
The Mystery of EVP4593: Perspectives of the Quinazoline-Derived Compound in the Treatment of Huntington’s Disease and Other Human Pathologies


Abstract
:1. Introduction
2. The Molecular Mechanisms of EVP4593 Activity
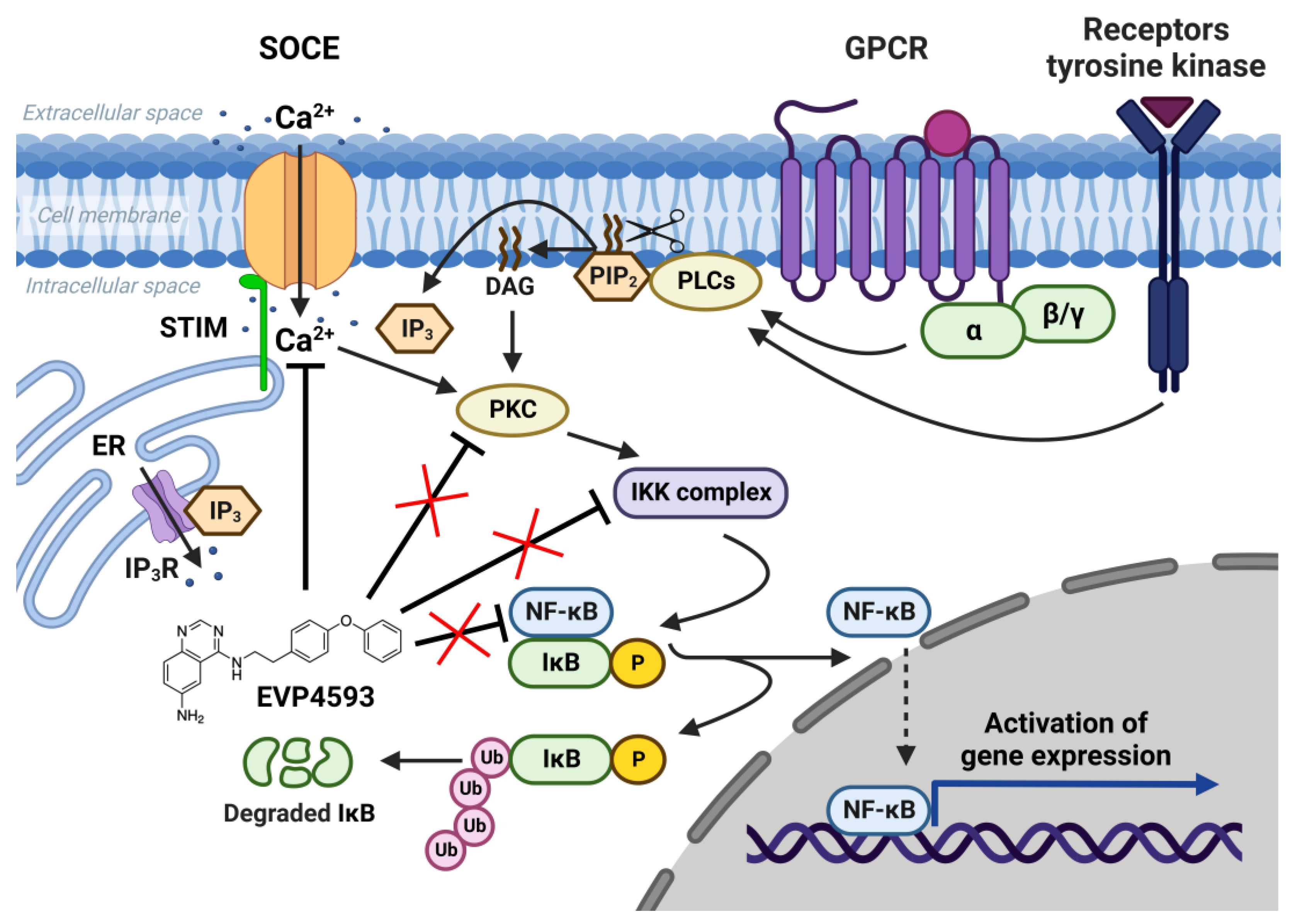
2.1. EVP4593 and NF-κB Signaling
2.2. EVP4593 and SOCE
2.3. EVP4593 and Mitochondrial Complex I
2.4. EVP4593 and mTOR Signaling
2.5. EVP4593 and Gene Expression
3. Perspectives of EVP4593 in Clinical Trials
3.1. Neurodegeneration
3.2. Other Pathologies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Mechanism of EVP4593 Activity | EVP4593 Concentration | Potential Treatment | References |
|---|---|---|---|---|
| Neurodegeneration | ||||
| Juvenile and adult-onset HD iPSCs-derived GABA neurons | EVP4593 attenuates pathologically enhanced huntingtin and STIM2 level. Pretreated-by-EVP4593 HD-specific neurons demonstrate reduced SOCE. | 300 nM | HD | [42] |
| Adult-onset HD iPSCs-derived GABA neurons | EVP4593 inhibits both ICRAC and ISOC channels. | 100 nM | HD | [19] |
| Adult-onset HD iPSCs-derived GABA neurons | EVP4593 attenuates SOCE both in wild-type and HD-specific neurons. EVP4593 reduces the number of lysosomes/ autophagosomes. EVP4593 reduces MG132-induced HD-specific neuronal death. | 100 nM | HD | [31] |
| The primary culture of mice MSNs with expressed Htt138Q-1exon | EVP4593 reduces SOCE to the normal level. | 300 nM | HD | [41] |
| YAC128 transgenic HD mice. Mixed cortical/striatal (MSNs) cultures | EVP4593 reduces synaptic neuronal SOCE and rescues spine loss. Intraventricular delivery of EVP4593 in YAC128 mice rescues age-dependent striatal spine loss in vivo. | 30 nM and 0.25 mg/mL in vivo | HD | [45] |
| YAC128 transgenic HD mice. The primary culture of mice MSNs. Fly HD model (Drosophila melanogaster) with expressed first 4 exons of human huntingtin (128Q) | EVP4593 attenuates enhanced SOCE. EVP4593 affects heteromeric channels containing the TRPC1 subunit but has no effect on homooligomer channels composed of TRPC1. EVP4593 delays a progression of a motor dysfunction phenotype in a transgenic fly HD model and protects YAC128 MSNs in a glutamate toxicity assay. | 300 nM and 100–400 µM in vivo | HD | [10] |
| R6/2 transgenic HD mice. The primary culture of cortical pyramidal neurons | EVP4593 reduces somatic calcium transient oscillations. | 3 µM | HD | [68] |
| The primary hippocampal cultures obtained from mice with PSEN1ΔE9 expression | EVP4593 decreases PSEN1ΔE9-mediated SOCE and rescues mushroom spines in PSEN1ΔE9-expressing neurons. | 30 nM | AD | [46] |
| Oncology | ||||
| Hepatoblastoma cell line (HepG2) and hepatocellular carcinoma cell lines (Huh7 and SNU-387) | EVP4593 affects NF-κB signaling and has an antitumor effect. | Huh7 and SNU-387—5 µM and HepG2—10 µM | Liver cancer | [60] |
| Head and neck squamous cell carcinoma (HNSCC) cell lines PCI1, PCI9, PCI13 and PCI52 | EVP4593 inhibits the NF-κB signaling. Antitumor effects. | 1–10 μM | Cancer | [61] |
| Breast cancer cell lineMCF7 (Michigan Cancer Foundation-7) | EVP4593 inhibits the NF-κB signaling and the mTOR pathway. Antitumor effects. | 1 mg/kg | Cancer | [29] |
| Nonsmall cell lung cancer (NSCLC) CL1-5-F4 cells | EVP4593 suppresses the NF-ĸB activation. EVP4593 inhibited the expression of metastasis-associated proteins. EVP4593 reduced cell migration and invasion. | 0.25 μM | Lung cancer | [62] |
| SK-HEP-1 cells | EVP4593 significantly inhibits the expression of antiapoptotic proteins and triggers extrinsic and intrinsic apoptosis pathways. EVP4593 reduces cell viability. EVP4593 inhibits the expression of NF-κB p65 and antiapoptotic proteins (XIAP, MCL-1 and c-FLIP) and increases levels of proapoptotic proteins (caspase-3 and -8 and cytochrome c). | 0.4 µM | Human hepatocellular carcinoma | [63] |
| Parasites invasion | ||||
| L.amazonensis-infected macrophages | EVP4593 inhibits amastigote growth and induces the production of high levels of NO and IL-1β. | 10 μM | Antileishmanial activity | [67] |
| L3 larvae of C. oncophora | Anthelmintic activity | 1.9–3.4 μM | Anthelmintic activity | [66] |
| Cardiovascular diseases | ||||
| Myocardial ischemia/reperfusion rat model | EVP4593 inhibits the NF-κB pathway, decreases the expression of NF-κB and has an anti-inflammatory effect. EVP4593 enhances morphine-induced cardio protection. | 1 mg/kg | Myocardial infarction | [64] |
| Diabetic retinopathy | ||||
| Streptozotocin-induced diabetes model rats | EVP4593 could alleviate the aggravation of retinopathy. EVP4593 decrease the endothelial cell proliferation and significantly reduces p65 expression. EVP4593 reduces blood glucose level. | 80 mg/kg | Diabetic retinopathy | [69] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asif, M. Chemical characteristics, synthetic methods, and biological potential of quinazoline and quinazolinone derivatives. Int. J. Med. Chem. 2014, 2014, 395637. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Ghoneim, O.A. Quinazoline Based HDAC Dual Inhibitors as Potential Anti-Cancer Agents. Molecules 2022, 27, 2294. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Hong, J. Recent advancements of 4-aminoquinazoline derivatives as kinase inhibitors and their applications in medicinal chemistry. Eur. J. Med. Chem. 2019, 170, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Bolteau, R.; Duroux, R.; Laversin, A.; Vreulz, B.; Shiriaeva, A.; Stauch, B.; Han, G.W.; Cherezov, V.; Renault, N.; Barczyk, A.; et al. High ligand efficiency quinazoline compounds as novel A2A adenosine receptor antagonists. Eur. J. Med. Chem. 2022, 241, 114620. [Google Scholar] [CrossRef] [PubMed]
- Bolteau, R.; Descamps, F.; Ettaoussi, M.; Caignard, D.H.; Delagrange, P.; Melnyk, P.; Yous, S. Quinazoline and phthalazine derivatives as novel melatonin receptor ligands analogues of agomelatine. Eur. J. Med. Chem. 2020, 189, 112078. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Gomez, K.; Moutal, A.; Patek, M.; Perez-Miller, S.; Khanna, R. Comparison of quinazoline and benzoylpyrazoline chemotypes targeting the CaVα-β interaction as antagonists of the N-type CaV2.2 channel. Channels 2021, 15, 128–135. [Google Scholar] [CrossRef]
- Darwish, S.S.; Chen, P.J.; Hamed, M.M.; Wagdy, R.A.; Chen, S.H.; Abadi, A.H.; Abdel-Halim, M.; Hwang, T.L.; Engel, M. Development of (4-Phenylamino)quinazolineAlkylthiourea Derivatives as Novel NF-κB Inhibitors. Pharmaceuticals 2022, 15, 778. [Google Scholar] [CrossRef]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Nagasaki, T.; Takahashi, H.; Hayashi, H. A novel structural class of potent inhibitors of NF-kappa B activation: Structure-activity relationships and biological effects of 6-aminoquinazoline derivatives. Bioorg. Med. Chem. 2003, 11, 3869–3878. [Google Scholar] [CrossRef]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Nagasaki, T.; Takahashi, H.; Fukazawa, T.; Hayashi, H. Discovery of quinazolines as a novel structural class of potent inhibitors of NF-kappa B activation. Bioorg. Med. Chem. 2003, 11, 383–391. [Google Scholar] [CrossRef]
- Wu, J.; Shih, H.P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef]
- Choi, S.; Kim, J.H.; Roh, E.J.; Ko, M.J.; Jung, J.E.; Kim, H.J. Nuclear factor-kappaB activated by capacitative Ca2+ entry enhances muscarinic receptor-mediated soluble amyloid precursor protein (sAPPalpha) release in SH-SY5Y cells. J. Biol. Chem. 2006, 281, 12722–12728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankunas, K.; Graef, I.A.; Neilson, J.R.; Park, S.H.; Crabtree, G.R. Signaling through calcium, calcineurin, and NF-AT in lymphocyte activation and development. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Rao, A. Transcriptional regulation in lymphocytes. Curr. Opin. Cell. Biol. 2001, 13, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamornwan, S.; Chokpanuwat, T.; Uppakara, K.; Soodvilai, S.; Saengsawang, W. Anti-Inflammatory Activity of Panduratin A against LPS-Induced Microglial Activation. Biomedicines 2022, 10, 2587. [Google Scholar] [CrossRef]
- Srinivasan, M.; Lahiri, D.K. Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert. Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Lv, B.; Ge, W.; Cui, Z.; Zhao, K.; Feng, Y.; Yang, X. Suppression of store-operated Ca2+ entry regulated by silencing Orai1 inhibits C6 glioma cell motility via decreasing Pyk2 activity and promoting focal adhesion. Cell Cycle 2020, 19, 3468–3479. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef]
- Vigont, V.; Nekrasov, E.; Shalygin, A.; Gusev, K.; Klushnikov, S.; Illarioshkin, S.; Lagarkova, M.; Kiselev, S.L.; Kaznacheyeva, E. Patient-Specific iPSC-Based Models of Huntington’s Disease as a Tool to Study Store-Operated Calcium Entry Drug Targeting. Front. Pharmacol. 2018, 9, 696. [Google Scholar] [CrossRef]
- Grekhnev, D.A.; Novikova, I.V.; Krisanova, A.V.; Yuskovets, V.N.; Chernov, N.M.; Yakovlev, I.P.; Kaznacheyeva, E.V.; Vigont, V.A. Dithiadiazole derivative 3-(4-nitrophenyl)-5-phenyl-3H-1,2,3,4-dithiadiazole-2-oxide—Novel modulator of store-operated calcium entry. Biochem. Biophys. Res. Commun. 2022, 626, 38–43. [Google Scholar] [CrossRef]
- Krishnathas, R.; Bonke, E.; Dröse, S.; Zickermann, V.; Nasiri, H.R. Identification of 4-N-[2-(4-phenoxyphenyl)ethyl]quinazoline-4,6-diamine as a novel, highly potent and specific inhibitor of mitochondrial complex I. Medchemcomm 2017, 8, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Kurelac, I.; Cavina, B.; Sollazzo, M.; Miglietta, S.; Fornasa, A.; De Luise, M.; Iorio, M.; Lama, E.; Traversa, D.; Nasiri, H.R.; et al. NDUFS3 knockout cancer cells and molecular docking reveal specificity and mode of action of anti-cancer respiratory complex I inhibitors. Open Biol. 2022, 12, 220198. [Google Scholar] [CrossRef] [PubMed]
- Bassal, M.A.; Samaraweera, S.E.; Lim, K.; Benard, B.A.; Bailey, S.; Kaur, S.; Leo, P.; Toubia, J.; Thompson-Peach, C.; Nguyen, T.; et al. Germline mutations in mitochondrial complex I reveal genetic and targetable vulnerability in IDH1-mutant acute myeloid leukaemia. Nat. Commun. 2022, 13, 2614. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Hu, C.; Kluza, J.; Ke, W.; Tian, G.; Giurgiu, M.; Bleilevens, A.; Campos, A.R.; Charbono, A.; Stickeler, E.; et al. Metabolic targeting of cancer by a ubiquinone uncompetitive inhibitor of mitochondrial complex I. Cell Chem. Biol. 2022, 29, 436–450. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.; Lee, H.K. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol. Neurodegener. 2021, 16, 44. [Google Scholar] [CrossRef]
- Occhiuzzi, M.A.; Lico, G.; Ioele, G.; De Luca, M.; Garofalo, A.; Grande, F. Recent advances in PI3K/PKB/mTOR inhibitors as new anticancer agents. Eur. J. Med. Chem. 2022, 246, 114971. [Google Scholar] [CrossRef]
- Marciano, R.; Prasad, M.; Ievy, T.; Tzadok, S.; Leprivier, G.; Elkabets, M.; Rotblat, B. High-Throughput Screening Identified Compounds Sensitizing Tumor Cells to Glucose Starvation in Culture and VEGF Inhibitors In Vivo. Cancers 2019, 11, 156. [Google Scholar] [CrossRef] [Green Version]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-KB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Lu, L.; Wang, H.; Hou, Y.; Dou, H. Hirsutella sinensis mycelium regulates autophagy of alveolar macrophages via TLR4/NF-κB signaling pathway. Int. J. Med. Sci. 2021, 18, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Firth, A.L.; Smith, K.A.; Maliakal, M.V.; Yuan, J.X. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2012, 302, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Liu, J.; Sun, Q.; Chen, R.; Wang, Y.; Duan, J.; Li, C.; Li, B.; Jing, Y.; Chen, X.; et al. mTORC1 enhancement of STIM1-mediated store-operated Ca2+ entry constrains tuberous sclerosis complex-related tumor development. Oncogene 2013, 32, 4702–4711. [Google Scholar] [CrossRef] [Green Version]
- Vaeth, M.; Maus, M.; Klein-Hessling, S.; Freinkman, E.; Yang, J.; Eckstein, M.; Cameron, S.; Turvey, S.E.; Serfling, E.; Berberich-Siebelt, F.; et al. Store-Operated Ca2+ Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity 2017, 47, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef]
- Chen, Y.W.; Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 22142. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, L.; Dong, L.; Yang, Z.W.; Zhang, J.; Zhang, S.L.; Niu, M.J.; Xia, J.W.; Gong, Y.; Zhu, N. Crosstalk between the Akt/mTORC1 and NF-κB signaling pathways promotes hypoxia-induced pulmonary hypertension by increasing DPP4 expression in PASMCs. Acta Pharmacol. Sin. 2019, 40, 1322–1333. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca2+ in autophagy regulation. Cell Calcium. 2011, 50, 242–250. [Google Scholar] [CrossRef]
- Vigont, V.; Kolobkova, Y.; Skopin, A.; Zimina, O.; Zenin, V.; Glushankova, L.; Kaznacheyeva, E. Both Orai1 and TRPC1 are Involved in Excessive Store-Operated Calcium Entry in Striatal Neurons Expressing Mutant Huntingtin Exon 1. Front. Physiol. 2015, 6, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigont, V.A.; Grekhnev, D.A.; Lebedeva, O.S.; Gusev, K.O.; Volovikov, E.A.; Skopin, A.Y.; Bogomazova, A.N.; Shuvalova, L.D.; Zubkova, O.A.; Khomyakova, E.A.; et al. STIM2 Mediates Excessive Store-Operated Calcium Entry in Patient-Specific iPSC-Derived Neurons Modeling a Juvenile Form of Huntington’s Disease. Front. Cell Dev. Biol. 2021, 9, 625231. [Google Scholar] [CrossRef] [PubMed]
- Bečanović, K.; Nørremølle, A.; Neal, S.J.; Kay, C.; Collins, J.A.; Arenillas, D.; Lilja, T.; Gaudenzi, G.; Manoharan, S.; Doty, C.N.; et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat. Neurosci. 2015, 18, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ryskamp, D.A.; Liang, X.; Egorova, P.; Zakharova, O.; Hung, G.; Bezprozvanny, I. Enhanced Store-Operated Calcium Entry Leads to Striatal Synaptic Loss in a Huntington’s Disease Mouse Model. J. Neurosci. 2016, 36, 125–141. [Google Scholar] [CrossRef] [Green Version]
- Chernyuk, D.; Zernov, N.; Kabirova, M.; Bezprozvanny, I.; Popugaeva, E. Antagonist of neuronal store-operated calcium entry exerts beneficial effects in neurons expressing PSEN1ΔE9 mutant linked to familial Alzheimer disease. Neuroscience 2019, 410, 118–127. [Google Scholar] [CrossRef]
- Sukkar, B.; Hauser, S.; Pelzl, L.; Hosseinzadeh, Z.; Sahu, I.; Al-Maghout, T.; Bhuyan, A.A.M.; Zacharopoulou, N.; Stournaras, C.; Schöls, L.; et al. Inhibition of Lithium Sensitive Orai1/ STIM1 Expression and Store Operated Ca2+ Entry in Chorea-Acanthocytosis Neurons by NF-κB Inhibitor Wogonin. Cell. Physiol. Biochem. 2018, 51, 278–289. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, J. Wogonin increases β-amyloid clearance and inhibits tau phosphorylation via inhibition of mammalian target of rapamycin: Potential drug to treat Alzheimer’s disease. Neurol. Sci. 2015, 36, 1181–1188. [Google Scholar] [CrossRef]
- Skobeleva, K.; Shalygin, A.; Mikhaylova, E.; Guzhova, I.; Ryazantseva, M.; Kaznacheyeva, E. The STIM1/2-Regulated Calcium Homeostasis Is Impaired in Hippocampal Neurons of the 5xFAD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 14810. [Google Scholar] [CrossRef]
- Grekhnev, D.A.; Kaznacheyeva, E.V.; Vigont, V.A. Patient-Specific iPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling. Int. J. Mol. Sci. 2022, 23, 624. [Google Scholar] [CrossRef]
- Kolobkova, Y.A.; Vigont, V.A.; Shalygin, A.V.; Kaznacheyeva, E.V. Huntington’s Disease: Calcium Dyshomeostasis and Pathology Models. Acta Naturae 2017, 9, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, P.P.; Cao, L.L.; Yang, Y.; Wang, P. Calcium Ions Aggravate Alzheimer’s Disease Through the Aberrant Activation of Neuronal Networks, Leading to Synaptic and Cognitive Deficits. Front. Mol. Neurosci. 2021, 14, 757515. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Langston, J.W. The Parkinson’s complex: Parkinsonism is just the tip of the iceberg. Ann. Neurol. 2006, 59, 591–596. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathologicalstageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Towards theories of brain aging. In Handbook of Studies on Psychiatry and Old Age; Kay, D.S., Burrows, G.W., Eds.; Elsevier: Amsterdam, The Netherlands, 1984; pp. 7–30. [Google Scholar]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.L.; Huang, J.; Zhen, L.L. Knockdown of otubain 2 inhibits liver cancer cell growth by suppressing NF-κB signaling. Kaohsiung J. Med. Sci. 2020, 36, 399–404. [Google Scholar] [CrossRef]
- Scheurer, M.J.J.; Brands, R.C.; El-Mesery, M.; Hartmann, S.; Müller-Richter, U.D.A.; Kübler, A.C.; Seher, A. The Selection of NFκB Inhibitors to Block Inflammation and Induce Sensitisation to FasL-Induced Apoptosis in HNSCC Cell Lines Is Critical for Their Use as a Prospective Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1306. [Google Scholar] [CrossRef]
- Wu, J.Y.; Lin, S.S.; Hsu, F.T.; Chung, J.G. Fluoxetine Inhibits DNA Repair and NF-ĸB-modulated Metastatic Potential in Non-small Cell Lung Cancer. Anticancer Res. 2018, 38, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.J.; Pan, P.J.; Hsu, F.T. Regorafenib induces extrinsic and intrinsic apoptosis through inhibition of ERK/NF-κB activation in hepatocellular carcinoma cells. Oncol. Rep. 2017, 37, 1036–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, R.H.; Wang, D.X.; Zhong, G.Q.; Meng, J.J.; Wen, H.; Feng, J.; Bi, Q.; He, Y. New targets of morphine postconditioning protection of the myocardium in ischemia/reperfusion injury: Involvement of HSP90/Akt and C5a/NF-κB. Open Med. 2021, 6, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Gusev, K.O.; Vigont, V.V.; Grekhnev, D.A.; Shalygin, A.V.; Glushankova, L.N.; Kaznacheeva, E.V. Store-Operated Calcium Entry in Mouse Cardiomyocytes. Bull. Exp. Biol. Med. 2019, 167, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Landuyt, B.; Klaassen, H.; Geldhof, P.; Luyten, W. Screening of a drug repurposing library with a nematode motility assay identifies promising anthelmintic hits against Cooperiaoncophora and other ruminant parasites. Vet. Parasitol. 2019, 265, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Macedo, S.R.; de FigueiredoNicolete, L.D.; Ferreira Ados, S.; de Barros, N.B.; Nicolete, R. The pentavalent antimonial therapy against experimental Leishmania amazonensis infection is more effective under the inhibition of the NF-κB pathway. Int. Immunopharmacol. 2015, 28, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yuan, H.; Zhao, T.; Yan, Y.; Liu, Z.; Cai, J.; Qiu, C.; Li, C. miR-874 ameliorates retinopathy in diabetic rats by NF-κB signaling pathway. Adv. Clin. Exp. Med. 2021, 30, 421–430. [Google Scholar] [CrossRef]
- Oikonomou, K.D.; Donzis, E.J.; Bui, M.T.N.; Cepeda, C.; Levine, M.S. Calcium dysregulation and compensation in cortical pyramidal neurons of the R6/2 mouse model of Huntington’s disease. J. Neurophysiol. 2021, 126, 1159–1171. [Google Scholar] [CrossRef]
- Garcia-Romero, E.M.; López-López, E.; Soriano-Correa, C.; Medina-Franco, J.L.; Barrientos-Salcedo, C. Polypharmacological drug design opportunities against Parkinson’s disease. F1000Research 2022, 11, 1176. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grekhnev, D.A.; Kruchinina, A.A.; Vigont, V.A.; Kaznacheyeva, E.V. The Mystery of EVP4593: Perspectives of the Quinazoline-Derived Compound in the Treatment of Huntington’s Disease and Other Human Pathologies. Int. J. Mol. Sci. 2022, 23, 15724. https://doi.org/10.3390/ijms232415724
Grekhnev DA, Kruchinina AA, Vigont VA, Kaznacheyeva EV. The Mystery of EVP4593: Perspectives of the Quinazoline-Derived Compound in the Treatment of Huntington’s Disease and Other Human Pathologies. International Journal of Molecular Sciences. 2022; 23(24):15724. https://doi.org/10.3390/ijms232415724
Chicago/Turabian StyleGrekhnev, Dmitriy A., Anna A. Kruchinina, Vladimir A. Vigont, and Elena V. Kaznacheyeva. 2022. "The Mystery of EVP4593: Perspectives of the Quinazoline-Derived Compound in the Treatment of Huntington’s Disease and Other Human Pathologies" International Journal of Molecular Sciences 23, no. 24: 15724. https://doi.org/10.3390/ijms232415724