
µ-Opioid Receptors Expressed by Intrinsically Photosensitive Retinal Ganglion Cells Contribute to Morphine-Induced Behavioral Sensitization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
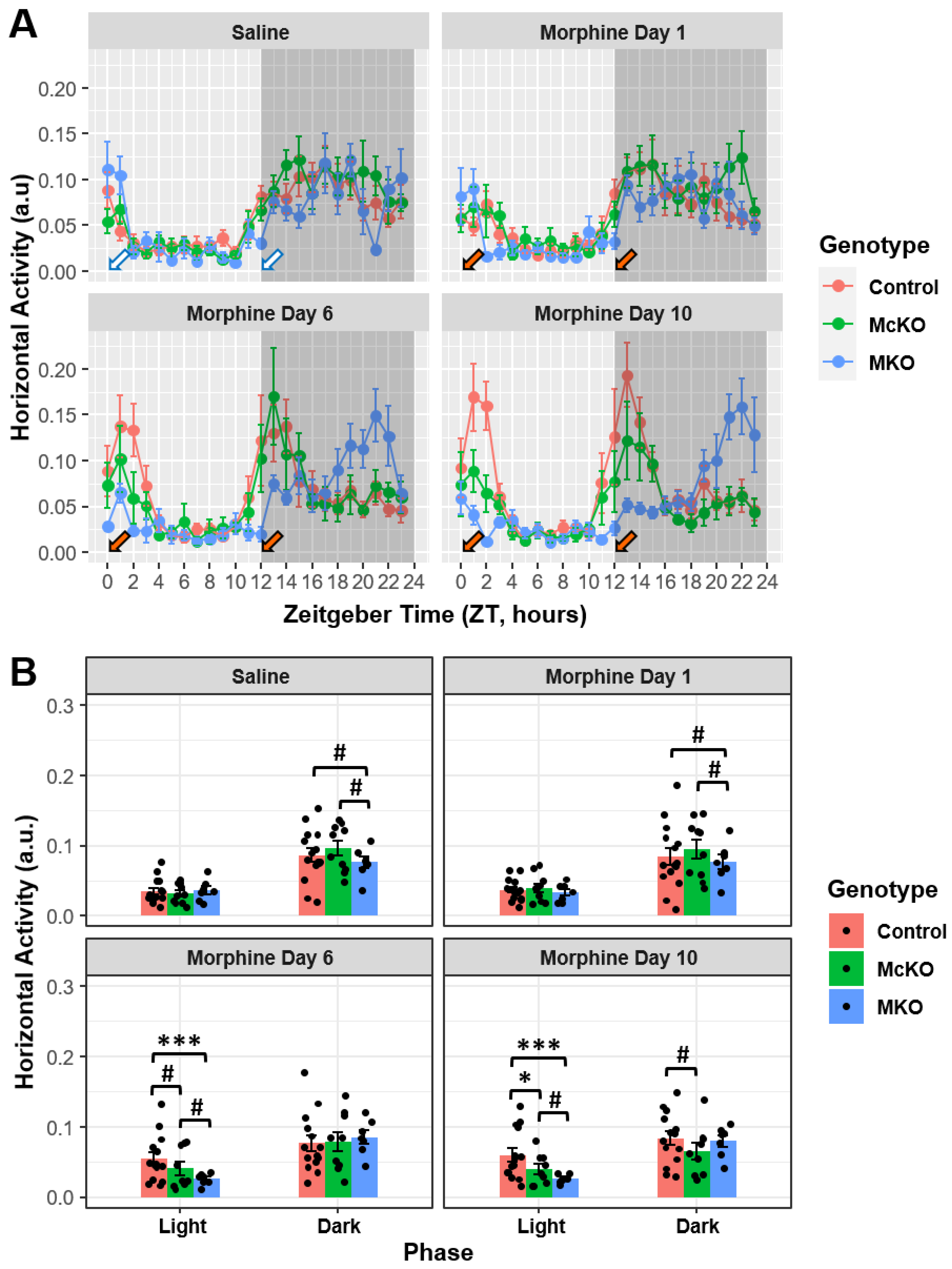
2.1. Prolonged Morphine Exposure Has Differential Effects on Locomotor Activity in Control, McKO, and MKO Mice
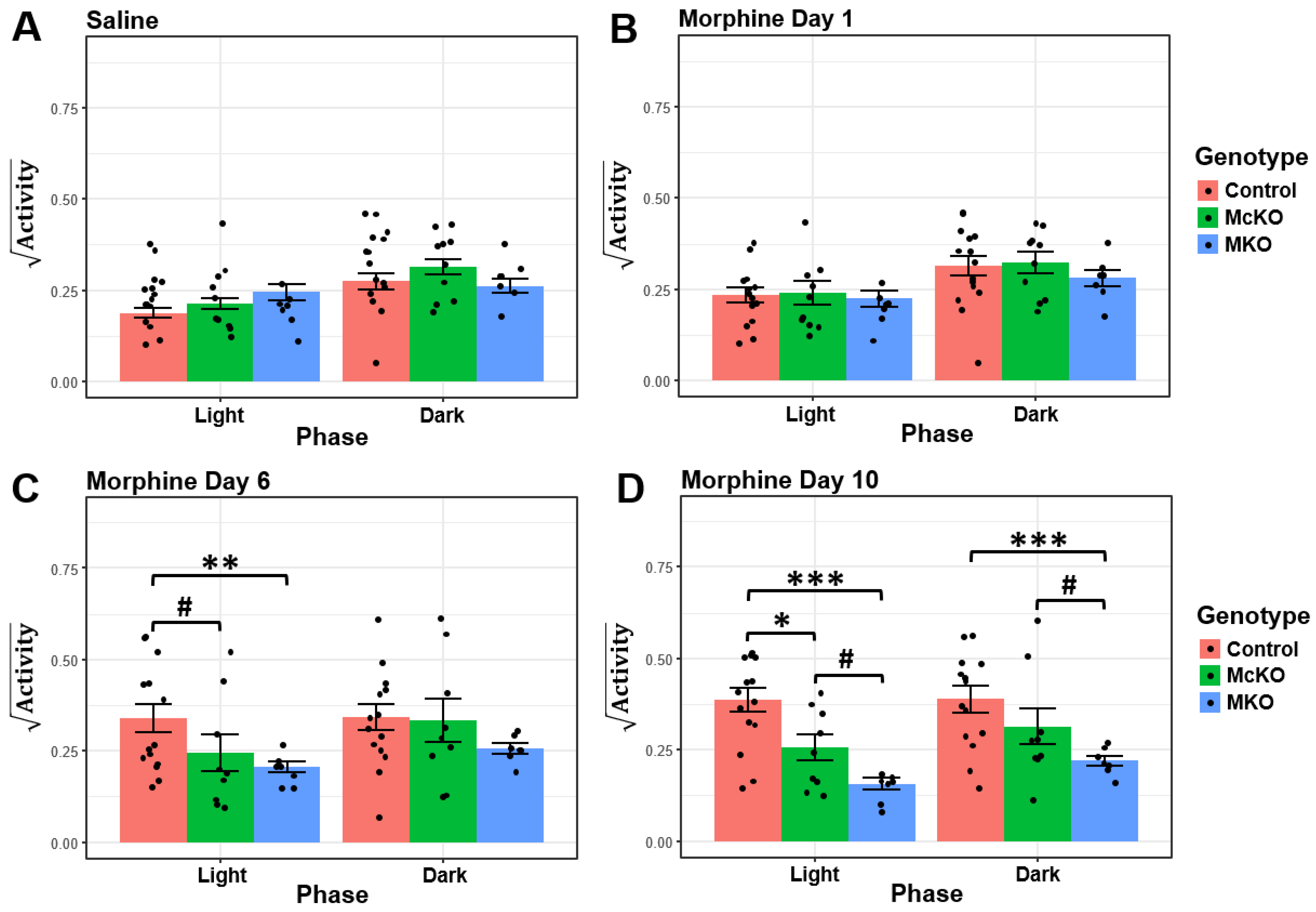
2.2. McKO Mice Display Diminished Morphine-Induced Behavioral Sensitization Compared with Control Littermates
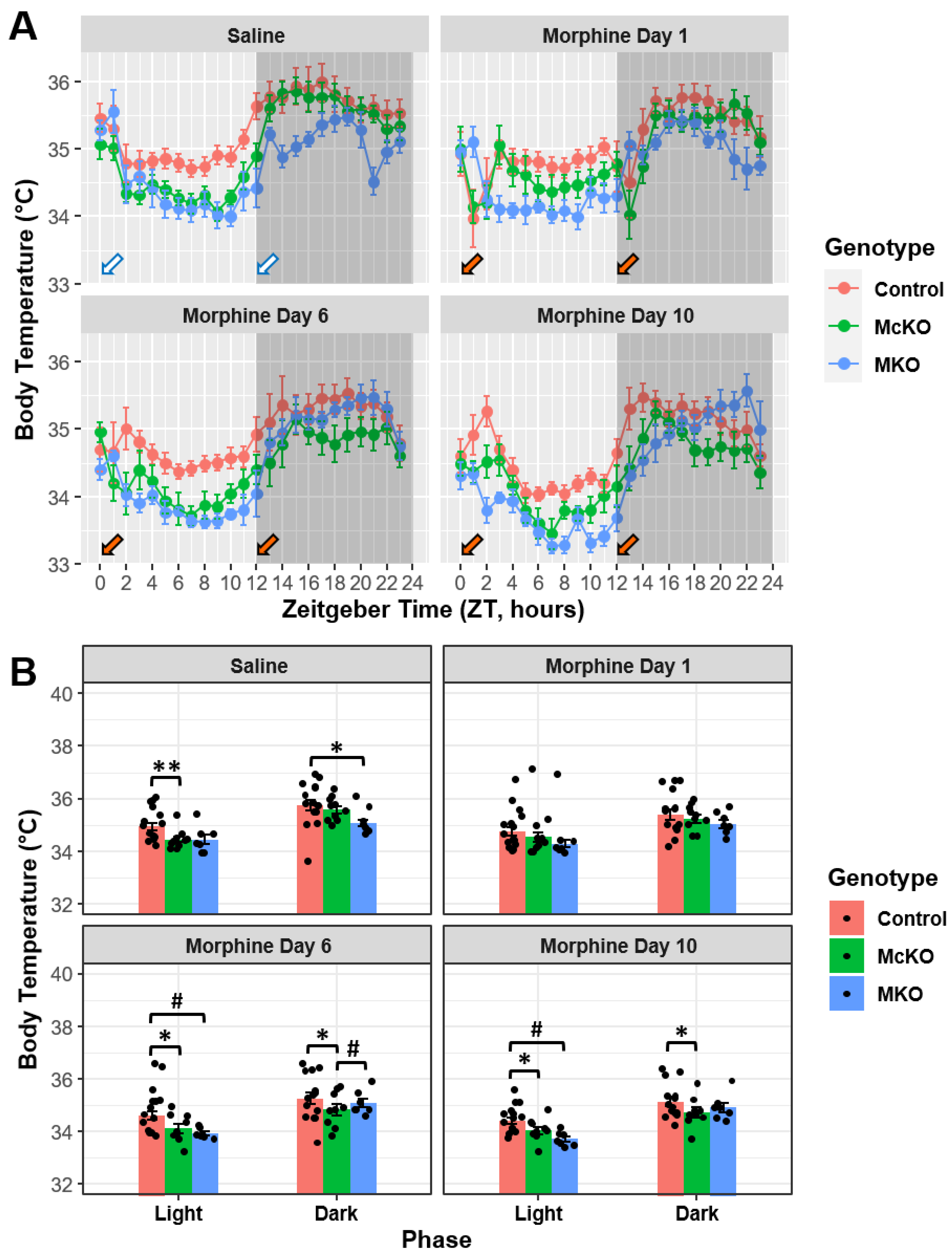
2.3. Morphine Has Disparate Effects on Body Temperature across the Chronic Treatment Paradigm in Mice Differentially Expressing the MOR
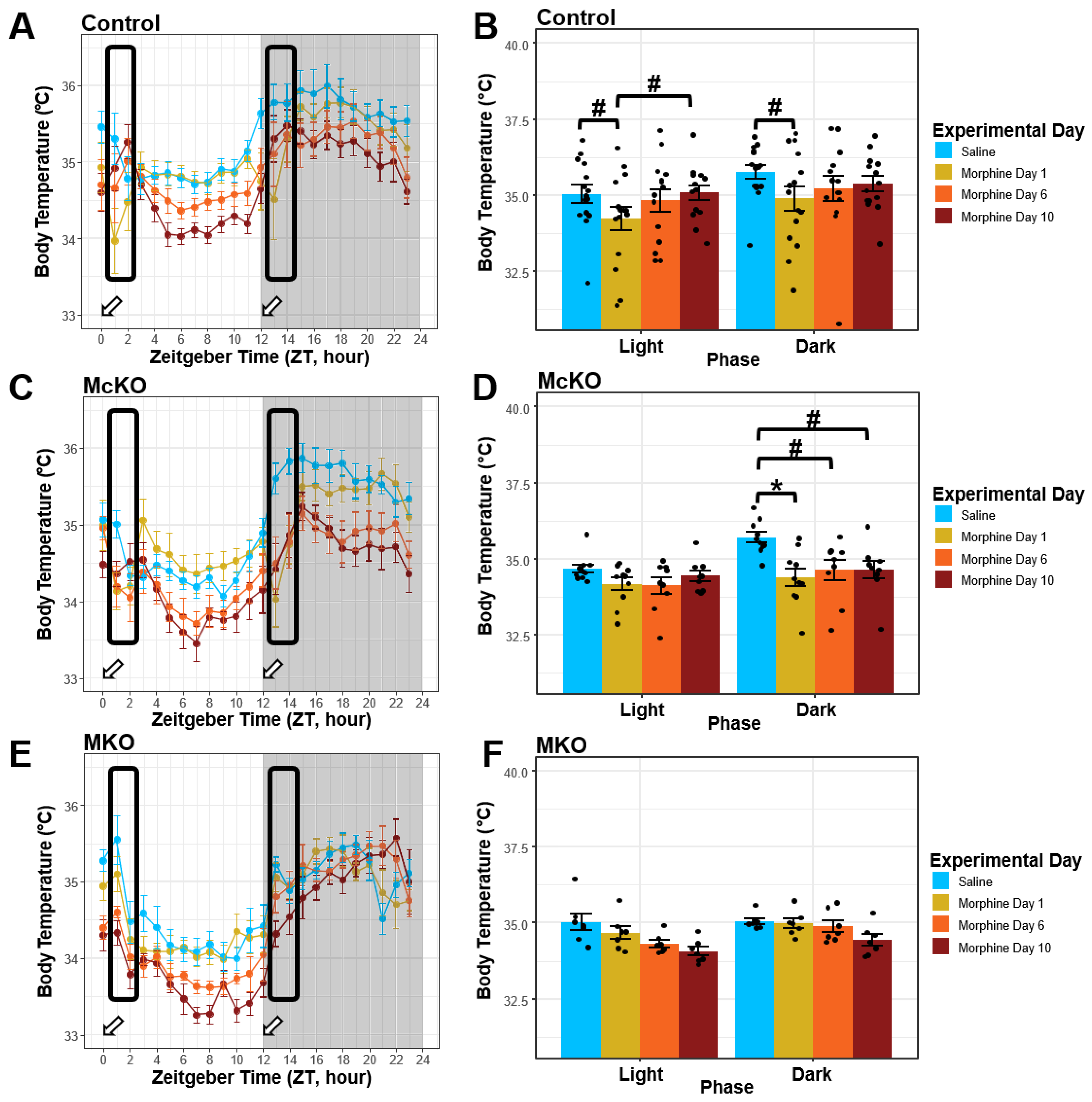
2.4. Control Mice, but Not McKO or MKO Mice, Appear to Develop Tolerance to the Hypothermic Effects of Morphine during Chronic Administration
2.5. Morphine-Induced Behavioral Alterations in McKO Animals Likely Arise from MOR Expression Changes within the Retina
3. Discussion
3.1. Systemic Morphine Affects Behavioral Activity via MORs Expressed by ipRGCs
3.2. Systemic Morphine Affects Thermoregulation via MORs Expressed by ipRGCs
3.3. Strengths and Limitations
4. Materials and Methods
4.1. Animals
4.2. Morphine Treatment
4.3. Surgery and Telemetry Recordings
4.4. Immunohistochemistry
4.5. Data and Statistrical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volkow, N.D.; Jones, E.B.; Einstein, E.B.; Wargo, E.M. Prevention and Treatment of Opioid Misuse and Addiction: A Review. JAMA Psychiatry 2019, 76, 208. [Google Scholar] [CrossRef]
- Chrobok, A.I.; Krause, D.; Winter, C.; Plörer, D.; Martin, G.; Koller, G.; Adorjan, K.; Canolli, M.; Adam, R.; Wagner, E.M.; et al. Sleeping Patterns in Patients with Opioid Use Disorder: Effects of Opioid Maintenance Treatment and Detoxification. J. Psychoact. Drugs 2020, 52, 203–210. [Google Scholar] [CrossRef]
- Dimsdale, J.E.; Norman, D.; DeJardin, D.; Wallace, M. The Effect of Opioids on Sleep Architecture. J. Clin. Sleep Med. 2007, 3, 4. [Google Scholar]
- Serdarevic, M.; Osborne, V.; Striley, C.W.; Cottler, L.B. The Association between Insomnia and Prescription Opioid Use: Results from a Community Sample in Northeast Florida. Sleep Health 2017, 3, 368–372. [Google Scholar] [CrossRef]
- Fathi, H.R.; Yoonessi, A.; Khatibi, A.; Rezaeitalab, F.; Rezaei-Ardani, A. Crosstalk between Sleep Disturbance and Opioid Use Disorder: A Narrative Review. Addict. Health 2020, 12, 140–158. [Google Scholar] [CrossRef]
- Kay, D.C. Human Sleep during Chronic Morphine Intoxication. Psychopharmacologia 1975, 44, 117–124. [Google Scholar] [CrossRef]
- Cheatle, M.D.; Webster, L.R. Opioid Therapy and Sleep Disorders: Risks and Mitigation Strategies. Pain Med. 2015, 16, S22–S26. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, E.E.; Pfeifer, J.G.; McCauley, J.L.; Moran-Santa Maria, M.; Back, S.E. Sleep Disturbances and Pain among Individuals with Prescription Opioid Dependence. Addict. Behav. 2014, 39, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Latif, Z.-H.; Šaltyte Benth, J.; Solli, K.K.; Opheim, A.; Kunoe, N.; Krajci, P.; Sharma-Haase, K.; Tanum, L. Anxiety, Depression, and Insomnia Among Adults With Opioid Dependence Treated With Extended-Release Naltrexone vs Buprenorphine-Naloxone: A Randomized Clinical Trial and Follow-up Study. JAMA Psychiatry 2019, 76, 127. [Google Scholar] [CrossRef] [Green Version]
- Huhn, A.S.; Finan, P.H. Sleep Disturbance as a Therapeutic Target to Improve Opioid Use Disorder Treatment. Exp. Clin. Psychopharmacol. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Simmons, Z.; Erickson, L.D.; Hedges, D.; Kay, D.B. Insomnia Is Associated With Frequency of Suicidal Ideation Independent of Depression: A Replication and Extension of Findings From the National Health and Nutrition Examination Survey. Front. Psychiatry 2020, 11, 561564. [Google Scholar] [CrossRef]
- Eacret, D.; Veasey, S.C.; Blendy, J.A. Bidirectional Relationship between Opioids and Disrupted Sleep: Putative Mechanisms. Mol. Pharm. 2020, 98, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Gillman, A.G.; Leffel, J.K.; Kosobud, A.E.K.; Timberlake, W. Fentanyl, but Not Haloperidol, Entrains Persisting Circadian Activity Episodes When Administered at 24- and 31-h Intervals. Behav. Brain Res. 2009, 205, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Glaser, A.M.; Reyes-Vázquez, C.; Prieto-Gómez, B.; Burau, K.; Dafny, N. Morphine Administration and Abrupt Cessation Alter the Behavioral Diurnal Activity Pattern. Pharmacol. Biochem. Behav. 2012, 101, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Meijer, J.H.; Ruijs, A.C.J.; Albus, H.; van de Geest, B.; Duindam, H.; Zwinderman, A.H.; Dahan, A. Fentanyl, a Mu-Opioid Receptor Agonist, Phase Shifts the Hamster Circadian Pacemaker. Brain Res. 2000, 868, 135–140. [Google Scholar] [CrossRef]
- Vansteensel, M.J.; Magnone, M.C.; van Oosterhout, F.; Baeriswyl, S.; Albrecht, U.; Albus, H.; Dahan, A.; Meijer, J.H. The Opioid Fentanyl Affects Light Input, Electrical Activity and Per Gene Expression in the Hamster Suprachiasmatic Nuclei. Eur. J. Neurosci. 2005, 21, 2958–2966. [Google Scholar] [CrossRef] [Green Version]
- Bergum, N.; Berezin, C.-T.; Vigh, J. A Retinal Contribution to Opioid-Induced Sleep Disorders? Front. Neurosci. 2022, 16, 981939. [Google Scholar] [CrossRef]
- Greco, M.-A.; Fuller, P.M.; Jhou, T.C.; Martin-Schild, S.; Zadina, J.E.; Hu, Z.; Shiromani, P.; Lu, J. Opioidergic Projections to Sleep-Active Neurons in the Ventrolateral Preoptic Nucleus. Brain Res. 2008, 1245, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Saper, C.B.; Fuller, P.M.; Pedersen, N.P.; Lu, J.; Scammell, T.E. Sleep State Switching. Neuron 2010, 68, 1023–1042. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yue, X.-F.; Qu, W.-M.; Tan, R.; Zheng, P.; Urade, Y.; Huang, Z.-L. Morphine Inhibits Sleep-Promoting Neurons in the Ventrolateral Preoptic Area Via Mu Receptors and Induces Wakefulness in Rats. Neuropsychopharmacology 2013, 38, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Lupi, D.; Oster, H.; Thompson, S.; Foster, R.G. The Acute Light-Induction of Sleep Is Mediated by OPN4-Based Photoreception. Nat. Neurosci. 2008, 11, 1068–1073. [Google Scholar] [CrossRef]
- Cleymaet, A.M.; Gallagher, S.K.; Tooker, R.E.; Lipin, M.Y.; Renna, J.M.; Sodhi, P.; Berg, D.; Hartwick, A.T.E.; Berson, D.M.; Vigh, J. μ-Opioid Receptor Activation Directly Modulates Intrinsically Photosensitive Retinal Ganglion Cells. Neuroscience 2019, 408, 400–417. [Google Scholar] [CrossRef]
- LeGates, T.A.; Fernandez, D.C.; Hattar, S. Light as a Central Modulator of Circadian Rhythms, Sleep and Affect. Nat. Rev. Neurosci. 2014, 15, 443–454. [Google Scholar] [CrossRef]
- Berezin, C.-T.; Bergum, N.; Luchini, K.A.; Curdts, S.; Korkis, C.; Vigh, J. Endogenous Opioid Signaling in the Retina Modulates Sleep/Wake Activity in Mice. Neurobiol. Sleep Circadian Rhythm. 2022, 13, 100078. [Google Scholar] [CrossRef]
- Wyman, J.; Bultman, S. Postmortem Distribution of Heroin Metabolites in Femoral Blood, Liver, Cerebrospinal Fluid, and Vitreous Humor. J. Anal. Toxicol. 2004, 28, 260–263. [Google Scholar] [CrossRef]
- Bergum, N.; Berezin, C.-T.; Dooley, G.; Vigh, J. Morphine Accumulates in the Retina Following Chronic Systemic Administration. Pharmaceuticals 2022, 15, 527. [Google Scholar] [CrossRef]
- Matthes, H.W.D.; Maldonadot, R.; Simonin, F.; Valverdet, O.; Slowet, S.; Kitchen, I.; Befort, K.; Dierich, A.; Le Meur, M.; Dolle, P.; et al. Loss of Morphine-Induced Analgesia, Reward Effect and Withdrawal Symptoms in Mice Lacking the Mu-Opioid-Receptor Gene. Nature 1996, 383, 819–823. [Google Scholar] [CrossRef]
- Weibel, R.; Reiss, D.; Karchewski, L.; Gardon, O.; Matifas, A.; Filliol, D.; Becker, J.A.J.; Wood, J.N.; Kieffer, B.L.; Gaveriaux-Ruff, C. Mu Opioid Receptors on Primary Afferent Nav1.8 Neurons Contribute to Opiate-Induced Analgesia: Insight from Conditional Knockout Mice. PLoS ONE 2013, 8, e74706. [Google Scholar] [CrossRef] [Green Version]
- Severino, A.L.; Mittal, N.; Hakimian, J.K.; Velarde, N.; Minasyan, A.; Albert, R.; Torres, C.; Romaneschi, N.; Johnston, C.; Tiwari, S.; et al. μ-Opioid Receptors on Distinct Neuronal Populations Mediate Different Aspects of Opioid Reward-Related Behaviors. eNeuro 2020, 7, 0146-20. [Google Scholar] [CrossRef]
- Kalivas, P.W.; Duffy, P. Sensitization to Repeated Morphine Injection in the Rat: Possible Involvement of A10 Dopamine Neurons. J. Pharm. Exp. 1987, 241, 204–212. [Google Scholar]
- Tian, M.; Broxmeyer, H.E.; Fan, Y.; Lai, Z.; Zhang, S.; Aronica, S.; Cooper, S.; Bigsby, R.M.; Steinmetz, R.; Engle, S.J.; et al. Altered Hematopoiesis, Behavior, and Sexual Function in Mu Opioid Receptor–Deficient Mice. J. Exp. Med. 1997, 185, 1517–1522. [Google Scholar] [CrossRef]
- Sora, I.; Li, X.-F. Mu Opiate Receptor Gene Dose Effects on Different Morphine Actions: Evidence for Differential in Vivo Mu Receptor Reserve. Neuropsychopharmacology 2001, 25, 14. [Google Scholar] [CrossRef]
- Yoo, J.-H.; Yang, E.-M.; Lee, S.-Y.; Loh, H.H.; Ho, I.K.; Jang, C.-G. Differential Effects of Morphine and Cocaine on Locomotor Activity and Sensitization in μ-Opioid Receptor Knockout Mice. Neurosci. Lett. 2003, 344, 37–40. [Google Scholar] [CrossRef]
- McLane, V.D.; Bergquist, I.; Cormier, J.; Barlow, D.J.; Houseknecht, K.L.; Bilsky, E.J.; Cao, L. Long-Term Morphine Delivery via Slow Release Morphine Pellets or Osmotic Pumps: Plasma Concentration, Analgesia, and Naloxone-Precipitated Withdrawal. Life Sci. 2017, 185, 1–7. [Google Scholar] [CrossRef]
- Smith, M.A.; Greene-Naples, J.L.; Lyle, M.A.; Iordanou, J.C.; Felder, J.N. The Effects of Repeated Opioid Administration on Locomotor Activity: I. Opposing Actions of μ and κ Receptors. J. Pharm. Exp. 2009, 330, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Hecht, A.; Schiorring, E. Behavioral Effects of Low and High Acute Doses of Morphine in Solitary Mice. Psychopharmacology 1979, 64, 73–79. [Google Scholar] [CrossRef]
- Itoh, T.; Murai, S.; Yoshida, H.; Masuda, Y.; Saito, H.; Chen, C.H. Effects of Methamphetamine and Morphine on the Vertical and Horizontal Motor Activities in Mice. Pharmacol. Biochem. Behav. 1987, 27, 193–197. [Google Scholar] [CrossRef]
- Szkeley, J.I.; Miglecz, E.; Ronai, Z. Biphasic Effects of a Potent Enkephalin Analogue (D-Met2,ProS)-Enkephalinamide and Morphine on Locomotor Activity in Mice. Psychopharmacology 1980, 71, 299–301. [Google Scholar]
- Rawls, S.M.; Benamar, K. Effects of Opioids, Cannabinoids, and Vanilloids on Body Temperature. Front. Biosci. 2011, S3, 822. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Autry, A.E.; Bergan, J.F.; Watabe-Uchida, M.; Dulac, C.G. Galanin Neurons in the Medial Preoptic Area Govern Parental Behaviour. Nature 2014, 509, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Moreton, J.E.; Roehrs, T.; Khazan, N. Drug Self-Administration and Sleep-Awake Activity in Rats Dependent on Morphine, Methadone, or/-Alpha-Acetylmethadol. Psychopharmacology 1976, 47, 237–241. [Google Scholar] [CrossRef]
- Andersen, G.; Christrup, L.; Sjøgren, P. Relationships Among Morphine Metabolism, Pain and Side Effects During Long-Term Treatment: An Update. J. Pain Symptom Manag. 2003, 25, 18. [Google Scholar] [CrossRef]
- Byku, M.; Gannon, R.L. Opioid Induced Non-Photic Phase Shifts of Hamster Circadian Activity Rhythms. Brain Res. 2000, 873, 189–196. [Google Scholar] [CrossRef]
- Kerdelhue, B.; Karteszi, M.; Pasqualini, C.; Reinberg, A.; Mezey, E.; Palkovits, M. Circadian Variations in β-Endorphin Concentrations in Pituitary and in Some Brain Nuclei of the Adult Male Rat. Brain Res. 1983, 261, 243–248. [Google Scholar] [CrossRef]
- Koch, C.E.; Leinweber, B.; Drengberg, B.C.; Blaum, C.; Oster, H. Interaction between Circadian Rhythms and Stress. Neurobiol. Stress 2017, 6, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Robinson, T.E.; Berridge, K.C. The Neural Basis of Drug Craving: An Incentive-Sensitization Theory of Addiction. Brain Res. Rev. 1993, 18, 247–291. [Google Scholar] [CrossRef]
- Babbini, M.; Davis, W.M. Time-Dose Relationships for Locomotor Activity Effects of Morphine after Acute or Repeated Treatment. Br. J. Pharmacol. 1972, 46, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Contarino, A.; Picetti, R.; Matthes, H.W.; Koob, G.F.; Kieffer, B.L.; Gold, L.H. Lack of Reward and Locomotor Stimulation Induced by Heroin in μ-Opioid Receptor-Deficient Mice. Eur. J. Pharmacol. 2002, 446, 103–109. [Google Scholar] [CrossRef]
- Rupp, A.C.; Ren, M.; Altimus, C.M.; Fernandez, D.C.; Richardson, M.; Turek, F.; Hattar, S.; Schmidt, T.M. Distinct IpRGC Subpopulations Mediate Light’s Acute and Circadian Effects on Body Temperature and Sleep. eLife 2019, 8, e44358. [Google Scholar] [CrossRef]
- Morrison, S.F.; Nakamura, K. Central Neural Pathways for Thermoregulation. Front. Biosci. 2011, 16, 74. [Google Scholar] [CrossRef] [Green Version]
- Refinetti, R. Relationship between the Daily Rhythms of Locomotor Activity and Body Temperature in Eight Mammalian Species. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 1999, 277, R1493–R1500. [Google Scholar] [CrossRef] [PubMed]
- Koek, W. Effects of Repeated Exposure to Morphine in Adolescent and Adult Male C57BL/6J Mice: Age-Dependent Differences in Locomotor Stimulation, Sensitization, and Body Weight Loss. Psychopharmacology 2014, 231, 1517–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, D.; Waterhouse, J. Diurnally Changing Effects of Locomotor Activity on Body Temperature in Laboratory Mice. Physiol. Behav. 1998, 63, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Meert, T. Morphine and D-Amphetamine Nullify Each Others’ Hypothermic Effects in Mice. Pharmacol. Toxicol. 2003, 92, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-H.; Geller, E.B.; Kim DeRiel, J.; Liu-Chen, L.-Y.; Adler, M.W. Antisense Confirmation of μ- and κ-Opioid Receptor Mediation of Morphine’s Effects on Body Temperature in Rats. Drug Alcohol Depend. 1996, 43, 119–124. [Google Scholar] [CrossRef]
- Zarrindast, M.-R.; Barghi-Lashkari, S.; Shafizadeh, M. The Possible Cross-Tolerance between Morphine- and Nicotine-Induced Hypothermia in Mice. Pharmacol. Biochem. Behav. 2001, 68, 283–289. [Google Scholar] [CrossRef]
- Baker, A.K.; Meert, T.F. Functional Effects of Systemically Administered Agonists and Antagonists of μ, δ, and κ Opioid Receptor Subtypes on Body Temperature in Mice. J. Pharm. Exp. 2002, 302, 1253–1264. [Google Scholar] [CrossRef]
- Becker, A.; Grecksch, G.; Kraus, J.; Peters, B.; Schroeder, H.; Schulz, S.; Höllt, V. Loss of Locomotor Sensitisation in Response to Morphine in D1 Receptor Deficient Mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 363, 562–568. [Google Scholar] [CrossRef]
- Koek, W.; France, C.P.; Javors, M.A. Morphine-Induced Motor Stimulation, Motor Incoordination, and Hypothermia in Adolescent and Adult Mice. Psychopharmacology 2012, 219, 1027–1037. [Google Scholar] [CrossRef] [Green Version]
- Le Marec, T.; Marie-Claire, C.; Noble, F.; Marie, N. Chronic and Intermittent Morphine Treatment Differently Regulates Opioid and Dopamine Systems: A Role in Locomotor Sensitization. Psychopharmacology 2011, 216, 297–303. [Google Scholar] [CrossRef]
- Zarrindast, M.-R.; Zarghi, A. Morphine Stimulates Locomotor Activity by an Indirect Dopaminergic Mechanism: Possible D-1 and D-2 Receptor Involvement. Gen. Pharmacol. Vasc. Syst. 1992, 23, 1221–1225. [Google Scholar] [CrossRef]
- Novak, C.M.; Burghardt, P.R.; Levine, J.A. The Use of a Running Wheel to Measure Activity in Rodents: Relationship to Energy Balance, General Activity, and Reward. Neurosci. Biobehav. Rev. 2012, 36, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Ecker, J.L.; Dumitrescu, O.N.; Wong, K.Y.; Alam, N.M.; Chen, S.-K.; LeGates, T.; Renna, J.M.; Prusky, G.T.; Berson, D.M.; Hattar, S. Melanopsin-Expressing Retinal Ganglion-Cell Photoreceptors: Cellular Diversity and Role in Pattern Vision. Neuron 2010, 67, 49–60. [Google Scholar] [CrossRef]
- Maloney, R.; Quattrochi, L.; Yoon, J.; Souza, R.; Berson, D. Efficacy and Specificity of Melanopsin Reporters for Retinal Ganglion Cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lax, P.; Esquiva, G.; Fuentes-Broto, L.; Segura, F.; Sánchez-Cano, A.; Cuenca, N.; Pinilla, I. Age-Related Changes in Photosensitive Melanopsin-Expressing Retinal Ganglion Cells Correlate with Circadian Rhythm Impairments in Sighted and Blind Rats. Chronobiol. Int. 2016, 33, 374–391. [Google Scholar] [CrossRef] [Green Version]
- Lax, P.; Ortuño-Lizarán, I.; Maneu, V.; Vidal-Sanz, M.; Cuenca, N. Photosensitive Melanopsin-Containing Retinal Ganglion Cells in Health and Disease: Implications for Circadian Rhythms. IJMS 2019, 20, 3164. [Google Scholar] [CrossRef] [Green Version]
- Moraes, M.N.; de Assis, L.V.M.; Provencio, I.; Castrucci, A.M. de L. Opsins Outside the Eye and the Skin: A More Complex Scenario than Originally Thought for a Classical Light Sensor. Cell Tissue Res. 2021, 385, 519–538. [Google Scholar] [CrossRef]
- Flyktman, A.; Jernfors, T.; Manttari, S.; Nissila, J.; Timonen, M.; Saarela, S. Transcranial Light Alters Melanopsin and Monoamine Production in Mouse ( Mus Musculus ) Brain. J. Neurol. Res. 2017, 7, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef] [Green Version]
- Storch, C.; Höhne, A.; Holsboer, F.; Ohl, F. Activity Patterns as a Correlate for Sleep–Wake Behaviour in Mice. J. Neurosci. Methods 2004, 133, 173–179. [Google Scholar] [CrossRef]
- Mogil, J.S.; Chesler, E.J.; Wilson, S.G.; Juraska, J.M.; Sternberg, W.F. Sex Differences in Thermal Nociception and Morphine Antinociception in Rodents Depend on Genotype. Neurosci. Biobehav. Rev. 2000, 24, 375–389. [Google Scholar] [CrossRef]
- Craft, R.M.; Clark, J.L.; Hart, S.P.; Pinckney, M.K. Sex Differences in Locomotor Effects of Morphine in the Rat. Pharmacol. Biochem. Behav. 2006, 85, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Zubieta, J.-K.; Smith, Y.R.; Bueller, J.A.; Xu, Y.; Kilbourn, M.R.; Jewett, D.M.; Meyer, C.R.; Koeppe, R.A.; Stohler, C.S. μ-Opioid Receptor-Mediated Antinociceptive Responses Differ in Men and Women. J. Neurosci. 2002, 22, 5100–5107. [Google Scholar] [CrossRef] [Green Version]
- Cleymaet, A.M.; Berezin, C.-T.; Vigh, J. Endogenous Opioid Signaling in the Mouse Retina Modulates Pupillary Light Reflex. IJMS 2021, 22, 554. [Google Scholar] [CrossRef]
- Peart, J.N.; Gross, G.J. Morphine-Tolerant Mice Exhibit a Profound and Persistent Cardioprotective Phenotype. Circulation 2004, 109, 1219–1222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Beier, C.; Weil, T.; Hattar, S. The Retinal IpRGC-Preoptic Circuit Mediates the Acute Effect of Light on Sleep. Nat. Commun. 2021, 12, 5115. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Bolte, S.; Cordelières, F.P. A Guided Tour into Subcellular Colocalization Analysis in Light Microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Ollion, J.; Cochennec, J.; Loll, F.; Escudé, C.; Boudier, T. TANGO: A Generic Tool for High-Throughput 3D Image Analysis for Studying Nuclear Organization. Bioinformatics 2013, 29, 1840–1841. [Google Scholar] [CrossRef] [Green Version]
- Cook, C. The Modulatory Actions of Dopamine D2/3 Agonists and Antagonists on the Locomotor-Activating Effects of Morphine and Caffeine in Mice. Pharmacol. Biochem. Behav. 2003, 75, 363–371. [Google Scholar] [CrossRef]
- Tzschentke, T.M. Pharmacology and Behavioral Pharmacology of the Mesocortical Dopamine System. Prog. Neurobiol. 2001, 63, 241–320. [Google Scholar] [CrossRef]
- Wise, R.A.; Bozarth, M.A. A Psychomotor Stimulant Theory of Addiction. Psychol. Rev. 1987, 94, 469–492. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergum, N.; Berezin, C.-T.; King, C.M.; Vigh, J. µ-Opioid Receptors Expressed by Intrinsically Photosensitive Retinal Ganglion Cells Contribute to Morphine-Induced Behavioral Sensitization. Int. J. Mol. Sci. 2022, 23, 15870. https://doi.org/10.3390/ijms232415870
Bergum N, Berezin C-T, King CM, Vigh J. µ-Opioid Receptors Expressed by Intrinsically Photosensitive Retinal Ganglion Cells Contribute to Morphine-Induced Behavioral Sensitization. International Journal of Molecular Sciences. 2022; 23(24):15870. https://doi.org/10.3390/ijms232415870
Chicago/Turabian StyleBergum, Nikolas, Casey-Tyler Berezin, Connie M. King, and Jozsef Vigh. 2022. "µ-Opioid Receptors Expressed by Intrinsically Photosensitive Retinal Ganglion Cells Contribute to Morphine-Induced Behavioral Sensitization" International Journal of Molecular Sciences 23, no. 24: 15870. https://doi.org/10.3390/ijms232415870