Sci. Rep. 2015, 5, 12228. [Google Scholar] [CrossRef] [Green Version]
- Telias, A.; Lin-Wang, K.; Stevenson, D.E.; Cooney, J.M.; Hellens, R.P.; Allan, A.C.; Hoover, E.E.; Bradeen, J.M. Apple skin patterning is associated with differential expression of MYB10. BMC Plant Biol. 2011, 11, 93. [Google Scholar] [CrossRef]
- Ruiz-Sola, M.; Rodríguez-Concepción, M. Carotenoid biosynthesis in Arabidopsis: A colorful pathway. Arab. Book 2012, 10, e0158. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.Y.; Vrebalov, J.; Alba, R.; Lee, J.; McQuinn, R.; Chung, J.D.; Klein, P.; Giovannoni, J. A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J. 2010, 64, 936–947. [Google Scholar] [CrossRef]
- Dong, X.; Chen, W.; Wang, W.; Zhang, H.; Liu, X.; Luo, J. Comprehensive profiling and natural variation of flavonoids in rice. J. Integr. Plant Biol. 2014, 56, 876–886. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [Green Version]
- Pujana, M.A.; Han, J.D.; Starita, L.M.; Stevens, K.N.; Tewari, M.; Ahn, J.S.; Rennert, G.; Moreno, V.; Kirchhoff, T.; Gold, B.; et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 2007, 39, 1338–1349. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Y.; He, J.; Li, M.; Du, K.; Lang, H.; Gao, P.; Xie, Y. Integrative Analysis of Metabolome and Transcriptome Reveals the Mechanism of Color Formation in Yellow-Fleshed Kiwifruit. Int. J. Mol. Sci. 2023, 24, 1573. https://doi.org/10.3390/ijms24021573
Xiong Y, He J, Li M, Du K, Lang H, Gao P, Xie Y. Integrative Analysis of Metabolome and Transcriptome Reveals the Mechanism of Color Formation in Yellow-Fleshed Kiwifruit. International Journal of Molecular Sciences. 2023; 24(2):1573. https://doi.org/10.3390/ijms24021573
Chicago/Turabian StyleXiong, Yun, Junya He, Mingzhang Li, Kui Du, Hangyu Lang, Ping Gao, and Yue Xie. 2023. "Integrative Analysis of Metabolome and Transcriptome Reveals the Mechanism of Color Formation in Yellow-Fleshed Kiwifruit" International Journal of Molecular Sciences 24, no. 2: 1573. https://doi.org/10.3390/ijms24021573