
Histological Methods to Assess Skeletal Muscle Degeneration and Regeneration in Duchenne Muscular Dystrophy

 , , ,
, , ,
Abstract
:
1. Introduction
2. Animal Models
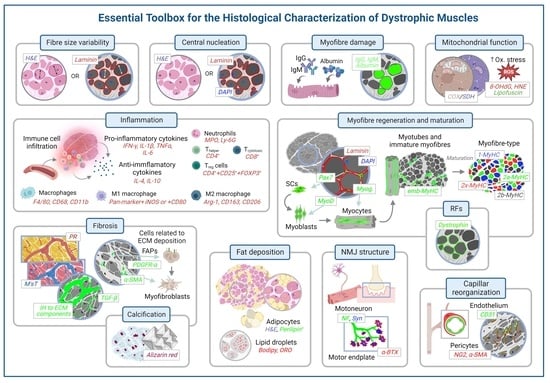
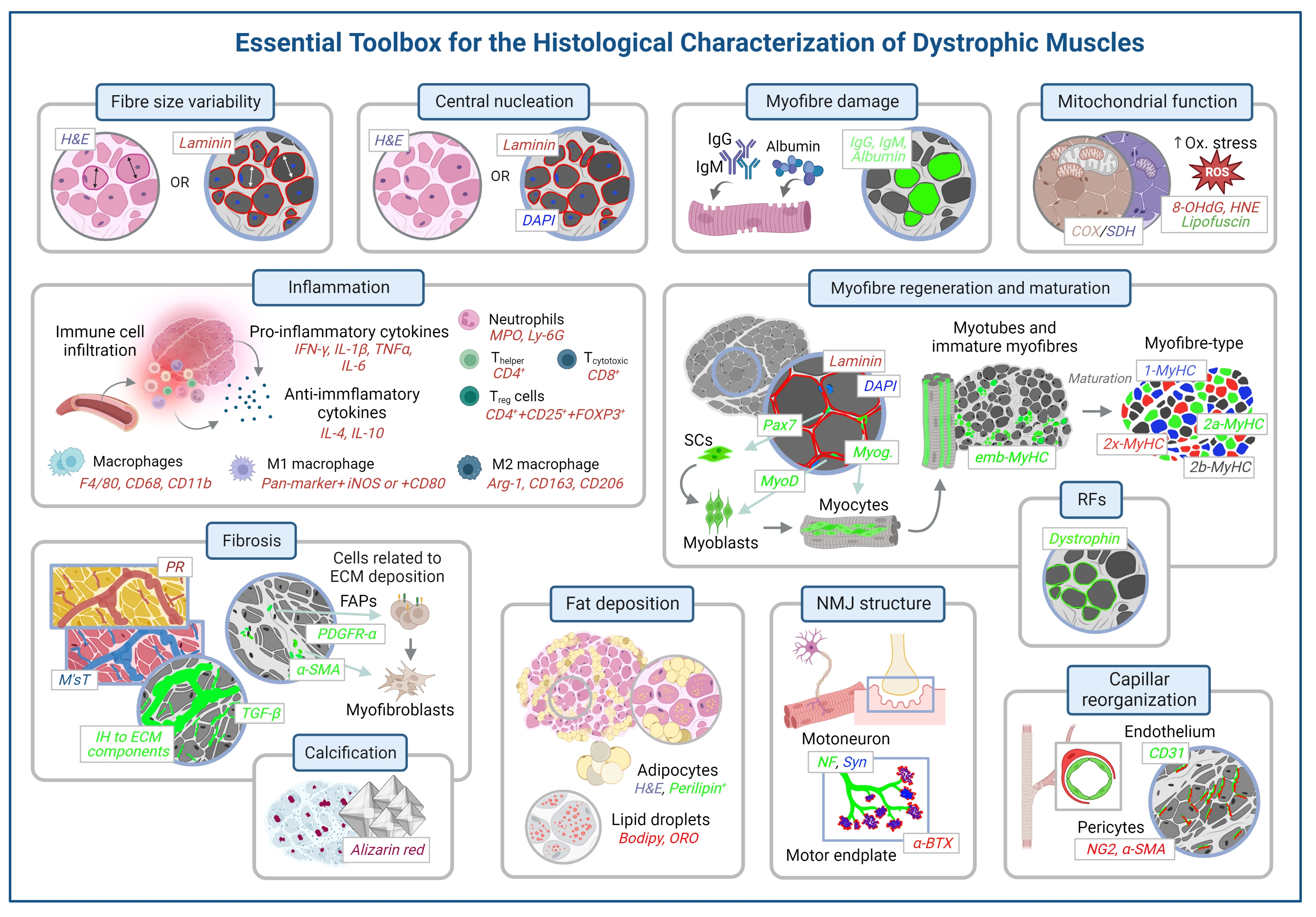
3. Histological Methods to Assess Degeneration/Regeneration in Dystrophic Muscles
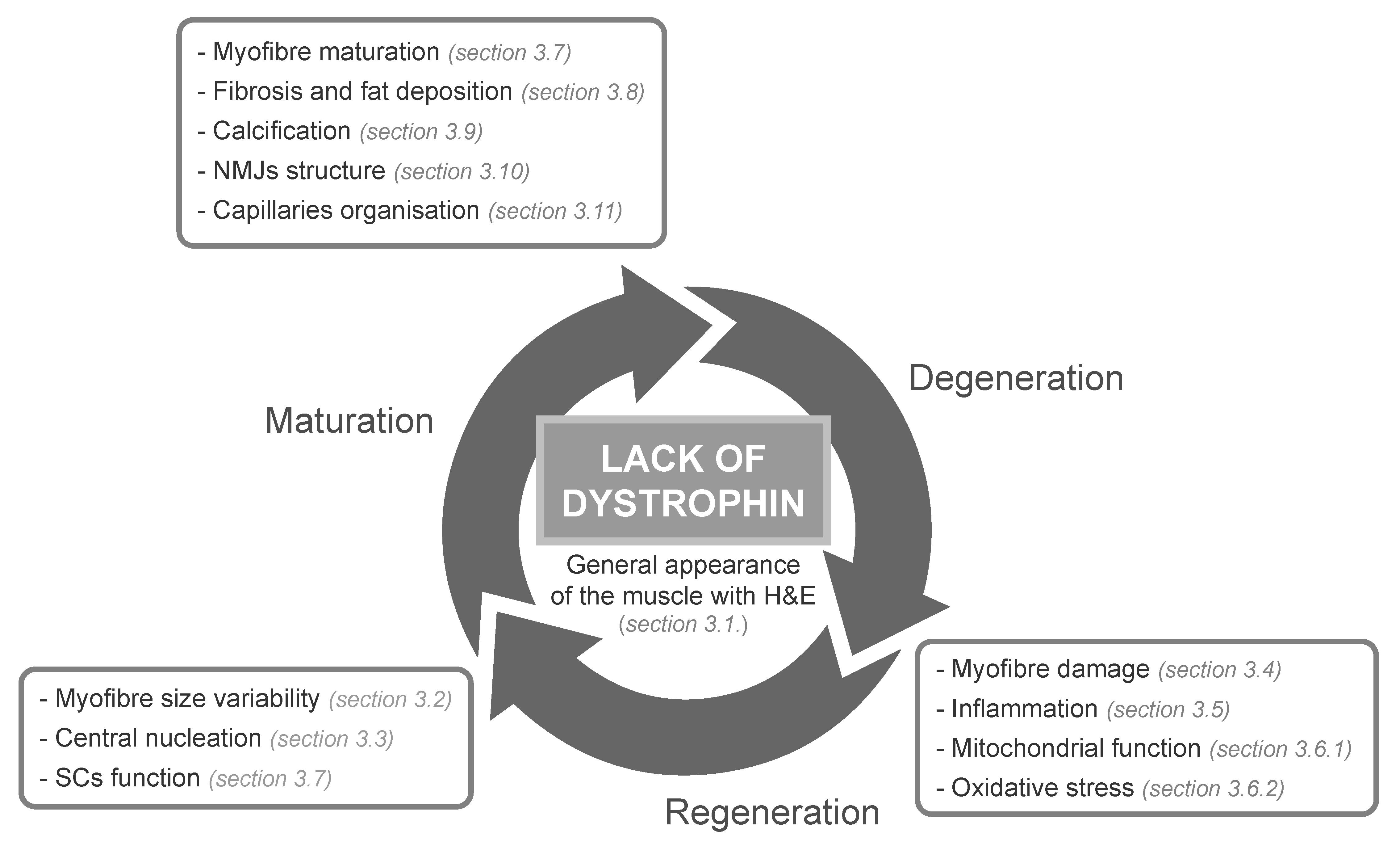
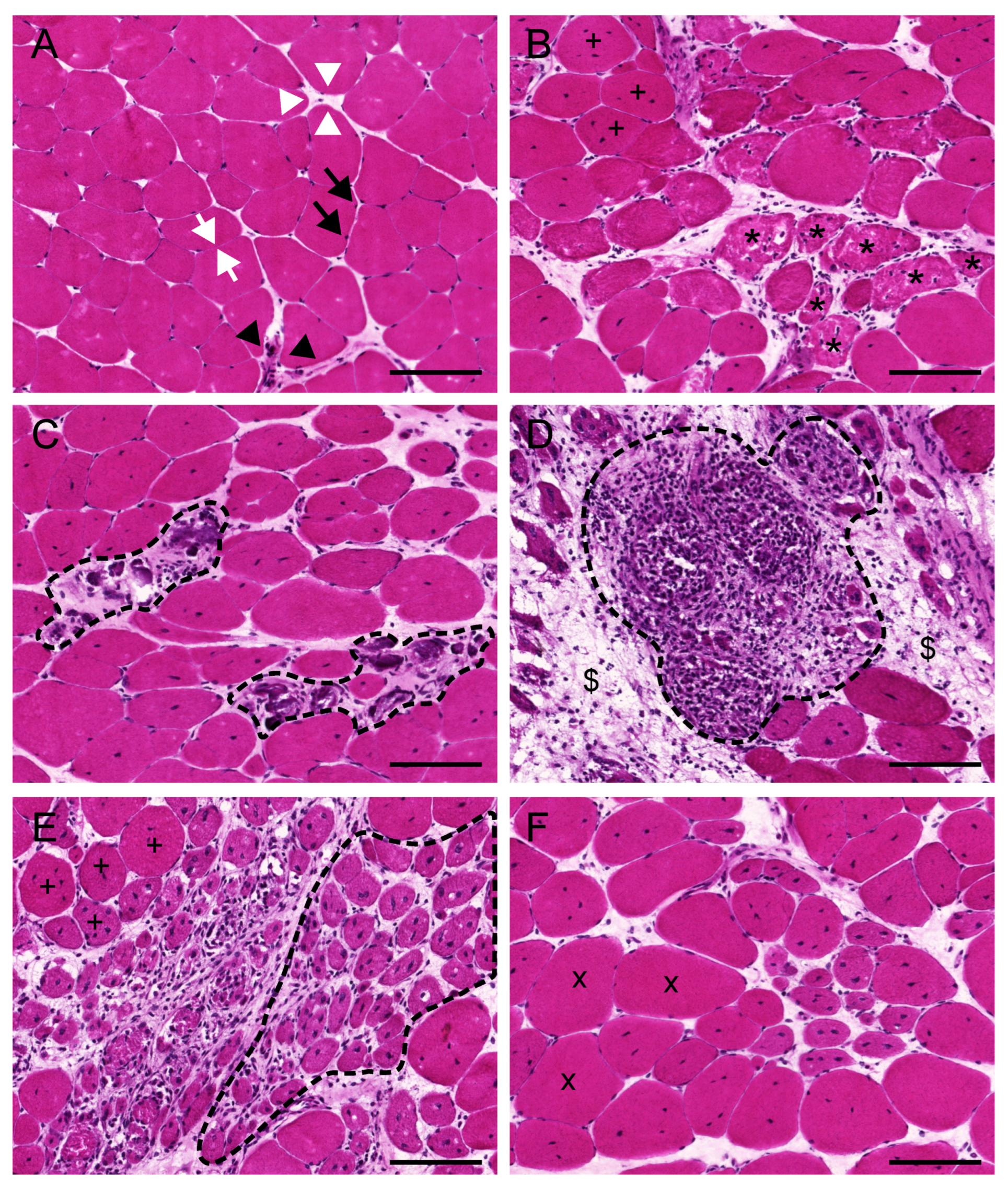
3.1. Evaluation of the General Appearance of the Muscle with Haematoxylin and Eosin
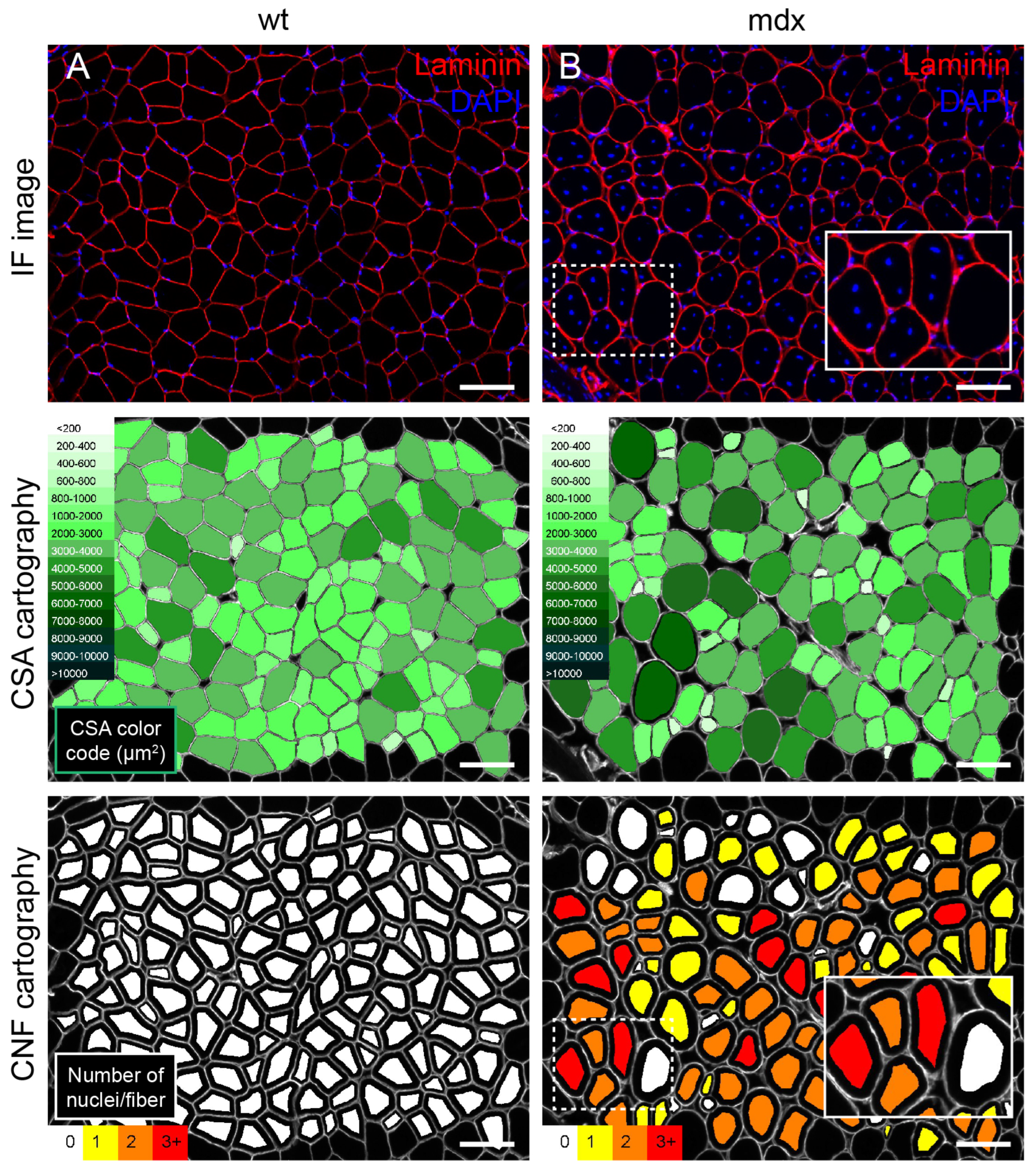
3.2. Evaluation of the Morphometric Features of the Myofibre

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Myofibre/ ECM Location | Technique | Subunits or Types | Morphometry Measurements on DMD | Reference for Full Protocol |
|---|---|---|---|---|---|
| Laminin-211 | BL | IHC, IF | α, β, γ | + | [55] |
| Spectrin | SM | IHC, IF | - | + | [52] |
| Perlecan | BL | IHC, IF | - | + | [56,57] |
| Dystroglycans | SM/BL | IHC, IF | α, β | - | [58] |
| Sarcoglycans | SM | IHC, IF | α, β, δ, γ | - | [58] |
| Dystrophin | SM | IHC, IF | - | - | [52,58] |
| Collagens | ECM | IHC, IF | I, IV, VI | - | [59,60] |
| Decorin | ECM | IF | - | - | [60] |
| Biglycan | ECM | IF | - | - | [60] |
| WGA | SM | HC | - | + | [54,61] |
3.3. Evaluation of Centrally Nucleated Fibres

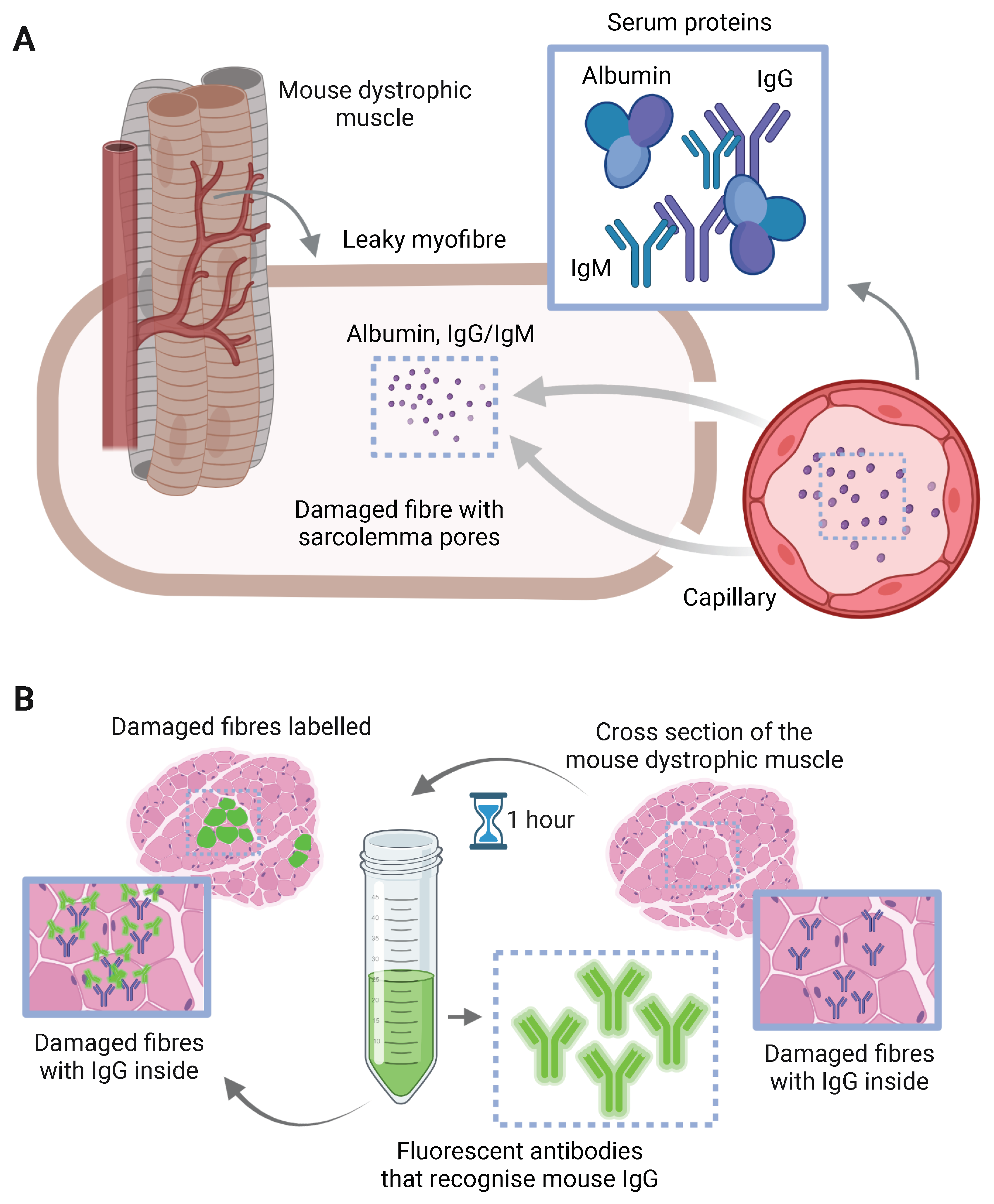
3.4. Myofibre Damage and Cell Death

3.5. Inflammation
3.5.1. Immune System Cell Infiltration
- Neutrophils:
- Macrophages:
- Lymphocytes:
3.5.2. Cytokines
- Proinflammatory Cytokines:
| Cell/Cytokine | Type | Antigen/Marker | IH Method | Tissue | Animal | References for Protocol |
|---|---|---|---|---|---|---|
| Macrophages | Pan-marker | CD11b | IHC/IF | Skeletal muscle | mdx mice | [107,108] |
| CD11c | IHC | [107] | ||||
| CD68 | IHC/IF | [109] | ||||
| F4/80 | IHC/IF | [110] | ||||
| M1 | CCR2 | IHC/IF | [109,111] | |||
| CCR7 | IHC | CCL19KL mice | [112] | |||
| IF | Rat | [113] | ||||
| CD80 | IHC | Rat | [114] | |||
| iNOS | IHC/IF | mdx mice | [110] | |||
| COX-2 | IHC | [115] | ||||
| M2 | Arginase-1 | IF | [116] | |||
| CD206 | IHC/IF | [109] | ||||
| CD163 | IHC/IF | [109,117] | ||||
| T cells | Pan-marker | CD3 | IHC/IF | Skeletal muscle | mdx mice | [118,119] |
| T cell helper | CD4 | IHC/IF | [109,120] | |||
| T cell cytotoxic | CD8 | IHC/IF | [107,120] | |||
| Treg | FOXP3 | IHC/IF | [121] | |||
| CD25 | IF | [120] | ||||
| Neutrophils | / | Ly6B2 | IHC | Skeletal muscle | mdx mice | [109] |
| Ly6G | IHC/IF | [110,119] | ||||
| MPO | IF | SOD mice | [122] | |||
| Cytokines | Proinflammatory | TNFα | IHC/IF | Skeletal muscle | mdx mice | [118,123] |
| IL-6 | IF | [106] | ||||
| IFNγ | IHC | [110] | ||||
| IL-18 | IHC | [124] | ||||
| IL-1β | IHC/IF | [124,125] | ||||
| Anti-inflammatory | IL-10 | IF | Peripheral nerve | [126] | ||
| IL-4 | IHC | Skeletal muscle | [110] |
- Anti-inflammatory cytokines:
3.6. Mitochondrial and Sarcoplasmic Reticulum Function and Oxidative Stress
3.6.1. Mitochondrial Function
3.6.2. Oxidative Stress
3.6.3. Sarcoplasmic Reticulum
3.7. Myofibre Regeneration and Maturation
3.7.1. Satellite Cells and Early Regeneration
3.7.2. Early Maturation of Muscle Fibres
3.7.3. Late Maturation of Muscle Fibres
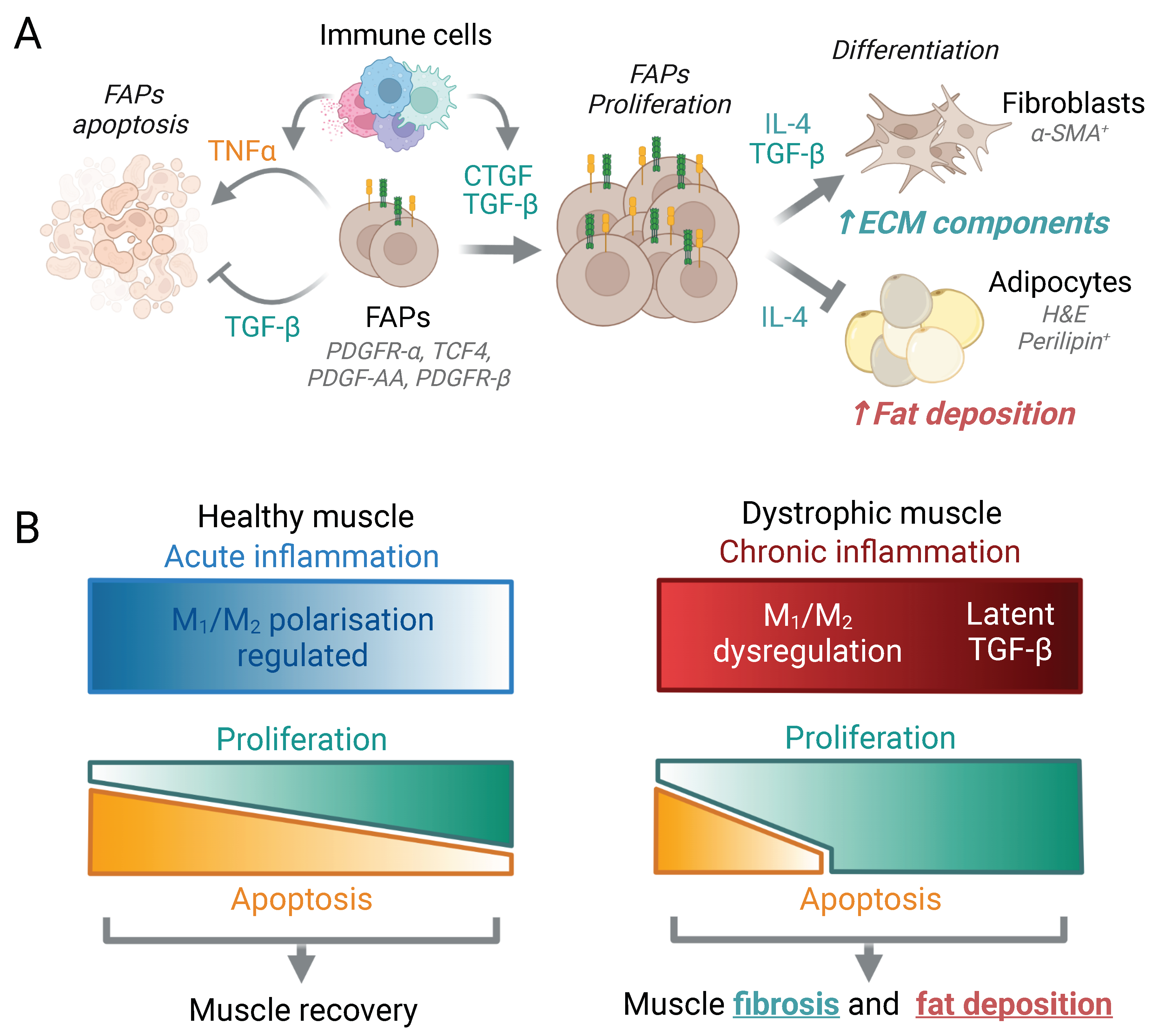
3.8. Fibrosis and Fat Deposition
3.8.1. Fibrosis

3.8.2. Fat Deposition
3.9. Calcification
3.10. Changes in Neuromuscular Junction
3.11. Changes in Capillarisation
3.12. Revertant Fibres and Detection of Dystrophin in Gene-Editing Therapies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| α-BTX | alpha bungarotoxin |
| α-SMA | alpha smooth muscle actin |
| ALP | alkaline phospatase |
| APC | antigen-presenting cells |
| CCR | C-C chemokine receptor |
| CD | cluster of differentiation |
| CNF | centrally nucleated fibres |
| COX | cytochrome C oxidase |
| CSA | cross sectional area |
| CTGF | connective tissue growth factor |
| DAMPs | damage-associated molecular patterns |
| DAPC | dystrophin-associated protein complex |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DHE | dihydroethidium |
| DMD | Duchenne muscular dystrophy |
| EBD | Evan’s blue dye |
| ECM | extracellular matrix |
| emb-MyHC | embryonic myosin heavy chain |
| FAPs | fibro-adipogenic progenitors |
| FOXP3 | Forkhead box protein 3 |
| GT | Gömöri trichrome |
| H&E | haemotoxylin and eosin |
| HNE | 4-hydroxynonenal |
| IB4 | isolectin B4 from Griffonia Simplicifolia |
| IFNγ | interferon gamma |
| Ig | immunoglobulin |
| IH | immunohistology/immunohistological |
| IL-10 | interleukin 10 |
| IL-1β | interleukin 1 beta |
| IL-4 | interleukin 4 |
| IL-6 | interleukin 6 |
| iNOS | inducible nitric oxide synthase |
| M’sT | Masson’s trichrome |
| MF’sD | minimum Feret’s diameter |
| MPO | myeloperoxidase |
| MyHC | myosin heavy chain |
| MyoD | myoblast determination protein 1 |
| Myog. | myogenin |
| NF | neurofilament |
| NG2 | neuron-glial antigen 2 |
| NMJ | neuromuscular junction |
| NO | nitric oxide |
| nNOS | neuronal nitric oxide synthase |
| ORO | Oil red O |
| Pax7 | paired box protein 7 |
| PDGFR-α | platelet-derived growth factor receptor-alpha |
| PDGFR-β | platelet-derived growth factor receptor-beta |
| PR | Picrosirius red |
| RF | revertant fibre |
| ROS | reactive oxygen species |
| RT-qPCR | real-time quantitative polymerase chain reaction |
| SC | satellite cell |
| SDH | succinate dehydrogenase |
| SR | sarcoplasmic reticulum |
| Syn | synaptophysin |
| TEM | transmission electron microscopy |
| TGF-β | transforming growth factor-beta |
| TCF4 | transcription factor 4 |
| Treg | T regulatory cell |
| TNFα | tumour necrosis factor alpha |
| UEA 1 | Ulex europaeus agglutinin 1 |
| WB | Western blotting |
| WGA | wheat germ agglutinin |
| WT | wild type |
References
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne Muscular Dystrophy: Myonecrosis, Inflammation and Oxidative Stress. DMM Dis. Model. Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Paepe, B.; de Bleecker, J.L. Cytokines and Chemokines as Regulators of Skeletal Muscle Inflammation: Presenting the Case of Duchenne Muscular Dystrophy. Mediat. Inflamm. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabłocka, B.; Górecki, D.C.; Zabłocki, K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. Int. J. Mol. Sci. 2021, 22, 11040. [Google Scholar] [CrossRef]
- Petrof, B.J.; Stedman, H.H.; Shrager, J.B.; Eby, J.; Sweeney, H.L.; Kelly, A.M. Adaptations in Myosin Heavy Chain Expression and Contractile Function in Dystrophic Mouse Diaphragm. Am. J. Physiol. Physiol. 1993, 265, C834–C841. [Google Scholar] [CrossRef]
- Morgan, J.E.; Prola, A.; Mariot, V.; Pini, V.; Meng, J.; Hourde, C.; Dumonceaux, J.; Conti, F.; Relaix, F.; Authier, F.J.; et al. Necroptosis Mediates Myofibre Death in Dystrophin-Deficient Mice. Nat. Commun. 2018, 9, 3655. [Google Scholar] [CrossRef]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; López de Munain, A. Dysregulation of Calcium Homeostasis in Muscular Dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef] [Green Version]
- Deconinck, N.; Dan, B. Pathophysiology of Duchenne Muscular Dystrophy: Current Hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Carlson, C.G. Does the Pathogenic Sequence of Skeletal Muscle Degeneration in Duchenne Muscular Dystrophy Begin and End with Unrestrained Satellite Cell Activation? Muscles 2022, 1, 75–81. [Google Scholar] [CrossRef]
- Latroche, C.; Matot, B.; Martins-Bach, A.; Briand, D.; Chazaud, B.; Wary, C.; Carlier, P.G.; Chrétien, F.; Jouvion, G. Structural and Functional Alterations of Skeletal Muscle Microvasculature in Dystrophin-Deficient Mdx Mice. Am. J. Pathol. 2015, 185, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J. Treat-NMD SOP MDX-DMD_M.1.1_001. Available online: https://treat-nmd.org/wp-content/uploads/2016/08/MDX-DMD_M.1.1_001-21.pdf (accessed on 17 November 2022).
- Aartsma-Rus, A.; van Putten, M. Assessing Functional Performance in the Mdx Mouse Model. J. Vis. Exp. 2014, 85, e51303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treat-NMD SOP DMD_M.2.1.004. Available online: https://treat-nmd.org/wp-content/uploads/2021/06/uncategorized-Wire-test.pdf (accessed on 17 November 2022).
- Treat-NMD SOP DMD_M.2.1.001. Available online: https://treat-nmd.org/wp-content/uploads/2016/08/MDX-DMD_M.2.1.001.pdf (accessed on 17 November 2022).
- Treat-NMD SOP DMD_M.2.2.001. Available online: https://treat-nmd.org/wp-content/uploads/2016/08/MDX-DMD_M.2.2.001.pdf (accessed on 17 November 2022).
- Zweyer, M.; Sabir, H.; Dowling, P.; Gargan, S.; Murphy, S.; Swandulla, D.; Ohlendieck, K. Histopathology of Duchenne Muscular Dystrophy in Correlation with Changes in Proteomic Biomarkers. Histol. Histopathol. 2022, 37, 101–116. [Google Scholar] [CrossRef]
- Dubowitz, V.; Sewry, C.; Oldfors, A. Muscle Biopsy a Practical Approach, 5th ed.; Dubowitz, V., Sewry, C., Oldfors, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780702074714. [Google Scholar]
- Nonaka, I. Animal Models of Muscular Dystrophies. Lab. Anim. Sci. 1998, 48, 8–17. [Google Scholar]
- Bulfield, G.; Siller, W.G.; Wight, P.A.L.; Mooret, K.J. X Chromosome-Linked Muscular Dystrophy (Mdx) in the Mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, J.M.; Blake, D.J.; Roche, A.; Fairbrother, U.; Riss, J.; Byth, B.C.; Knight, A.E.; Kendrick-Jones, J.; Suthers, G.K.; Love, D.R.; et al. Primary Structure of Dystrophin-Related Protein. Nature 1992, 360, 591–593. [Google Scholar] [CrossRef]
- Hodges, B.L.; Hayashi, Y.K.; Nonaka, I.; Wang, W.; Arahata, K.; Kaufman, S.J. Altered Expression of the A7β1 Integrin in Human and Murine Muscular Dystrophies. J. Cell Sci. 1997, 110, 2873–2881. [Google Scholar] [CrossRef]
- Burkin, D.J.; Wallace, G.Q.; Nicol, K.J.; Kaufman, D.J.; Kaufman, S.J. Enhanced Expression of the A7β1 Integrin Reduces Muscular Dystrophy and Restores Viability in Dystrophic Mice. J. Cell Biol. 2001, 152, 1207–1218. [Google Scholar] [CrossRef]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; des Rosiers, C.; Burelle, Y. Stress-Induced Opening of the Permeability Transition Pore in the Dystrophin-Deficient Heart Is Attenuated by Acute Treatment with Sildenafil. Am. J. Physiol. Circ. Physiol. 2011, 300, 144–153. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Heart Mitochondria in the Early Stages of Duchenne Muscular Dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Panel, M.; Lacôte, M.; Rieusset, J.; Lacampagne, A.; Fauconnier, J. Metformin Reverses the Enhanced Myocardial SR/ER–Mitochondria Interaction and Impaired Complex I-Driven Respiration in Dystrophin-Deficient Mice. Front. Cell Dev. Biol. 2021, 8, 609493. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Serov, D.A.; Tenkov, K.S.; Belosludtseva, E.V.; Belosludtsev, K.N. Effect of the Non-Immunosuppressive Mpt Pore Inhibitor Alisporivir on the Functioning of Heart Mitochondria in Dystrophin-Deficient Mdx Mice. Biomedicines 2021, 9, 1232. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Yue, Y.; Long, C.; Marschalk, N.; Fine, D.M.; Chen, J.; Duan, D. Cardiac Expression of a Mini-Dystrophin That Normalizes Skeletal Muscle Force Only Partially Restores Heart Function in Aged Mdx Mice. Mol. Ther. 2009, 17, 253–261. [Google Scholar] [CrossRef]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-Dystrophin-Deficient Mice as a Model for Duchenne Muscular Dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Yucel, N.; Chang, A.C.; Day, J.W.; Rosenthal, N.; Blau, H.M. Humanizing the Mdx Mouse Model of DMD: The Long and the Short of It. NPJ Regen. Med. 2018, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Vulin, A.; Wein, N.; Simmons, T.R.; Rutherford, A.M.; Findlay, A.R.; Yurkoski, J.A.; Kaminoh, Y.; Flanigan, K.M. The First Exon Duplication Mouse Model of Duchenne Muscular Dystrophy: A Tool for Therapeutic Development. Neuromuscul. Disord. 2015, 25, 827–834. [Google Scholar] [CrossRef]
- Kornegay, J.N. The Golden Retriever Model of Duchenne Muscular Dystrophy. Skelet. Muscle 2017, 7, 9. [Google Scholar] [CrossRef]
- Chey, Y.C.J.; Arudkumar, J.; Aartsma-Rus, A.; Adikusuma, F.; Thomas, P.Q. CRISPR Applications for Duchenne Muscular Dystrophy: From Animal Models to Potential Therapies. WIREs Mech. Dis. 2022, e1580. [Google Scholar] [CrossRef]
- Egorova, T.V.; Galkin, I.I.; Ivanova, Y.V.; Polikarpova, A.V. Duchenne Muscular Dystrophy Animal Models. In Preclinical Animal Modeling in Medicine; Purevjav, E., Pierre, J., Lu, L., Eds.; IntechOpen: London, UK, 2022. [Google Scholar]
- Meng, H.; Janssen, P.M.L.; Grange, R.W.; Yang, L.; Beggs, A.H.; Swanson, L.C.; Cossette, S.A.; Frase, A.; Childers, M.K.; Granzier, H.; et al. Tissue Triage and Freezing for Models of Skeletal Muscle Disease. J. Vis. Exp. 2014, e51586. [Google Scholar] [CrossRef] [Green Version]
- Roelofs, A.J.; de Bari, C. Immunostaining of Skeletal Tissues. In Bone Research Protocols, Methods in Molecular Biology, 3rd ed.; Idris, A.I., Ed.; Humana Press Inc.: New York, NY, USA, 2019; Volume 1914, pp. 437–450. [Google Scholar] [CrossRef]
- Johnson, C.D.; Zhou, L.Y.; Kopinke, D. A Guide to Examining Intramuscular Fat Formation and Its Cellular Origin in Skeletal Muscle. J. Vis. Exp. 2022, 2022, e63996. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.L.; Wells, D.J. Histopathological Evaluation of Skeletal Muscle with Specific Reference to Mouse Models of Muscular Dystrophy. Curr. Protoc. Mouse Biol. 2016, 6, 343–363. [Google Scholar] [CrossRef] [PubMed]
- Scalia, C.R.; Boi, G.; Bolognesi, M.M.; Riva, L.; Manzoni, M.; DeSmedt, L.; Bosisio, F.M.; Ronchi, S.; Leone, B.E.; Cattoretti, G. Antigen Masking During Fixation and Embedding, Dissected. J. Histochem. Cytochem. 2017, 65, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Accorsi, A.; Rhee, Y.; Girgenrath, M. Do’s and Don’ts in the Preparation of Muscle Cryosections for Histological Analysis. J. Vis. Exp. 2015, 2015, e52793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruegg, M. Treat-NMD SOP MDC1A_M.1.2.004. Available online: https://treat-nmd.org/sop/mdc1a_m-1-2-004/ (accessed on 17 November 2022).
- Duddy, W.; Duguez, S.; Johnston, H.; Cohen, T.V.; Phadke, A.; Gordish-Dressman, H.; Nagaraju, K.; Gnocchi, V.; Low, S.H.; Partridge, T. Muscular Dystrophy in the Mdx Mouse Is a Severe Myopathy Compounded by Hypotrophy, Hypertrophy and Hyperplasia. Skelet. Muscle 2015, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindi, S.; Tajrishi, M.; Kumar, A. Signaling Mechanisms in Mammalian Myoblast Fusion. Sci. Signal. 2013, 6, re2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemionow, M.; Langa, P.; Brodowska, S.; Kozlowska, K.; Zalants, K.; Budzynska, K.; Heydemann, A. Long-Term Protective Effect of Human Dystrophin Expressing Chimeric (DEC) Cell Therapy on Amelioration of Function of Cardiac, Respiratory and Skeletal Muscles in Duchenne Muscular Dystrophy. Stem. Cell Rev. Rep. 2022, 18, 2872–2892. [Google Scholar] [CrossRef]
- Briguet, A.; Courdier-Fruh, I.; Foster, M.; Meier, T.; Magyar, J.P. Histological Parameters for the Quantitative Assessment of Muscular Dystrophy in the Mdx-Mouse. Neuromuscul. Disord. 2004, 14, 675–682. [Google Scholar] [CrossRef]
- Rüegg, M.A.; Biozentrum, S.M. Treat-NMD SOP MDC1A_M.1.2.002. Available online: https://treat-nmd.org/sop/mdc1a_m-1-2-002/ (accessed on 17 November 2022).
- Wang, Q.Q.; Jing, X.M.; Bi, Y.Z.; Cao, X.F.; Wang, Y.Z.; Li, Y.X.; Qiao, B.J.; Chen, Y.; Hao, Y.L.; Hu, J. Human Umbilical Cord Wharton’s Jelly Derived Mesenchymal Stromal Cells May Attenuate Sarcopenia in Aged Mice Induced by Hindlimb Suspension. Med. Sci. Monit. 2018, 24, 9272–9281. [Google Scholar] [CrossRef]
- Kim, Y.J.; Brox, T.; Feiden, W.; Weickert, J. Fully Automated Segmentation and Morphometrical Analysis of Muscle Fiber Images. Cytom. Part A 2007, 71, 8–15. [Google Scholar] [CrossRef]
- Liu, F.; Mackey, A.L.; Srikuea, R.; Esser, K.A.; Yang, L. Automated Image Segmentation of Haematoxylin and Eosin Stained Skeletal Muscle Cross-Sections. J. Microsc. 2013, 252, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmberg, J.; Durbeej, M. Laminin-211 in Skeletal Muscle Function. Cell Adh. Migr. 2013, 7, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, L.E.; Kaminoh, Y.J.; Rodesch, C.K.; Flanigan, K.M. Quantification of Dystrophin Immunofluorescence in Dystrophinopathy Muscle Specimens. Neuropathol. Appl. Neurobiol. 2012, 38, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Feng, L.; Anthony, K.; Arechavala-Gomeza, V.; Torelli, S.; Sewry, C.; Morgan, J.E.; Muntoni, F. Restoration of the Dystrophin-Associated Glycoprotein Complex after Exon Skipping Therapy in Duchenne Muscular Dystrophy. Mol. Ther. 2012, 20, 462–467. [Google Scholar] [CrossRef] [Green Version]
- Kostrominova, T.Y. Application of WGA Lectin Staining for Visualization of the Connective Tissue in Skeletal Muscle, Bone, and Ligament/Tendon Studies. Microsc. Res. Tech. 2011, 74, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Mayeuf-Louchart, A.; Hardy, D.; Thorel, Q.; Roux, P.; Gueniot, L.; Briand, D.; Mazeraud, A.; Bouglé, A.; Shorte, S.L.; Staels, B.; et al. MuscleJ: A High-Content Analysis Method to Study Skeletal Muscle with a New Fiji Tool. Skelet. Muscle 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Casar, J.C.; Cabello-Verrugio, C.; Olguin, H.; Aldunate, R.; Inestrosa, N.C.; Brandan, E. Heparan Sulfate Proteoglycans Are Increased during Skeletal Muscle Regeneration: Requirement of Syndecan-3 for Successful Fiber Formation. J. Cell Sci. 2004, 117, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Barateau, A.; Vadrot, N.; Agbulut, O.; Vicart, P.; Batonnet-Pichon, S.; Buendia, B. Distinct Fiber Type Signature in Mouse Muscles Expressing a Mutant Lamin a Responsible for Congenital Muscular Dystrophy in a Patient. Cells 2017, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Omairi, S.; Hau, K.L.; Collins-Hooper, H.; Scott, C.; Vaiyapuri, S.; Torelli, S.; Montanaro, F.; Matsakas, A.; Patel, K. Regulation of the Dystrophin-Associated Glycoprotein Complex Composition by the Metabolic Properties of Muscle Fibres. Sci. Rep. 2019, 9, 2770. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Muñoz, P.; Torrella, J.R.; Serres, X.; Rizo-Roca, D.; de La Varga, M.; Viscor, G.; Martínez-Ibáñez, V.; Peiró, J.L.; Järvinen, T.A.H.; Rodas, G.; et al. Postinjury Exercise and Platelet-Rich Plasma Therapies Improve Skeletal Muscle Healing in Rats but Are Not Synergistic When Combined. Am. J. Sports Med. 2017, 45, 2131–2141. [Google Scholar] [CrossRef]
- Zanotti, S.; Negri, T.; Cappelletti, C.; Bernasconi, P.; Canioni, E.; di Blasi, C.; Pegoraro, E.; Angelini, C.; Ciscato, P.; Prelle, A.; et al. Decorin and Biglycan Expression Is Differentially Altered in Several Muscular Dystrophies. Brain 2005, 128, 2546–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markworth, J.F.; Brown, L.A.; Lim, E.; Floyd, C.; Larouche, J.; Castor-Macias, J.A.; Sugg, K.B.; Sarver, D.C.; Macpherson, P.C.D.; Davis, C.; et al. Resolvin D1 Supports Skeletal Myofiber Regeneration via Actions on Myeloid and Muscle Stem Cells. JCI Insight 2020, 5, e137713. [Google Scholar] [CrossRef]
- Folker, E.S.; Baylies, M.K. Nuclear Positioning in Muscle Development and Disease. Front. Physiol. 2013, 4, 363. [Google Scholar] [CrossRef] [Green Version]
- Selvais, C.M.; de Cock, L.L.; Brichard, S.M.; Davis-López de Carrizosa, M.A. Fiber Type and Subcellular-Specific Analysis of Lipid Droplet Content in Skeletal Muscle. J. Vis. Exp. 2022, e63718. [Google Scholar] [CrossRef] [PubMed]
- Capers, C.R. Multinucleation of Skeletal Muscle in Vitro. J. Biophys. Biochem. Cytol. 1960, 7, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite Cells and the Muscle Stem Cell Niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, M.; Echigoya, Y.; Maruyama, R.; Lim, K.R.Q.; Fukada, S.I.; Yokota, T. Impaired Regenerative Capacity and Lower Revertant Fibre Expansion in Dystrophin-Deficient Mdx Muscles on DBA/2 Background. Sci. Rep. 2016, 6, 38371. [Google Scholar] [CrossRef] [Green Version]
- Hakim, C.H.; Wasala, N.B.; Pan, X.; Kodippili, K.; Yue, Y.; Zhang, K.; Yao, G.; Haffner, B.; Duan, S.X.; Ramos, J.; et al. A Five-Repeat Micro-Dystrophin Gene Ameliorated Dystrophic Phenotype in the Severe DBA/2J-Mdx Model of Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2017, 6, 216–230. [Google Scholar] [CrossRef] [Green Version]
- Podkalicka, P.; Mucha, O.; Bronisz-Budzyńska, I.; Kozakowska, M.; Pietraszek-Gremplewicz, K.; Cetnarowska, A.; Głowniak-Kwitek, U.; Bukowska-Strakova, K.; Cieśla, M.; Kulecka, M.; et al. Lack of MiR-378 Attenuates Muscular Dystrophy in Mdx Mice. JCI Insight 2020, 5, 38371. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Lee, J.; Rodrigues, M.; Nagata, T.; Tanihata, J.; Nozohourmehrabad, A.; Panesar, D.; Miskew, B.; Aoki, Y.; Yokota, T. Mutation Types and Aging Differently Affect Revertant Fiber Expansion in Dystrophic Mdx and Mdx52 Mice. PLoS ONE 2013, 8, e69194. [Google Scholar] [CrossRef]
- Miller, J.B.; Girgenrath, M. The Role of Apoptosis in Neuromuscular Diseases and Prospects for Anti-Apoptosis Therapy. Trends Mol. Med. 2006, 12, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, N.; Versele, R.; Davis-López de Carrizosa, M.A.; Selvais, C.M.; Brichard, S.M.; Abou-Samra, M. Walking down Skeletal Muscle Lane: From Inflammasome to Disease. Cells 2021, 10, 3023. [Google Scholar] [CrossRef]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef] [PubMed]
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced Necrosis of Dystrophic Muscle by Depletion of Host Neutrophils, or Blocking TNFα Function with Etanercept in Mdx Mice. Neuromuscul. Disord. 2006, 16, 591–602. [Google Scholar] [CrossRef]
- Matsuda, R.; Nishikawa, A.; Tanaka, H. Visualization of Dystrophic Muscle Fibers in Mdx Mouse by Vital Staining with Evans Blue: Evidence of Apoptosis in Dystrophin-Deficient Muscle 1. J. Biochem. 1995, 118, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Hamer, P.W.; Mcgeachie, J.M.; Davies, M.J.; Grounds, M.D. Evans Blue Dye as an in Vivo Marker of Myofibre Damage: Optimising Parameters for Detecting Initial Myofibre Membrane Permeability. J. Anat. 2002, 200, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Puebla, C.; Cisterna, B.A.; Escamilla, R.; Vargas, A.A.; Frank, M.; Martínez-Montero, P.; Prior, C.; Molano, J.; Esteban-Rodríguez, I.; et al. Fast Skeletal Myofibers of Mdx Mouse, Model of Duchenne Muscular Dystrophy, Express Connexin Hemichannels That Lead to Apoptosis. Cell. Mol. Life Sci. 2016, 73, 2583–2599. [Google Scholar] [CrossRef]
- Cornelio, F.; Dones, I. Muscle Fiber Degeneration and Necrosis in Muscular Dystrophy and Other Muscle Diseases: Cytochemical and Immunocytochemical Data. Ann. Neurol. 1984, 16, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Bencze, M.; Periou, B.; Baba-Amer, Y.; Authier, F.J. Immunolabelling Myofiber Degeneration in Muscle Biopsies. J. Vis. Exp. 2019, 2019, e59754. [Google Scholar] [CrossRef] [PubMed]
- Bronisz-Budzyńska, I.; Chwalenia, K.; Mucha, O.; Podkalicka, P.; Bukowska-Strakova, K.; Józkowicz, A.; Łoboda, A.; Kozakowska, M.; Dulak, J. MiR-146a Deficiency Does Not Aggravate Muscular Dystrophy in Mdx Mice. Skelet. Muscle 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-Mediated Pathology in Duchenne Muscular Dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hu, P. Skeletal Muscle Regeneration Is Modulated by Inflammation. J. Orthop. Translat. 2018, 13, 25–32. [Google Scholar] [CrossRef]
- Larouche, J.A.; Fraczek, P.M.; Kurpiers, S.J.; Yang, B.A.; Davis, C.; Castor-Macias, J.A.; Sabin, K.; Anderson, S.; Harrer, J.; Hall, M.; et al. Neutrophil and Natural Killer Cell Imbalances Prevent Muscle Stem Cell-Mediated Regeneration Following Murine Volumetric Muscle Loss. Proc. Natl. Acad. Sci. USA 2022, 119, e2111445119. [Google Scholar] [CrossRef] [PubMed]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019, 2019, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; DeNardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, C.M.; Phillips, A.R.J.; Cooper, G.J.S.; Dunbar, P.R. Three-Colour Fluorescence Immunohistochemistry Reveals the Diversity of Cells Staining for Macrophage Markers in Murine Spleen and Liver. J. Immunol. Methods 2008, 334, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Kosmac, K.; Peck, B.; Walton, R.; Mula, J.; Kern, P.; Bamman, M.; Dennis, R.; Jacobs, C.; Lattermann, C.; Johnson, D.; et al. Immunohistochemical Identification of Human Skeletal Muscle Macrophages. Bio-Protoc 2018, 8, e2883. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, G.; Egner, I.; Raastad, T.; Reinholt, F.; Owe, S.; Lauritzen, F.; Brorson, S.H.; Koskinen, S. Inflammatory Markers CD11b, CD16, CD66b, CD68, Myeloperoxidase and Neutrophil Elastase in Eccentric Exercised Human Skeletal Muscles. Histochem. Cell Biol. 2013, 139, 691–715. [Google Scholar] [CrossRef] [PubMed]
- Mcknight, A.J.; Gordon, S. EGF-TM7: A Novel Subfamily of Seven-Transmembrane-Region Leukocyte Cell-Surface Molecules. Immunol. Today 1996, 17, 283–287. [Google Scholar] [CrossRef]
- Austyn, J.M.; Gordon, S. F4/80, a Monoclonal Antibody Directed Specifically against the Mouse Macrophage. Eur. J. Immunol. 1981, 11, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Gordon, S. Mononuclear Phagocyte System of the Mouse Defined by Immunohistochemical Localization of Antigen F4/80. Identification of Resident Macrophages in Renal Medullary and Cortical Interstitium and the Juxtaglomerular Complex. J. Exp. Med. 1983, 157, 1704–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassado, A.d.A.; de Albuquerque, J.A.T.; Sardinha, L.R.; de Buzzo, C.L.; Faustino, L.; Nascimento, R.; Ghosn, E.E.B.; Lima, M.R.; Alvarez, J.M.M.; Bortoluci, K.R. Cellular Renewal and Improvement of Local Cell Effector Activity in Peritoneal Cavity in Response to Infectious Stimuli. PLoS ONE 2011, 6, e22141. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.A.; Krausz, S.; van Eijk, M.; Hamann, J.; Radstake, T.R.D.J.; Reedquist, K.A.; Tak, P.P.; Baeten, D.L.P. Systematic Validation of Specific Phenotypic Markers for in Vitro Polarized Human Macrophages. J. Immunol. Methods 2012, 375, 196–206. [Google Scholar] [CrossRef]
- Jansen, K.M.; Pavlath, G.K. Mannose Receptor Regulates Myoblast Motility and Muscle Growth. J. Cell Biol. 2006, 174, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto-Mariz, F.; Rodrigues Carvalho, L.; de Mello, W.; de Queiroz Campos Araújo, A.; Gonçalves Ribeiro, M.; do Carmo Soares Alves Cunha, M.; Voit, T.; Butler-Browne, G.; Silva-Barbosa, S.D.; Savino, W. Differential Integrin Expression by T Lymphocytes: Potential Role in DMD Muscle Damage. J. Neuroimmunol. 2010, 223, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, L.; Villa, C.; Molinaro, D.; Torrente, Y.; Farini, A. The Immune System in Duchenne Muscular Dystrophy Pathogenesis. Biomedicines 2021, 9, 1447. [Google Scholar] [CrossRef]
- Mohseni, Y.R.; Tung, S.L.; Dudreuilh, C.; Lechler, R.I.; Fruhwirth, G.O.; Lombardi, G. The Future of Regulatory T Cell Therapy: Promises and Challenges of Implementing CAR Technology. Front. Immunol. 2020, 11, 1608. [Google Scholar] [CrossRef]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. XA Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Butcher, S.K.; O’Carroll, C.E.; Wells, C.A.; Carmody, R.J. Toll-like Receptors Drive Specific Patterns of Tolerance and Training on Restimulation of Macrophages. Front. Immunol. 2018, 9, 933. [Google Scholar] [CrossRef]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome Up-Regulation and Activation in Dysferlin-Deficient Skeletal Muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- de Senzi Moraes Pinto, R.; Ferretti, R.; Moraes, L.H.R.; Neto, H.S.; Marques, M.J.; Minatel, E. N-Acetylcysteine Treatment Reduces TNF-α Levels and Myonecrosis in Diaphragm Muscle of Mdx Mice. Clin. Nutr. 2013, 32, 472–475. [Google Scholar] [CrossRef] [PubMed]
- de Paepe, B.; Creus, K.K.; Martin, J.J.; de Bleecker, J.L. Upregulation of Chemokines and Their Receptors in Duchenne Muscular Dystrophy: Potential for Attenuation of Myofiber Necrosis. Muscle Nerve 2012, 46, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Bansal, Y.; Kumar, R.; Bansal, G. A Panoramic Review of IL-6: Structure, Pathophysiological Roles and Inhibitors. Bioorg. Med. Chem. 2020, 28, 115327. [Google Scholar] [CrossRef]
- Messina, S.; Vita, G.L.; Aguennouz, M.; Sframeli, M.; Romeo, S.; Rodolico, C.; Vita, G. Activation of NF-KappaB Pathway in Duchenne Muscular Dystrophy: Relation to Age. Acta Myol. 2011, 30, 16–23. [Google Scholar]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Spelta, E.; Rizzuto, E.; Nicoletti, C.; Camilli, C.; Testa, E.; Catizone, A.; de Benedetti, F.; et al. Increased Levels of Interleukin-6 Exacerbate the Dystrophic Phenotype in Mdx Mice. Hum. Mol. Genet. 2015, 24, 6041–6053. [Google Scholar] [CrossRef] [Green Version]
- Pelosi, L.; Berardinelli, M.G.; de Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; de Benedetti, F.; et al. Functional and Morphological Improvement of Dystrophic Muscle by Interleukin 6 Receptor Blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Yuasa, K.; Sakamoto, M.; Miyagoe-Suzuki, Y.; Tanouchi, A.; Yamamoto, H.; Li, J.; Chamberlain, J.S.; Xiao, X.; Takeda, S. Adeno-Associated Virus Vector-Mediated Gene Transfer into Dystrophin-Deficient Skeletal Muscles Evokes Enchanced Immune Response against the Transgene Product. Gene Ther. 2002, 9, 1576–1588. [Google Scholar] [CrossRef] [Green Version]
- Shiba, N.; Miyazaki, D.; Yoshizawa, T.; Fukushima, K.; Shiba, Y.; Inaba, Y.; Imamura, M.; Takeda, S.; Koike, K.; Nakamura, A. Differential Roles of MMP-9 in Early and Late Stages of Dystrophic Muscles in a Mouse Model of Duchenne Muscular Dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2170–2182. [Google Scholar] [CrossRef] [Green Version]
- Welc, S.S.; Flores, I.; Wehling-Henricks, M.; Ramos, J.; Wang, Y.; Bertoni, C.; Tidball, J.G. Targeting a Therapeutic LIF Transgene to Muscle via the Immune System Ameliorates Muscular Dystrophy. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidbal, J.G. Shifts in Macrophage Phenotypes and Macrophage Competition for Arginine Metabolism Affect the Severity of Muscle Pathology in Muscular Dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Hulderman, T.; Mishra, D.; Gao, X.; Millecchia, L.; O’Farrell, L.; Kuziel, W.A.; Simeonova, P.P. Chemokine Receptor CCR2 Involvement in Skeletal Muscle Regeneration. FASEB J. 2005, 19, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Iwashita, M.; Nishimura, Y.; Shinjo, T.; Sano, T.; Yamashita, A.; Fukuda, T.; Sanui, T.; Asano, T.; Nishimura, F. Adipose-Specific C-C Motif Chemokine Ligand (CCL) 19 Overexpression Drives the Mice to Both Insulin Resistance and Weight Gain. BMJ Open Diabetes Res. Care 2021, 9, e001871. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Fonseca, A.; Santiago Maniega, S.; Gorbenko del Blanco, D.; Catalán Bernardos, B.; Vega Castrillo, A.; Álvarez Barcia, Á.J.; Alonso, M.; Aguado, H.J.; Rodríguez-Cabello, J.C. Elastin-Like Recombinamer Hydrogels for Improved Skeletal Muscle Healing Through Modulation of Macrophage Polarization. Front. Bioeng. Biotechnol. 2020, 8, 413. [Google Scholar] [CrossRef]
- Oliveira, K.M.C.; Barker, J.H.; Berezikov, E.; Pindur, L.; Kynigopoulos, S.; Eischen-Loges, M.; Han, Z.; Bhavsar, M.B.; Henrich, D.; Leppik, L. Electrical Stimulation Shifts Healing/Scarring towards Regeneration in a Rat Limb Amputation Model. Sci. Rep. 2019, 9, 11433. [Google Scholar] [CrossRef] [Green Version]
- Lazzarin, M.C.; Quintana, H.T.; de Araújo Baptista, V.I.; de Oliveira, F. Lack of Dystrophin Influences Muscle Inflammation but Not Myogenic Regulatory Factors after Eccentric Exercise in Mdx Mice. Motriz. Rev. Educ. Fis. 2020, 26. [Google Scholar] [CrossRef]
- Morotti, M.; Garofalo, S.; Cocozza, G.; Antonangeli, F.; Bianconi, V.; Mozzetta, C.; de Stefano, M.E.; Capitani, R.; Wulff, H.; Limatola, C.; et al. Muscle Damage in Dystrophic Mdx Mice Is Influenced by the Activity of Ca2+-Activated KCa 3.1 Channels. Life 2022, 12, 538. [Google Scholar] [CrossRef]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-γ Promotes Muscle Damage in the Mdx Mouse Model of Duchenne Muscular Dystrophy by Suppressing M2 Macrophage Activation and Inhibiting Muscle Cell Proliferation. J. Immunol. 2011, 187, 5419–5428. [Google Scholar] [CrossRef] [Green Version]
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of Adiponectin in the Pathogenesis of Dystrophinopathy. Skelet Muscle 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Dort, J.; Orfi, Z.; Fabre, P.; Molina, T.; Conte, T.C.; Greffard, K.; Pellerito, O.; Bilodeau, J.F.; Dumont, N.A. Resolvin-D2 Targets Myogenic Cells and Improves Muscle Regeneration in Duchenne Muscular Dystrophy. Nat. Commun. 2021, 12, 6264. [Google Scholar] [CrossRef]
- Morrison, J.; Palmer, D.B.; Cobbold, S.; Partridge, T.; Bou-Gharios, G. Effects of T-Lymphocyte Depletion on Muscle Fibrosis in the Mdx Mouse. Am. J. Pathol. 2005, 166, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Rosenthal, W.; Martinez, L.; Kaur, A.; Sparwasser, T.; Tidball, J.G.; Margeta, M.; Spencer, M.J.; Bluestone, J.A. Regulatory T Cells Suppress Muscle Inflammation and Injury in Muscular Dystrophy. Sci. Transl. Med. 2014, 6, 258ra142. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast Cells and Neutrophils Mediate Peripheral Motor Pathway Degeneration in ALS. JCI Insight 2018, 3, e123249. [Google Scholar] [CrossRef] [PubMed]
- Saclier, M.; Larbi, S.B.; Ly, H.M.; Moulin, E.; Mounier, R.; Chazaud, B.; Juban, G. Interplay between Myofibers and Pro-Inflammatory Macrophages Controls Muscle Damage in Mdx Mice. J. Cell Sci. 2021, 134, jcs258429. [Google Scholar] [CrossRef]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 Inflammasome by Adiponectin Rescues Duchenne Muscular Dystrophy. BMC Biol. 2018, 16, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubuisson, N.; Davis-López de Carrizosa, M.A.; Versele, R.; Selvais, C.M.; Noel, L.; van den Bergh, P.; Brichard, S.; Abou-Samra, M. Inhibiting the Inflammasome with MCC950 Counteracts Muscle Pyroptosis and Improves Duchenne Muscular Dystrophy. Front Immunol. 2022, 13, 1049076. [Google Scholar] [CrossRef]
- Mietto, B.S.; Kroner, A.; Girolami, E.I.; Santos-Nogueira, E.; Zhang, J.; David, S. Role of IL-10 in Resolution of Inflammation and Functional Recovery after Peripheral Nerve Injury. J. Neurosci. 2015, 35, 16431–16442. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, D.F.; Bond, M.W.; Mosmann, T.R. Two Types of Mouse T Helper Cell. IV. Th2 Clones Secrete a Factor That Inhibits Cytokine Production by Th1 Clones. J. Exp. Med. 1989, 170, 2081–2095. [Google Scholar] [CrossRef] [Green Version]
- Lang, R.; Rutschman, R.L.; Greaves, D.R.; Murray, P.J. Autocrine Deactivation of Macrophages in Transgenic Mice Constitutively Overexpressing IL-10 Under Control of the Human CD68 Promoter. J. Immunol. 2002, 168, 3402–3411. [Google Scholar] [CrossRef] [Green Version]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal Amplification of ROS and Ca2+ Signals in Stressed Mdx Dystrophic Skeletal Muscle Fibers. Pflugers Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and Pharmacologic Inhibition of Mitochondrial-Dependent Necrosis Attenuates Muscular Dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wojciechowska, S.; Szczepanowska, J.; Fronk, J.; Lochmüller, H.; Górecki, D.C.; Zabłocki, K. Mutation in Dystrophin-Encoding Gene Affects Energy Metabolism in Mouse Myoblasts. Biochem. Biophys. Res. Commun. 2009, 386, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria Mediate Cell Membrane Repair and Contribute to Duchenne Muscular Dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardero Therapeutics, Inc. Plus Epicatechin Duchenne Muscular Dystrophy in Non-Ambulatory Adolescents (ClinicalTrials.gov Identifier: NCT02964377). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02964377 (accessed on 17 November 2022).
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of Mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef] [PubMed]
- Rovira Gonzalez, Y.I.; Moyer, A.L.; LeTexier, N.J.; Bratti, A.D.; Feng, S.; Peña, V.; Sun, C.; Pulcastro, H.; Liu, T.; Iyer, S.R.; et al. Mss51 Deletion Increases Endurance and Ameliorates Histopathology in the Mdx Mouse Model of Duchenne Muscular Dystrophy. FASEB J. 2021, 35, e21276. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Miura, P.; Burt, M.; Boudreault, L.; Khogali, S.; Lunde, J.A.; Renaud, J.M.; Jasmin, B.J. Chronic AMPK Activation Evokes the Slow, Oxidative Myogenic Program and Triggers Beneficial Adaptations in Mdx Mouse Skeletal Muscle. Hum. Mol. Genet. 2011, 20, 3478–3493. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in Mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in Mitochondrial Localization and ATP Synthesis in the Mdx Mouse Model of Duchenne Muscular Dystrophy Are Not Alleviated by PDE5 Inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfín, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic Dysfunction and Altered Mitochondrial Dynamics in the Utrophin-Dystrophin Deficient Mouse Model of Duchenne Muscular Dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.; Nederveen, J.P.; Gillen, J.B.; Joanisse, S.; Parise, G.; Tarnopolsky, M.A.; Gibala, M.J. Skeletal Muscle Fiber-Type-Specific Changes in Markers of Capillary and Mitochondrial Content after Low-Volume Interval Training in Overweight Women. Physiol. Rep. 2018, 6, e13597. [Google Scholar] [CrossRef]
- Sato, Y.; Ohtsubo, H.; Nihei, N.; Kaneko, T.; Sato, Y.; Adachi, S.I.; Kondo, S.; Nakamura, M.; Mizunoya, W.; Iida, H.; et al. Apobec2 Deficiency Causes Mitochondrial Defects and Mitophagy in Skeletal Muscle. FASEB J. 2018, 32, 1428–1439. [Google Scholar] [CrossRef]
- Selvais, C.M.; de Carrizosa, M.A.D.-L.; Nachit, M.; Versele, R.; Dubuisson, N.; Noel, L.; Gillard, J.; Leclercq, I.A.; Brichard, S.M.; Abou-Samra, M. AdipoRon Enhances Healthspan in Middle-Aged Obese Mice: Striking Alleviation of Myosteatosis and Muscle Degenerative Markers. J. Cachexia Sarcopenia Muscle 2022. [Google Scholar] [CrossRef]
- Ross, J.M. Visualization of Mitochondrial Respiratory Function Using Cytochrome C Oxidase/Succinate Dehydrogenase (COX/SDH) Double-Labeling Histochemistry. J. Vis. Exp. 2011, 23, e3266. [Google Scholar] [CrossRef] [Green Version]
- Garvey, W.; Bigelow, F.; Fathi, A.; Jimenez, C.; Carpenter, B. Modified Gomori Trichrome Stain for Frozen Skeletal Muscle and Paraffin Embedded Sections. J. Histotechnol. 1996, 19, 329–333. [Google Scholar] [CrossRef]
- Joseph, G.A.; Hung, M.; Goel, A.J.; Hong, M.; Rieder, M.K.; Beckmann, N.D.; Serasinghe, M.N.; Chipuk, J.E.; Devarakonda, P.M.; Goldhamer, D.J.; et al. Late-Onset Megaconial Myopathy in Mice Lacking Group i Paks. Skelet. Muscle 2019, 9, 5. [Google Scholar] [CrossRef]
- Sohal, R.S.; Brunk, U.T. Lipofuscin as an Indicator of Oxidative Stress and Aging. Adv. Exp. Med. Biol. 1989, 266, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Tohma, H.; Hepworth, A.R.; Shavlakadze, T.; Grounds, M.D.; Arthur, P.G. Quantification of Ceroid and Lipofuscin in Skeletal Muscle. J. Histochem. Cytochem. 2011, 59, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Hermes, T.d.A.; Mizobuti, D.S.; da Rocha, G.L.; da Silva, H.N.M.; Covatti, C.; Pereira, E.C.L.; Ferretti, R.; Minatel, E. Tempol Improves Redox Status in Mdx Dystrophic Diaphragm Muscle. Int. J. Exp. Pathol. 2020, 101, 289–297. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Pham, C.; Gervasio, O.L.; Allen, D.G. N-Acetylcysteine Ameliorates Skeletal Muscle Pathophysiology in Mdx Mice. J. Physiol. 2008, 586, 2003–2014. [Google Scholar] [CrossRef]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulère, L.; Lagarde, M.; Soulage, C.O. The Lipid Peroxidation By-Product 4-Hydroxy-2-Nonenal (4-HNE) Induces Insulin Resistance in Skeletal Muscle through Both Carbonyl and Oxidative Stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, B.; Ranjit, R.; Kneis, P.; Xu, H.; Piekarz, K.M.; Freeman, W.M.; Kinter, M.; Richardson, A.; Ran, Q.; Brooks, S.V.; et al. Scavenging Mitochondrial Hydrogen Peroxide by Peroxiredoxin 3 Overexpression Attenuates Contractile Dysfunction and Muscle Atrophy in a Murine Model of Accelerated Sarcopenia. Aging Cell 2022, 21, e13569. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-Hydroxy-2′ -Deoxyguanosine (8-OHdG): A Critical Biomarker of Oxidative Stress and Carcinogenesis. J. Environ. Sci. Heal. Part C 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A Versatile Oxidative Stress Biomarker for Major Neurodegenerative Diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol Ameliorates Muscular Pathology in the Dystrophic Mdx Mouse, a Model for Duchenne Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, C.E.; Novo, D.; DiFranco, M.; Vergara, J.L. The Action Potential-Evoked Sarcoplasmic Reticulum Calcium Release Is Impaired in Mdx Mouse Muscle Fibres. J. Physiol. 2004, 557, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Divet, A.; Huchet-Cadiou, C. Sarcoplasmic Reticulum Function in Slow- and Fast-Twitch Skeletal Muscles from Mdx Mice. Pflugers Arch. 2002, 444, 634–643. [Google Scholar] [CrossRef]
- Cleverdon, R.E.G.; Braun, J.L.; Geromella, M.S.; Whitley, K.C.; Marko, D.M.; Hamstra, S.I.; Roy, B.D.; MacPherson, R.E.K.; Fajardo, V.A. Sarco(Endo)Plasmic Reticulum Ca2+-ATPase Function Is Impaired in Skeletal and Cardiac Muscles from Young DBA/2J Mdx Mice. iScience 2022, 25, 104972. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated Ryanodine Receptor Calcium Release Channels Are Leaky in Dystrophic Muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Chambers, P.J.; Juracic, E.S.; Rietze, B.A.; Gamu, D.; Bellissimo, C.; Kwon, F.; Quadrilatero, J.; Russell Tupling, A. Sarcolipin Deletion in Mdx Mice Impairs Calcineurin Signalling and Worsens Dystrophic Pathology. Hum. Mol. Genet. 2018, 27, 4094–4102. [Google Scholar] [CrossRef] [PubMed]
- Tanihata, J.; Nagata, T.; Ito, N.; Saito, T.; Nakamura, A.; Minamisawa, S.; Aoki, Y.; Ruegg, U.T.; Takeda, S. Truncated Dystrophin Ameliorates the Dystrophic Phenotype of Mdx Mice by Reducing Sarcolipin-Mediated SERCA Inhibition. Biochem. Biophys. Res. Commun. 2018, 505, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Chevessier, F.; Bauché-Godard, S.; Leroy, J.P.; Koenig, J.; Paturneau-Jouas, M.; Eymard, B.; Hantaï, D.; Verdière-Sahuqué, M. The Origin of Tubular Aggregates in Human Myopathies. J. Pathol. 2005, 207, 313–323. [Google Scholar] [CrossRef]
- Jain, D.; Sharma, M.C.; Sarkar, C.; Suri, V.; Sharma, S.K.; Singh, S.; Das, T.K. Tubular Aggregate Myopathy: A Rare Form of Myopathy. J. Clin. Neurosci. 2008, 15, 1222–1226. [Google Scholar] [CrossRef]
- Griffin, D.A.; Johnson, R.W.; Whitlock, J.M.; Pozsgai, E.R.; Heller, K.N.; Grose, W.E.; Arnold, W.D.; Sahenk, Z.; Hartzell, H.C.; Rodino-Klapac, L.R. Defective Membrane Fusion and Repair in Anoctamin5-Deficient Muscular Dystrophy. Hum. Mol. Genet. 2016, 25, 1900–1911. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.S.; Shanmugam, M.; Gonzalez, J.P.; Lopez, H.; Gordan, R.; Fraidenraich, D.; Babu, G.J. Increased Sarcolipin Expression and Decreased Sarco(Endo)Plasmic Reticulum Ca2+ Uptake in Skeletal Muscles of Mouse Models of Duchenne Muscular Dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, R.J.; Mundhenk, K.; Becker, T.; Hallerdei, J.; Waheed, A.; Shah, G.N.; Sly, W.S.; Gros, G.; Wetzel, P. Carbonic Anhydrases IV and IX: Subcellular Localization and Functional Role in Mouse Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2008, 294, C402–C412. [Google Scholar] [CrossRef]
- Cusimano, V.; Pampinella, F.; Giacomello, E.; Sorrentino, V. Assembly and Dynamics of Proteins of the Longitudinal and Junctional Sarcoplasmic Reticulum in Skeletal Muscle Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 4695–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadipour-Lakmehsari, S.; Driouchi, A.; Lee, S.H.; Kuzmanov, U.; Callaghan, N.I.; Heximer, S.P.; Simmons, C.A.; Yip, C.M.; Gramolini, A.O. Nanoscale Reorganization of Sarcoplasmic Reticulum in Pressure-Overload Cardiac Hypertrophy Visualized by DSTORM. Sci. Rep. 2019, 9, 7837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcina, L.; Cosentino, M.; Musarò, A. Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing. Cells 2020, 9, 1297. [Google Scholar] [CrossRef]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short Telomeres and Stem Cell Exhaustion Model Duchenne Muscular Dystrophy in Mdx/MTR Mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin Expression in Muscle Stem Cells Regulates Their Polarity and Asymmetric Division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef]
- Cooper, S.T.; Head, S.I. Membrane Injury and Repair in the Muscular Dystrophies. Neuroscientist 2015, 21, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Kiriaev, L.; Kueh, S.; Morley, J.W.; North, K.N.; Houweling, P.J.; Head, S.I. Branched Fibers from Old Fast-Twitch Dystrophic Muscles Are the Sites of Terminal Damage in Muscular Dystrophy. Am. J. Physiol. Cell Physiol. 2018, 314, C662–C674. [Google Scholar] [CrossRef] [Green Version]
- Filippelli, R.L.; Chang, N.C. Empowering Muscle Stem Cells for the Treatment of Duchenne Muscular Dystrophy. Cells Tissues Organs 2021, 211, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Naz, F.; Juan, A.H.; Dell’orso, S.; Sartorelli, V. Identification of Skeletal Muscle Satellite Cells by Immunofluorescence with Pax7 and Laminin Antibodies. J. Vis. Exp. 2018, 2018, e57212. [Google Scholar] [CrossRef]
- Ribeiro, A.F.; Souza, L.S.; Almeida, C.F.; Ishiba, R.; Fernandes, S.A.; Guerrieri, D.A.; Santos, A.L.F.; Onofre-Oliveira, P.C.G.; Vainzof, M. Muscle Satellite Cells and Impaired Late Stage Regeneration in Different Murine Models for Muscular Dystrophies. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, F.; Bi, P.; Wang, C.; Li, J.; Liu, X.; Kuang, S. Conditional Loss of Pten in Myogenic Progenitors Leads to Postnatal Skeletal Muscle Hypertrophy but Age-Dependent Exhaustion of Satellite Cells. Cell Rep. 2016, 17, 2340–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Schüler, S.C.; Hüttner, S.S.; von Eyss, B.; von Maltzahn, J. Adult Stem Cells at Work: Regenerating Skeletal Muscle. Cell. Mol. Life Sci. 2019, 76, 2559–2570. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Rudnicki, M.A. Characterizing Satellite Cells and Myogenic Progenitors during Skeletal Muscle Regeneration. In Histochemistry of Single Molecules: Methods and Protocols, Methods in Molecular Biology; Pelliciari, C., Biggiogera, M., Eds.; Humana: New York, NY, USA, 2017; Volume 1560, pp. 179–188. [Google Scholar]
- Schiaffino, S.; Rossi, A.C.; Smerdu, V.; Leinwand, L.A.; Reggiani, C. Developmental Myosins: Expression Patterns and Functional Significance. Skelet. Muscle 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S. Muscle Fiber Type Diversity Revealed by Anti-Myosin Heavy Chain Antibodies. FEBS J. 2018, 285, 3688–3694. [Google Scholar] [CrossRef]
- Sartore, S.; Gorza, L.; Schiaffino, S. Fetal Myosin Heavy Chains in Regenerating Muscle. Nature 1982, 298, 294–296. [Google Scholar] [CrossRef]
- Schiaffino, S.; Gorza, L.; Dones, I.; Cornelio, F.; Sartore, S. Fetal Myosin Immunoreactivity in Human Dystrophic Muscle. Muscle Nerve 1986, 9, 51–58. [Google Scholar] [CrossRef]
- DiMario, J.X.; Uzman, A.; Strohman, R.C. Fiber Regeneration Is Not Persistent in Dystrophic (Mdx) Mouse Skeletal Muscle. Dev. Biol. 1991, 148, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Edwards, B.; Squire, S.E.; Moir, L.; Berg, A.; Babbs, A.; Ramadan, N.; Wood, M.J.; Davies, K.E. Embryonic Myosin Is a Regeneration Marker to Monitor Utrophin-Based Therapies for DMD. Hum. Mol. Genet. 2019, 28, 307–319. [Google Scholar] [CrossRef]
- Lanfossi, M.; Cozzi, F.; Bugini, D.; Colombo, S.; Scarpa, P.; Morandi, L.; Galbiati, S.; Cornelio, F.; Pozza, O.; Mora, M. Development of Muscle Pathology in Canine X-Linked Muscular Dystrophy. I. Delayed Postnatal Maturation of Affected and Normal Muscle as Revealed by Myosin Isoform Analysis and Utrophin Expression. Acta Neuropathol. 1999, 97, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Janghra, N.; Morgan, J.E.; Sewry, C.A.; Wilson, F.X.; Davies, K.E.; Muntoni, F.; Tinsley, J. Correlation of Utrophin Levels with the Dystrophin Protein Complex and Muscle Fibre Regeneration in Duchenne and Becker Muscular Dystrophy Muscle Biopsies. PLoS ONE 2016, 11, e0150818. [Google Scholar] [CrossRef]
- Bucelli, R.C.; Pestronk, A. Immune Myopathies with Perimysial Pathology: Clinical and Laboratory Features. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C. Alkaline Phosphatase. PathologyOutlines.Com Website. Available online: https://www.pathologyoutlines.com/topic/stainsalkphos.html (accessed on 7 December 2022).
- Cai, C.; Anthony, D.C.; Pytel, P. A Pattern-Based Approach to the Interpretation of Skeletal Muscle Biopsies. Mod. Pathol. 2019, 32, 462–483. [Google Scholar] [CrossRef]
- Nguyen, M.; Do, V.; Yell, P.C.; Jo, C.; Liu, J.; Burns, D.K.; Wright, T.; Cai, C. Distinct Tissue Injury Patterns in Juvenile Dermatomyositis Auto-Antibody Subgroups. Acta Neuropathol. Commun. 2020, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Soblechero-Martín, P.; López-Martínez, A.; de la Puente-Ovejero, L.; Vallejo-Illarramendi, A.; Arechavala-Gomeza, V. Utrophin Modulator Drugs as Potential Therapies for Duchenne and Becker Muscular Dystrophies. Neuropathol. Appl. Neurobiol. 2021, 47, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gaschen, F.; Burgunder, J.-M. Utrophin Is a Regeneration-Associated Protein Transiently Present at the Sarcolemma of Regenerating Skeletal Muscle Fibers in Dystrophin-Deficient Hypertrophic Feline Muscular Dystrophy. J. Neuropathol. Exp. Neurol. 1998, 57, 780–790. [Google Scholar] [CrossRef]
- Squire, S.; Raymackers, J.M.; Vandebrouck, C.; Potter, A.; Tinsley, J.; Fisher, R.; Gillis, J.M.; Davies, K.E. Prevention of Pathology in Mdx Mice by Expression of Utrophin: Analysis Using an Inducible Transgenic Expression System. Hum. Mol. Genet. 2002, 11, 3333–3344. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, J.; Deconinck, N.; Fisher, R.; Kahn, D.; Phelps, S.; Gillis, J.-M.; Davies, K. Expression of Full-Length Utrophin Prevents Muscular Dystrophy in Mdx Mice. Nat. Med. 1998, 4, 1441–1444. [Google Scholar] [CrossRef]
- Guiraud, S.; Edwards, B.; Squire, S.E.; Babbs, A.; Shah, N.; Berg, A.; Chen, H.; Davies, K.E. Identification of Serum Protein Biomarkers for Utrophin Based DMD Therapy. Sci. Rep. 2017, 7, 43697. [Google Scholar] [CrossRef] [Green Version]
- Turk, R.; Sterrenburg, E.; de Meijer, E.J.; van Ommen, G.J.B.; den Dunnen, J.T.; ’t Hoen, P.A.C. Muscle Regeneration in Dystrophin-Deficient Mdx Mice Studied by Gene Expression Profiling. BMC Genom. 2005, 6, 98. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, Y.; Ikemoto-Uezumi, M.; Hitachi, K.; Fukada, S.I.; Uezumi, A. Methods for Accurate Assessment of Myofiber Maturity During Skeletal Muscle Regeneration. Front Cell Dev. Biol. 2020, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, G.; Pallavicini, A.; Comelli, A.; Salamon, M.; Bortoletto, G.; Ievolella, C.; Trevisan, S.; Kojić, S.; Vecchia, F.D.; Laveder, P.; et al. FATZ, a Filamin-, Actinin-, and Telethonin-Binding Protein of the Z-Disc of Skeletal Muscle. J. Biol. Chem. 2000, 275, 41234–41242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, F.; Woude, D.L.V.; Tong, H.-Q.; Thompson, T.G.; Watkins, S.C.; Kunkel, L.M.; Beggs, A.H. Myozenin: An α-Actinin- and γ-Filamin-Binding Protein of Skeletal Muscle Z Lines. Proc. Natl. Acad. Sci. USA 2001, 98, 1595–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, W.K. The Essentiality of Histo- and Cytochemical Studies of Skeletal Muscle in the Investigation of Neuromuscular Disease. Neurology 1962, 12, 778. [Google Scholar] [CrossRef] [Green Version]
- Doriguzzi, C.; Mongini, T.; Palmucci, L.; Schiffer, D. A New Method for Myofibrillar Ca++-ATPase Reaction Based on the Use of Metachromatic Dyes: Its Advantages in Muscle Fibre Typing. Histochemistry 1983, 79, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Sgarioto, N.; Norris, B.; Barbat-Artigas, S.; Aubertin-Leheudre, M.; Morais, J.A.; Burelle, Y.; Taivassalo, T.; Hepple, R.T. The Relationship between Muscle Fiber Type-Specific PGC-1α Content and Mitochondrial Content Varies between Rodent Models and Humans. PLoS ONE 2014, 9, e103044. [Google Scholar] [CrossRef] [PubMed]
- YANG, S.; LU, D.; LI, S.; CHEN, G. Histochemical Identification of Skeletal Muscle Fiber Type-Skeletal Muscle in Normal Men and Rats. Acta Histochem. Cytochem. 1990, 23, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Bottinelli, R.; Reggiani, C. Human Skeletal Muscle Fibres: Molecular and Functional Diversity. Prog. Biophys. Mol. Biol. 2000, 73, 195–262. [Google Scholar] [CrossRef] [PubMed]
- Bloemberg, D.; Quadrilatero, J. Rapid Determination of Myosin Heavy Chain Expression in Rat, Mouse, and Human Skeletal Muscle Using Multicolor Immunofluorescence Analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef]
- Anderson, J.E.; Bressler, B.H.; Ovalle, W.K. Functional Regeneration in the Hindlimb Skeletal Muscle of the Mdx Mouse. J. Muscle Res. Cell Motil. 1988, 9, 499–515. [Google Scholar] [CrossRef]
- Yuasa, K.; Nakamura, A.; Hijikata, T.; Takeda, S. Dystrophin Deficiency in Canine X-Linked Muscular Dystrophy in Japan (CXMDJ) Alters Myosin Heavy Chain Expression Profiles in the Diaphragm More Markedly than in the Tibialis Cranialis Muscle. BMC Musculoskelet Disord 2008, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast Muscle Fibers Are Preferentially Affected in Duchenne Muscular Dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Moens, P.; Baatsen, P.H.W.W.; Maréchal, G. Increased Susceptibility of EDL Muscles from Mdx Mice to Damage Induced by Contractions with Stretch. J. Muscle Res. Cell Motil. 1993, 14, 446–451. [Google Scholar] [CrossRef]
- Consolino, C.M.; Brooks, S.V. Susceptibility to Sarcomere Injury Induced by Single Stretches of Maximally Activated Muscles of Mdx Mice. J. Appl. Physiol. 2004, 96, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Kiriaev, L.; Kueh, S.; Morley, J.W.; North, K.N.; Houweling, P.J.; Head, S.I. Lifespan Analysis of Dystrophic Mdx Fast-Twitch Muscle Morphology and Its Impact on Contractile Function. Front Physiol. 2021, 12, 771499. [Google Scholar] [CrossRef]
- Kiriaev, L.; Kueh, S.; Morley, J.W.; Houweling, P.J.; Chan, S.; North, K.N.; Head, S.I. Dystrophin-Negative Slow-Twitch Soleus Muscles Are Not Susceptible to Eccentric Contraction Induced Injury over the Lifespan of the Mdx Mouse. Am. J. Physiol. Cell Physiol. 2021, 321, C704–C720. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Plant, D.R.; Lynch, G.S. Administration of Insulin-like Growth Factor-I Improves Fatigue Resistance of Skeletal Muscles from Dystrophicmdx Mice. Muscle Nerve 2004, 30, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Selsby, J.T.; Morine, K.J.; Pendrak, K.; Barton, E.R.; Sweeney, H.L. Rescue of Dystrophic Skeletal Muscle by PGC-1α Involves a Fast to Slow Fiber Type Shift in the Mdx Mouse. PLoS ONE 2012, 7, e30063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersini, S.; Gilardi, M.; Mora, M.; Krol, S.; Arrigoni, C.; Candrian, C.; Zanotti, S.; Moretti, M. Tackling Muscle Fibrosis: From Molecular Mechanisms to next Generation Engineered Models to Predict Drug Delivery. Adv. Drug Deliv. Rev. 2018, 129, 64–77. [Google Scholar] [CrossRef]
- Gillies, A.R.; Lieber, R.L. Structure and Function of the Skeletal Muscle Extracellular Matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and Adipogenesis Originate from a Common Mesenchymal Progenitor in Skeletal Muscle. J. Cell Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Gutpel, K.M.; Hrinivich, W.T.; Hoffman, L.M. Skeletal Muscle Fibrosis in the Mdx/Utrn+/-Mouse Validates Its Suitability as a Murine Model of Duchenne Muscular Dystrophy. PLoS ONE 2015, 10, e0117306. [Google Scholar] [CrossRef] [Green Version]
- Acuña, M.J.; Pessina, P.; Olguin, H.; Cabrera, D.; Vio, C.P.; Bader, M.; Muñoz-canoves, P.; Santos, R.A.; Cabello-verrugio, C.; Brandan, E. Restoration of Muscle Strength in Dystrophic Muscle by Angiotensin-1-7 through Inhibition of TGF-β Signalling. Hum. Mol. Genet. 2014, 23, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Milad, N.; White, Z.; Tehrani, A.Y.; Sellers, S.; Rossi, F.M.V.; Bernatchez, P. Increased Plasma Lipid Levels Exacerbate Muscle Pathology in the Mdx Mouse Model of Duchenne Muscular Dystrophy. Skelet Muscle 2017, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Ardite, E.; Perdiguero, E.; Vidal, B.; Gutarra, S.; Serrano, A.L.; Muñoz-Cánoves, P. PAI-1 Regulated MiR-21 Defines a Novel Age-Associated Fibrogenic Pathway in Muscular Dystrophy. J. Cell Biol. 2012, 196, 163–175. [Google Scholar] [CrossRef]
- Bernasconi, P.; Blasi, C.D.; Mora, M.; Morandi, L.; Galbiati, S.; Confalonieri, P.; Cornelio, F.; Mantegazza, R. Transforming Growth Factor-B1 and Fibrosis in Congenital Muscular Dystrophies. Neuromuscul. Disord. 1999, 9, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression Levels of TGF-Β1 and CTGF Are Associated with the Severity of Duchenne Muscular Dystrophy. Exp. Ther. Med. 2017, 13, 1209–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juban, G.; Saclier, M.; Yacoub-Youssef, H.; Kernou, A.; Arnold, L.; Boisson, C.; ben Larbi, S.; Magnan, M.; Cuvellier, S.; Théret, M.; et al. AMPK Activation Regulates LTBP4-Dependent TGF-Β1 Secretion by Pro-Inflammatory Macrophages and Controls Fibrosis in Duchenne Muscular Dystrophy. Cell Rep. 2018, 25, 2163–2176.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pessina, P.; Kharraz, Y.; Jardí, M.; Fukada, S.I.; Serrano, A.L.; Perdiguero, E.; Muñoz-Cánoves, P. Fibrogenic Cell Plasticity Blunts Tissue Regeneration and Aggravates Muscular Dystrophy. Stem Cell Rep. 2015, 4, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuña, M.J.; Salas, D.; Córdova-Casanova, A.; Cruz-Soca, M.; Céspedes, C.; Vio, C.P.; Brandan, E. Blockade of Bradykinin Receptors Worsens the Dystrophic Phenotype of Mdx Mice: Differential Effects for B1 and B2 Receptors. J. Cell Commun. Signal. 2018, 12, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the Process of Fibrosis in Duchenne Muscular Dystrophy. Biomed Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Haginoya, K.; Wu, Y.; Chiba, Y.; Nakanishi, T.; Onuma, A.; Sato, Y.; Takigawa, M.; Iinuma, K.; Tsuchiya, S. Connective Tissue Growth Factor Is Overexpressed in Muscles of Human Muscular Dystrophy. J. Neurol. Sci. 2008, 267, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; You, W.; Valencak, T.G.; Shan, T. Bidirectional Roles of Skeletal Muscle Fibro-Adipogenic Progenitors in Homeostasis and Disease. Ageing Res. Rev. 2022, 80, 101682. [Google Scholar] [CrossRef]
- Contreras, O.; Rebolledo, D.L.; Oyarzún, J.E.; Olguín, H.C.; Brandan, E. Connective Tissue Cells Expressing Fibro/Adipogenic Progenitor Markers Increase under Chronic Damage: Relevance in Fibroblast-Myofibroblast Differentiation and Skeletal Muscle Fibrosis. Cell Tissue Res. 2016, 364, 647–660. [Google Scholar] [CrossRef]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle Injury Activates Resident Fibro/Adipogenic Progenitors That Facilitate Myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Olson, L.E.; Soriano, P. Increased PDGFRα Activation Disrupts Connective Tissue Development and Drives Systemic Fibrosis. Dev. Cell 2009, 16, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ieronimakis, N.; Hays, A.; Prasad, A.; Janebodin, K.; Duffield, J.S.; Reyes, M. PDGFRα Signalling Promotes Fibrogenic Responses in Collagen-Producing Cells in Duchenne Muscular Dystrophy. J. Pathol. 2016, 240, 410–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Haginoya, K.; Sun, G.; Dai, H.; Onuma, A.; Iinuma, K. Platelet-Derived Growth Factor and Its Receptors Are Related to the Progression of Human Muscular Dystrophy: An Immunohistochemical Study. J. Pathol. 2003, 201, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Stratos, I.; Behrendt, A.K.; Anselm, C.; Gonzalez, A.; Mittlmeier, T.; Vollmar, B. Inhibition of TNF-α Restores Muscle Force, Inhibits Inflammation, and Reduces Apoptosis of Traumatized Skeletal Muscles. Cells 2022, 11, 2397. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.L.; Muñoz-Cánoves, P. Fibrosis Development in Early-Onset Muscular Dystrophies: Mechanisms and Translational Implications. Semin. Cell Dev. Biol. 2017, 64, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 Innate Signals Stimulate Fibro/Adipogenic Progenitors to Facilitate Muscle Regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Silva, K.A.S.; Dong, Y.; Zhang, L. Glucocorticoids Increase Adipocytes in Muscle by Affecting IL-4 Regulated FAP Activity. FASEB J. 2014, 28, 4123–4132. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.-H.; Chang, Y.; Reed, N.I.; Sheppard, D. Smooth Muscle Actin Is an Inconsistent Marker of Fibroblasts Responsible for Force-Dependent TGF Activation or Collagen Production across Multiple Models of Organ Fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L824–L836. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Wang, X.; Sun, K.H.; Zhou, L. A-Smooth Muscle Actin Is Not a Marker of Fibrogenic Cell Activity in Skeletal Muscle Fibrosis. PLoS ONE 2018, 13, e0191031. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Zhao, X.S.; Fields, M.; Ransohoff, R.M.; Zhou, L. Imatinib Attenuates Skeletal Muscle Dystrophy in Mdx Mice. FASEB J. 2009, 23, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhao, L.; Jiang, J.; Li, S.; Sun, Z.; Huang, X.; Li, C.; Wang, T.; Sun, L.; Li, X.; et al. A Potential Therapeutic Effect of Catalpol in Duchenne Muscular Dystrophy Revealed by Binding with TAK1. J. Cachexia Sarcopenia Muscle 2020, 11, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, P.; Gualandi, F.; Gara, S.K.; Grumati, P.; Zamparelli, A.; Martoni, E.; Pellegrini, C.; Merlini, L.; Ferlini, A.; Bonaldo, P.; et al. Expression of Collagen VI A5 and A6 Chains in Human Muscle and in Duchenne Muscular Dystrophy-Related Muscle Fibrosis. Matrix Biol. 2012, 31, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro–Adipogenic Progenitors Cross-Talk in Skeletal Muscle: The Social Network. Front. Physiol. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Mehlem, A.; Hagberg, C.E.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of Neutral Lipids by Oil Red O for Analyzing the Metabolic Status in Health and Disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, R.; Schaart, G.; Hesselink, M.K. Optimisation of Oil Red O Staining Permits Combination with Immunofluorescence and Automated Quantification of Lipids. Histochem. Cell Biol. 2001, 116, 63–68. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.I.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal Progenitors Distinct from Satellite Cells Contribute to Ectopic Fat Cell Formation in Skeletal Muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Manera, J.D.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic Progenitors Are Responsible for Muscle Loss in Limb Girdle Muscular Dystrophy 2B. Nat. Commun. 2019, 10, 2430. [Google Scholar] [CrossRef] [Green Version]
- Timpani, C.A.; Goodman, C.A.; Stathis, C.G.; White, J.D.; Mamchaoui, K.; Butler-Browne, G.; Gueven, N.; Hayes, A.; Rybalka, E. Adenylosuccinic Acid Therapy Ameliorates Murine Duchenne Muscular Dystrophy. Sci. Rep. 2020, 10, 1125. [Google Scholar] [CrossRef] [Green Version]
- Bonucci, E.; Sadun, R. Experimental Calcification of the Myocardium. Ultrastructural and Histochemical Investigations. Am. J. Pathol. 1973, 71, 167–192. [Google Scholar]
- BODENSTEINER, J.B.; ENGEL, A.G. Intracellular Calcium Accumulation in Duchenne Dystrophy and Other Myopathies: A Study of 567,000 Muscle Fibers in 114 Biopsies. Neurology 1978, 28, 439. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, N.; Ohno, T.; Nagata, Y.; Shiozuka, M.; Kogure, T.; Matsuda, R. Ectopic Calcification Is Caused by Elevated Levels of Serum Inorganic Phosphate in Mdx Mice. Cell Struct Funct. 2009, 34, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Wada, E.; Yoshida, M.; Kojima, Y.; Nonaka, I.; Ohashi, K.; Nagata, Y.; Shiozuka, M.; Date, M.; Higashi, T.; Nishino, I.; et al. Dietary Phosphorus Overload Aggravates the Phenotype of the Dystrophin-Deficient Mdx Mouse. Am. J. Pathol. 2014, 184, 3094–3104. [Google Scholar] [CrossRef]
- Young, C.N.J.; Gosselin, M.R.F.; Rumney, R.; Oksiejuk, A.; Chira, N.; Bozycki, L.; Matryba, P.; Łukasiewicz, K.; Kao, A.P.; Dunlop, J.; et al. Total Absence of Dystrophin Expression Exacerbates Ectopic Myofiber Calcification and Fibrosis and Alters Macrophage Infiltration Patterns. Am. J. Pathol. 2020, 190, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Barthélémy, I.; Uriarte, A.; Drougard, C.; Unterfinger, Y.; Thibaud, J.L.; Blot, S. Effects of an Immunosuppressive Treatment in the GRMD Dog Model of Duchenne Muscular Dystrophy. PLoS ONE 2012, 7, e48478. [Google Scholar] [CrossRef] [Green Version]
- Rumney, R.M.H.; Róg, J.; Chira, N.; Kao, A.P.; Al-Khalidi, R.; Górecki, D.C. P2X7 Purinoceptor Affects Ectopic Calcification of Dystrophic Muscles. Front. Pharmacol. 2022, 13, 2538. [Google Scholar] [CrossRef] [PubMed]
- Boulman, N.; Slobodin, G.; Rozenbaum, M.; Rosner, I. Calcinosis in Rheumatic Diseases. Semin. Arthritis Rheum. 2005, 34, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Mignemi, N.A.; Yuasa, M.; Baker, C.E.; Moore, S.N.; Ihejirika, R.C.; Oelsner, W.K.; Wallace, C.S.; Yoshii, T.; Okawa, A.; Revenko, A.S.; et al. Plasmin Prevents Dystrophic Calcification After Muscle Injury. J. Bone Miner. Res. 2017, 32, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Mázala, D.A.G.; Novak, J.S.; Hogarth, M.W.; Nearing, M.; Adusumalli, P.; Tully, C.B.; Habib, N.F.; Gordish-Dressman, H.; Chen, Y.W.; Jaiswal, J.K.; et al. TGF-β-Driven Muscle Degeneration and Failed Regeneration Underlie Disease Onset in a DMD Mouse Model. JCI Insight 2020, 5, e135703. [Google Scholar] [CrossRef]
- Puchtler, H.; Meloan, S.N.; Terry, M.S. On the history and mechanism of alizarin and alizarin red s stains for calcium. J. Histochem. Cytochem. 1969, 17, 110–124. [Google Scholar] [CrossRef]
- Meloan, S.N.; Puchtler, H. Chemical Mechanisms of Staining Methods Von Kossa’s Technique: What von Kossa Really Wrote and a Modified Reaction for Selective Demonstration of Inorganic Phosphates. J. Histotechnol. 2013, 8, 11–13. [Google Scholar] [CrossRef]
- Chen, Q.; Bei, J.J.; Liu, C.; Feng, S.B.; Zhao, W.B.; Zhou, Z.; Yu, Z.P.; Du, X.J.; Hu, H.Y. HMGB1 Induces Secretion of Matrix Vesicles by Macrophages to Enhance Ectopic Mineralization. PLoS ONE 2016, 11, e0156686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigueur, D.; Lyons, K.M. Whole-Mount Skeletal Staining. Methods Mol. Biol. 2014, 1130, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Ljubicic, V. Recent Insights into Neuromuscular Junction Biology in Duchenne Muscular Dystrophy: Impacts, Challenges, and Opportunities. EBioMedicine 2020, 61, 103032. [Google Scholar] [CrossRef] [PubMed]
- Banks, G.B.; Chamberlain, J.S.; Froehner, S.C. Truncated Dystrophins Can Influence Neuromuscular Synapse Structure. Mol. Cell. Neurosci. 2009, 40, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Yang, L.; Li, Q.; Cao, J.; Yang, J.; Chen, F.; Wang, Y.; Zhang, C. The Absence of Dystrophin Rather than Muscle Degeneration Causes Acetylcholine Receptor Cluster Defects in Dystrophic Muscle. Neuroreport 2012, 23, 82–87. [Google Scholar] [CrossRef]
- Kong, J.; Anderson, J.E. Dystrophin Is Required for Organizing Large Acetylcholine Receptor Aggregates. Brain Res. 1999, 839, 298–304. [Google Scholar] [CrossRef]
- Haddix, S.G.; Lee, Y.i.; Kornegay, J.N.; Thompson, W.J. Cycles of Myofiber Degeneration and Regeneration Lead to Remodeling of the Neuromuscular Junction in Two Mammalian Models of Duchenne Muscular Dystrophy. PLoS ONE 2018, 13, e0205926. [Google Scholar] [CrossRef]
- Li, Y.; Lee, Y.i.; Thompson, W.J. Changes in Aging Mouse Neuromuscular Junctions Are Explained by Degeneration and Regeneration of Muscle Fiber Segments at the Synapse. J. Neurosci. 2011, 31, 14910–14919. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Thompson, W.J. Nerve Terminal Growth Remodels Neuromuscular Synapses in Mice Following Regeneration of the Postsynaptic Muscle Fiber. J. Neurosci. 2011, 31, 13191–13203. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez Cruz, P.M.; Cossins, J.; Beeson, D.; Vincent, A. The Neuromuscular Junction in Health and Disease: Molecular Mechanisms Governing Synaptic Formation and Homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef]
- Grady, R.M.; Zhou, H.; Cunningham, J.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R. Maturation and Maintenance of the Neuromuscular Synapse: Genetic Evidence for Roles of the Dystrophin-Glycoprotein Complex. Neuron 2000, 25, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajanovska, S.; Ban, J.; Huang, J.; Gregorevic, P.; Morsch, M.; Allen, D.G.; Phillips, W.D. Muscle Specific Kinase Protects Dystrophic Mdx Mouse Muscles from Eccentric Contraction-Induced Loss of Force-Producing Capacity. J. Physiol. 2019, 597, 4831–4850. [Google Scholar] [CrossRef] [PubMed]
- Hui, T.; Jing, H.; Zhou, T.; Chen, P.; Liu, Z.; Dong, X.; Yan, M.; Ren, D.; Zou, S.; Wang, S.; et al. Increasing LRP4 Diminishes Neuromuscular Deficits in a Mouse Model of Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2021, 30, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, F.; Engel, A.G.; Gomez, M.R. Duchenne Dystrophy. II. Morphometric Study of Motor End-Plate Fine Structure. Brain 1974, 97, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Harriman, D.G.F. A comparison of the fine structure of motor end-plates in duchenne dystrophy and in human neurogenic diseases. J. Neurol. Sci. 1976, 28, 233–247. [Google Scholar] [CrossRef]
- Turney, S.G.; Walsh, M.K.; Lichtman, J.W. In Vivo Imaging of the Developing Neuromuscular Junction in Neonatal Mice. Cold Spring Harb. Protoc. 2012, 7, 1166–1176. [Google Scholar] [CrossRef]
- Pratt, S.J.P.; Valencia, A.P.; Le, G.K.; Shah, S.B.; Lovering, R.M. Pre- and Postsynaptic Changes in the Neuromuscular Junction in Dystrophic Mice. Front. Physiol. 2015, 6, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Bermedo-García, F.; Zelada, D.; Martínez, E.; Tabares, L.; Henríquez, J.P. Functional Regeneration of the Murine Neuromuscular Synapse Relies on Long-Lasting Morphological Adaptations. BMC Biol. 2022, 20, 1–18. [Google Scholar] [CrossRef]
- Pratt, S.J.P.; Iyer, S.R.; Shah, S.B.; Lovering, R.M. Imaging Analysis of the Neuromuscular Junction in Dystrophic Muscle. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1687, pp. 57–72. [Google Scholar]
- Wu, H.; Mei, L. Morphological Analysis of Neuromuscular Junctions by Immunofluorescent Staining of Whole-Mount Mouse Diaphragms. Methods Mol. Biol. 2013, 1018, 277–285. [Google Scholar] [CrossRef]
- Minty, G.; Hoppen, A.; Boehm, I.; Alhindi, A.; Gibb, L.; Potter, E.; Wagner, B.C.; Miller, J.; Skipworth, R.J.E.; Gillingwater, T.H.; et al. ANMJ-Morph: A Simple Macro for Rapid Analysis of Neuromuscular Junction Morphology. R. Soc. Open Sci. 2020, 7, 200128. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.A.; Reich, C.D.; Dissanayake, K.N.; Kristmundsdottir, F.; Findlater, G.S.; Ribchester, R.R.; Simmen, M.W.; Gillingwater, T.H. NMJ-Morph Reveals Principal Components of Synaptic Morphology Influencing Structure—Function Relationships at the Neuromuscular Junction. Open Biol. 2017, 6, 160240. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.A.; Harrison, C.; Eaton, S.L.; Llavero Hurtado, M.; Graham, L.C.; Alkhammash, L.; Oladiran, O.A.; Gale, A.; Lamont, D.J.; Simpson, H.; et al. Cellular and Molecular Anatomy of the Human Neuromuscular Junction. Cell Rep. 2017, 21, 2348–2356. [Google Scholar] [CrossRef]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins Carrying Spectrin-like Repeats 16 and 17 Anchor NNOS to the Sarcolemma and Enhance Exercise Performance in a Mouse Model of Muscular Dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Zhao, J.; Yue, Y.; Duan, D. A2 and A3 Helices of Dystrophin R16 and R17 Frame a Microdomain in the A1 Helix of Dystrophin R17 for Neuronal NOS Binding. Proc. Natl. Acad. Sci. USA 2013, 110, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Kodippili, K.; Hakim, C.H.; Yang, H.T.; Pan, X.; Yang, N.N.; Laughlin, M.H.; Terjung, R.L.; Duan, D. Nitric Oxide-Dependent Attenuation of Noradrenaline-Induced Vasoconstriction Is Impaired in the Canine Model of Duchenne Muscular Dystrophy. J. Physiol. 2018, 596, 5199–5216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.D.; Sander, M.; Lau, K.S.; Huang, P.L.; Stull, J.T.; Victor, R.G. Impaired Metabolic Modulation Of-Adrenergic Vasoconstriction in Dystrophin-Deficient Skeletal Muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 15090–15095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional Muscle Ischemia in Neuronal Nitric Oxide Synthase-Deficient Skeletal Muscle of Children with Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.-P.; Gherardi, R.K.; Christov, C. Endomysial Fibrosis in Duchenne Muscular Dystrophy: A Marker of Poor Outcome Associated With Macrophage Alternative Activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, M.; Asakura, Y.; Murakonda, B.S.R.; Pengo, T.; Latroche, C.; Chazaud, B.; McLoon, L.K.; Asakura, A. Muscle Satellite Cell Cross-Talk with a Vascular Niche Maintains Quiescence via VEGF and Notch Signaling. Cell Stem Cell 2018, 23, 530–543.e9. [Google Scholar] [CrossRef] [Green Version]
- Ennen, J.P.; Verma, M.; Asakura, A. Vascular-Targeted Therapies for Duchenne Muscular Dystrophy. Skelet Muscle 2013, 23, 9. [Google Scholar] [CrossRef] [Green Version]
- Leuchtmann, A.B.; Mueller, S.M.; Aguayo, D.; Petersen, J.A.; Ligon-Auer, M.; Flück, M.; Jung, H.H.; Toigo, M. Resistance Training Preserves High-Intensity Interval Training Induced Improvements in Skeletal Muscle Capillarization of Healthy Old Men: A Randomized Controlled Trial. Sci. Rep. 2020, 10, 6578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corliss, B.A.; Mathews, C.; Doty, R.; Rohde, G.; Peirce, S.M. Methods to Label, Image, and Analyze the Complex Structural Architectures of Microvascular Networks. Microcirculation 2019, 26, e12520. [Google Scholar] [CrossRef] [PubMed]
- Al-Shammari, A.A.; Kissane, R.W.P.; Holbek, S.; Mackey, A.L.; Andersen, T.R.; Gaffney, E.A.; Kjaer, M.; Egginton, S. Integrated Method for Quantitative Morphometry and Oxygen Transport Modeling in Striated Muscle. J. Appl. Physiol. 2019, 126, 544–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, A.; Trewin, A.J.; Sadler, K.J.; Laird, C.; della Gatta, P.A.; Russell, A.P. Sensitivity to Behavioral Stress Impacts Disease Pathogenesis in Dystrophin-Deficient Mice. FASEB J. 2021, 35, e12520. [Google Scholar] [CrossRef] [PubMed]
- Bailly, M.; Féasson, L.; Pereira, B.; Boileau, A.; Hourdé, C.; Germain, N.; Galusca, B.; Courteix, D.; Thivel, D.; Verney, J. Two New Reliable Immunohistochemical Methods for Simultaneous Identification of Capillaries, the Three Types of Fibers and Basal Lamina in Human Skeletal Muscle. Histochem. Cell Biol. 2020, 154, 327–337. [Google Scholar] [CrossRef]
- Woodfin, A.; Voisin, M.B.; Nourshargh, S. PECAM-1: A Multi-Functional Molecule in Inflammation and Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2514–2523. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, L.V.B.; Davison, K.; Johnson, M.A.; Slater, C.R.; Young, C.; Bhattacharya, S.; Gardner-Medwin, D.; Harris, J.B. Dystrophin in Skeletal Muscle II. Immunoreactivity in Patients with Xp21 Muscular Dystrophy. Neurol. Sci. 1989, 94, 137–146. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Morgan, J.E.; Watkins, S.C.; Partridge, T.A. Somatic Reversion/Suppression of the Mouse Mdx Phenotype in Vivo. J. Neurol. Sci. 1990, 99, 9–25. [Google Scholar] [CrossRef]
- Schatzberg, S.J.; Anderson, L.V.B.; Wilton, S.D.; Kornegay, J.N.; Mann, C.J.; Solomon, G.G.; Sharp, N.J.H. Alternative dystrophin gene transcripts in golden retriever muscular dystrophy. Muscle Nerve 1998, 21, 991–998. [Google Scholar] [CrossRef]
- Valentine, B.A.; Cooper, B.J.; Cummings, J.F.; de Lahunta, A.; Valentine, B.A. Canine X-Linked Muscular Dystrophy: Morphologic Lesions. J. Neurol. Sci. 1990, 97, 1–23. [Google Scholar] [CrossRef]
- Lu, Q.L. Revertant Phenomenon in DMD and LGMD2I and Its Therapeutic Implications: A Review of Study Under Mentorship of Terrence Partridge. J. Neuromuscul. Dis. 2021, 8, S359–S367. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J.; Coovert, D.D.; Bulman, D.E.; Ray, P.N.; Mendell, J.R.; Burghes, A.H.M. Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): Evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am. J. Hum. Genet. 1992, 50, 950–959. [Google Scholar] [PubMed]
- Lu, Q.L.; Morris, G.E.; Wilton, S.D.; Ly, T.; Artem’yeva, O.V.; Strong, P.; Partridge, T.A. Massive Idiosyncratic Exon Skipping Corrects the Nonsense Mutation in Dystrophic Mouse Muscle and Produces Functional Revertant Fibers by Clonal Expansion. J. Cell Biol. 2000, 148, 985–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, G.E.; Lu, Q.L.; Partridge, T.A.; Chamberlain, J.S. Suppression of Revertant Fibers in Mdx Mice by Expression of a Functional Dystrophin. Hum. Mol. Genet. 2001, 10, 2745–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallgren-Pettersson, C.; Jasani, B.; Rosser, L.G.; Lazarou, L.P.; Nicholson, L.V.; Clarke, A. Immunohistological Evidence for a Second or Somatic Mutations as the Underlying Cause of Dystrophin Expression by Isolated Fibres in Xp21 Muscular Dystrophy of Duchenne-Type Severity. J. Neurol. Sci. 1993, 118, 56–63. [Google Scholar] [CrossRef]
- Wilton, S.D.; Dye, D.E.; Blechynden, L.M.; Laing, N.G. Revertant fibres: A possible genetic therapy for Duchenne muscular dystrophy? Neuromuscul. Disord. 1997, 7, 329–335. [Google Scholar] [CrossRef]
- Fanin, M.; Danieli, A.; Cadaldini, M.; Miorin, M.; Angelini, C. Dystrophin-positive fibers in duchenne dystrophy: Origin and correlation to clinical course. Nerve Muscle 1995, 18, 1115–1120. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.L.; Morgan, J.E.; Davies, K.E.; Fisher, R.; Takeda, S.; Partridge, T.A. Expansion of Revertant Fibers in Dystrophic Mdx Muscles Reflects Activity of Muscle Precursor Cells and Serves as an Index of Muscle Regeneration. J. Cell Sci. 2006, 119, 2679–2687. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.A.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional Amounts of Dystrophin Produced by Skipping the Mutated Exon in the Mdx Dystrophic Mouse. Nat. Med. 2003, 9, 1009–1014. [Google Scholar] [CrossRef]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2004, 102, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Dzierlega, K.; Yokota, T. Optimization of Antisense-Mediated Exon Skipping for Duchenne Muscular Dystrophy. Gene Ther. 2020, 27, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Aoki, Y.; Miskew, B.; Panesar, D.; Touznik, A.; Nagata, T.; Tanihata, J.; Nakamura, A.; Nagaraju, K.; Yokota, T. Long-Term Efficacy of Systemic Multiexon Skipping Targeting Dystrophin Exons 45-55 with a Cocktail of Vivo-Morpholinos in Mdx52 Mice. Mol. Ther. Nucleic. Acids 2015, 4, e225. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Nguyen, G.H.; Man, N.T.; Sewry, C.A.; Morris, G.E. Exon-Specific Dystrophin Antibodies for Studies of Duchenne Muscular Dystrophy. Transl. Neurosci. 2010, 1, 233–237. [Google Scholar] [CrossRef]
- Kodippili, K.; Vince, L.; Shin, J.H.; Yue, Y.; Morris, G.E.; McIntosh, M.A.; Duan, D. Characterization of 65 Epitope-Specific Dystrophin Monoclonal Antibodies in Canine and Murine Models of Duchenne Muscular Dystrophy by Immunostaining and Western Blot. PLoS ONE 2014, 9, e88280. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.R.; Barton, E.R. SMASH—Semi-Automatic Muscle Analysis Using Segmentation of Histology: A MATLAB Application. Skelet. Muscle 2014, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Murach, K.A.; Vechetti, I.J., Jr.; Fry, C.S.; Vickery, C.; Peterson, C.A.; McCarthy, J.J.; Campbell, K.S. INNOVATIVE METHODOLOGY MyoVision: Software for Automated High-Content Analysis of Skeletal Muscle Immunohistochemistry. J. Appl. Physiol. 2018, 124, 40–51. [Google Scholar] [CrossRef]
- Mula, J.; Lee, J.D.; Liu, F.; Yang, L.; Peterson, C.A. Automated Image Analysis of Skeletal Muscle Fiber Cross-Sectional Area. J. Appl. Physiol. 2013, 114, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Fry, C.S.; Mula, J.; Jackson, J.R.; Lee, J.D.; Peterson, C.A.; Yang, L. Automated Fiber-Type-Specific Cross-Sectional Area Assessment and Myonuclei Counting in Skeletal Muscle. J. Appl. Physiol. 2013, 115, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Desgeorges, T.; Liot, S.; Lyon, S.; Bouvière, J.; Kemmel, A.; Trignol, A.; Rousseau, D.; Chapuis, B.; Gondin, J.; Mounier, R.; et al. Open-CSAM, a New Tool for Semi-Automated Analysis of Myofiber Cross-Sectional Area in Regenerating Adult Skeletal Muscle. Skelet. Muscle 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Lau, Y.S.; Xu, L.; Gao, Y.; Han, R. Automated Muscle Histopathology Analysis Using CellProfiler. Skelet. Muscle 2018, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Fernandez, P.C.; Periou, B.; Decrouy, X.; Relaix, F.; Authier, F.J. Automated Image-Analysis Method for the Quantification of Fiber Morphometry and Fiber Type Population in Human Skeletal Muscle. Skelet. Muscle 2019, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmeister, K.D.; Gröger, M.; Aman, M.; Willensdorfer, A.; Manzano-Szalai, K.; Salminger, S.; Aszmann, O.C. A Rapid Automated Protocol for Muscle Fiber Population Analysis in Rat Muscle Cross Sections Using Myosin Heavy Chain Immunohistochemistry. J. Vis. Exp. 2017, 2017, e55441. [Google Scholar] [CrossRef]
- Bergmeister, K.D.; Gröger, M.; Aman, M.; Willensdorfer, A.; Manzano-Szalai, K.; Salminger, S.; Aszmann, O.C. Automated Muscle Fiber Type Population Analysis with ImageJ of Whole Rat Muscles Using Rapid Myosin Heavy Chain Immunohistochemistry. Muscle Nerve 2016, 54, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Encarnacion-Rivera, L.; Foltz, S.; Hartzell, H.C.; Choo, H. Myosoft: An Automated Muscle Histology Analysis Tool Using Machine Learning Algorithm Utilizing FIJI/ImageJ Software. PLoS ONE 2020, 15, e0229041. [Google Scholar] [CrossRef] [PubMed]
- Kastenschmidt, J.M.; Ellefsen, K.L.; Mannaa, A.H.; Giebel, J.J.; Yahia, R.; Ayer, R.E.; Pham, P.; Rios, R.; Vetrone, S.A.; Mozaffar, T.; et al. QuantiMus: A Machine Learning-Based Approach for High Precision Analysis of Skeletal Muscle Morphology. Front. Physiol. 2019, 10, 1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miazaki, M.; Viana, M.P.; Yang, Z.; Comin, C.H.; Wang, Y.; da F Costa, L.; Xu, X. Automated High-Content Morphological Analysis of Muscle Fiber Histology. Comput. Biol. Med. 2015, 63, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Sanz, G.; Manuel Martínez-Aranda, L.; Tesch, P.A.; Fernandez-Gonzalo, R.; Tommy, X.; Lundberg, R. INNOVATIVE METHODOLOGY Muscle2View, a CellProfiler Pipeline for Detection of the Capillary-to-Muscle Fiber Interface and High-Content Quantification of Fiber Type-Specific Histology. J. Appl. Physiol. 2019, 127, 1698–1709. [Google Scholar] [CrossRef]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A Generalist Algorithm for Cellular Segmentation. Nat. Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Waisman, A.; Norris, A.M.; Elías Costa, M.; Kopinke, D. Automatic and Unbiased Segmentation and Quantification of Myofibers in Skeletal Muscle. Sci. Rep. 2021, 11, 11793. [Google Scholar] [CrossRef]
- Babcock, L.W.; Hanna, A.D.; Agha, N.H.; Hamilton, S.L. MyoSight—Semi-Automated Image Analysis of Skeletal Muscle Cross Sections. Skelet. Muscle 2020, 10, 33. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubuisson, N.; Versele, R.; Planchon, C.; Selvais, C.M.; Noel, L.; Abou-Samra, M.; Davis-López de Carrizosa, M.A. Histological Methods to Assess Skeletal Muscle Degeneration and Regeneration in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 16080. https://doi.org/10.3390/ijms232416080
Dubuisson N, Versele R, Planchon C, Selvais CM, Noel L, Abou-Samra M, Davis-López de Carrizosa MA. Histological Methods to Assess Skeletal Muscle Degeneration and Regeneration in Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2022; 23(24):16080. https://doi.org/10.3390/ijms232416080
Chicago/Turabian StyleDubuisson, Nicolas, Romain Versele, Chloé Planchon, Camille M. Selvais, Laurence Noel, Michel Abou-Samra, and María A. Davis-López de Carrizosa. 2022. "Histological Methods to Assess Skeletal Muscle Degeneration and Regeneration in Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 23, no. 24: 16080. https://doi.org/10.3390/ijms232416080