
Empagliflozin Preserves Skeletal Muscle Function in a HFpEF Rat Model
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Impact of Empaliflozin on Physiological and Echocardiographic Parameter
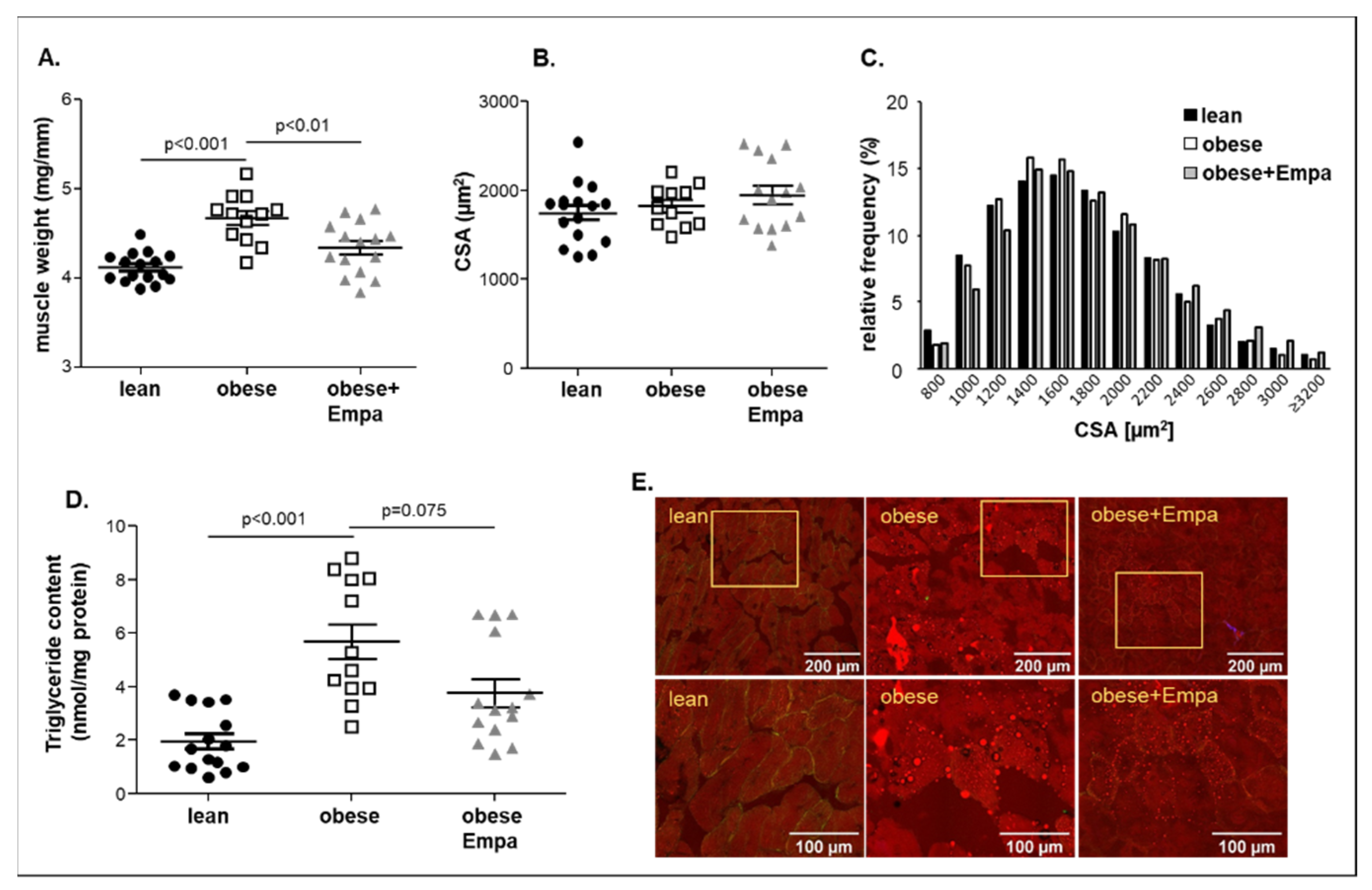
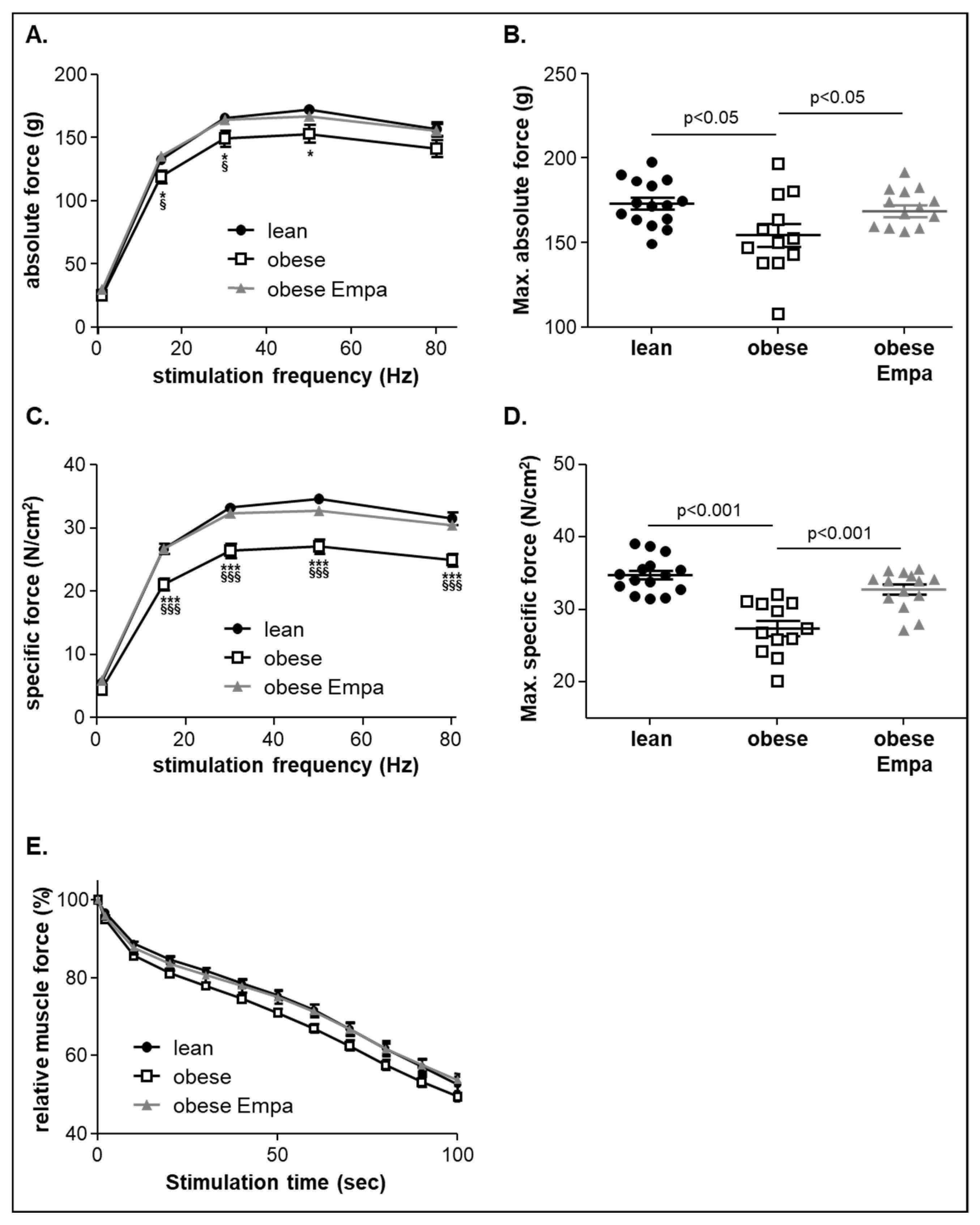
2.2. Impact of Empaliflozin on Muscle Trophicity and Function
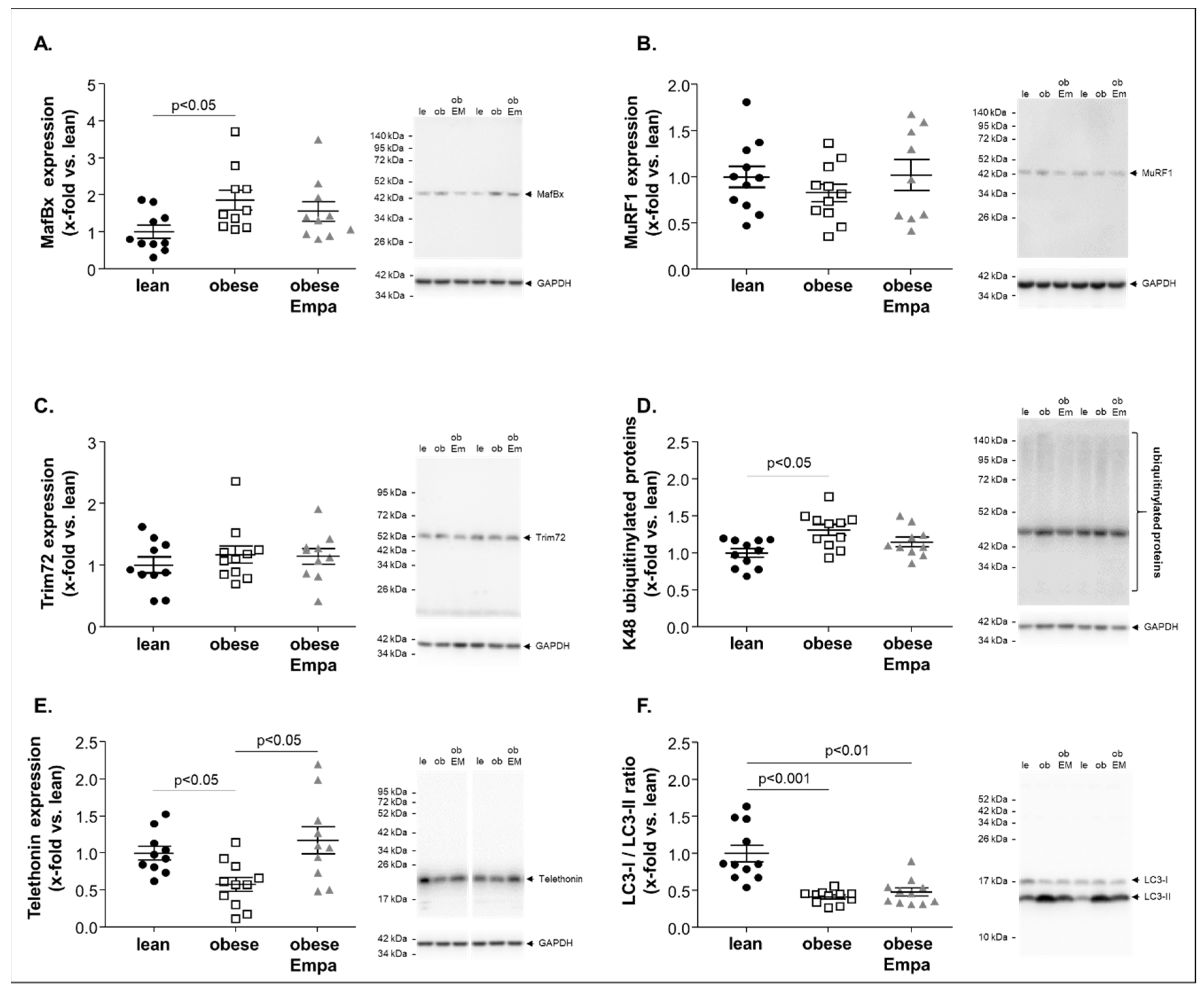
2.3. Impact of Empaliflozin on Muscle Atrophy Marker Expression
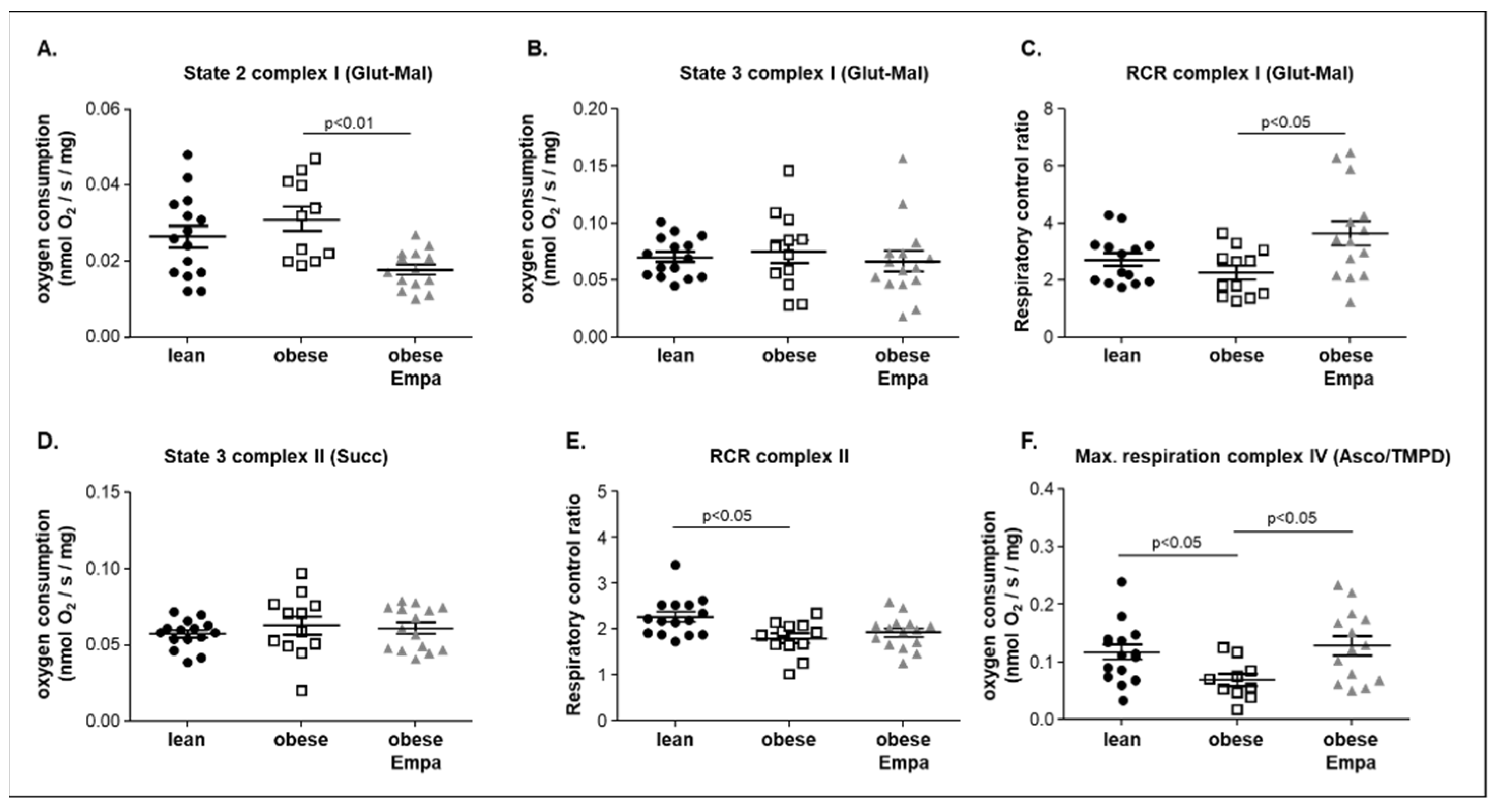
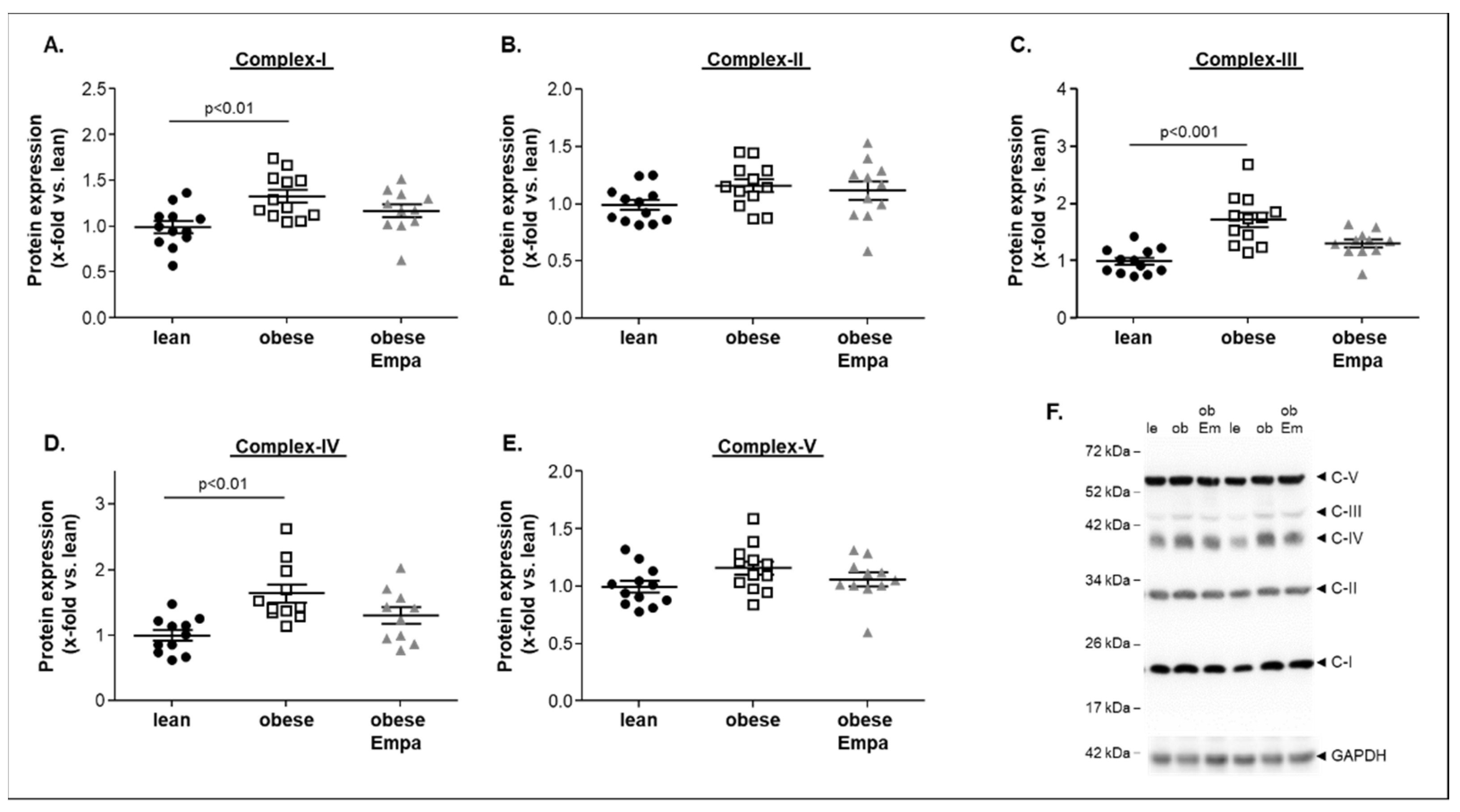
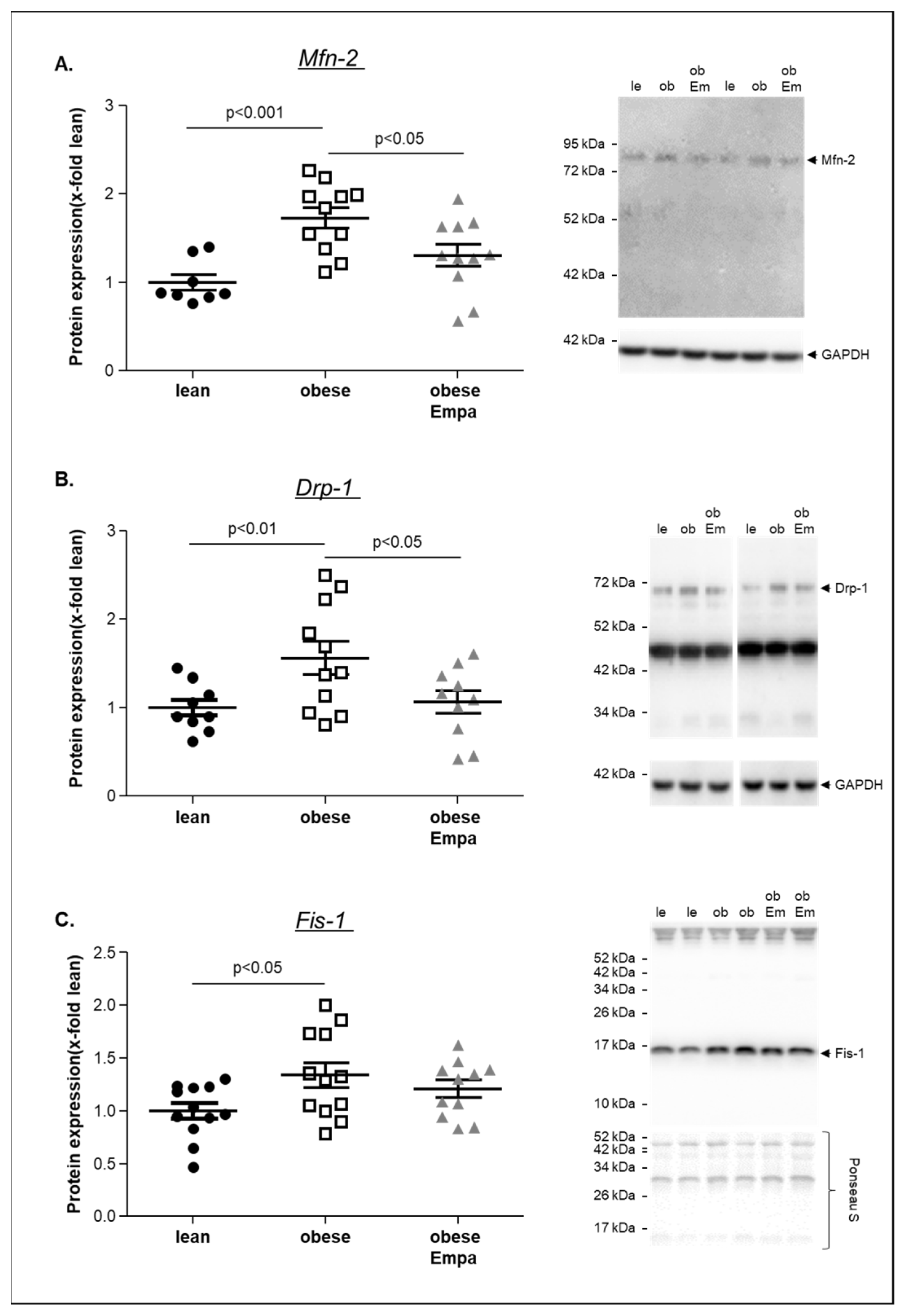
2.4. Impact of Empaliflozin on Mitochondrial Function
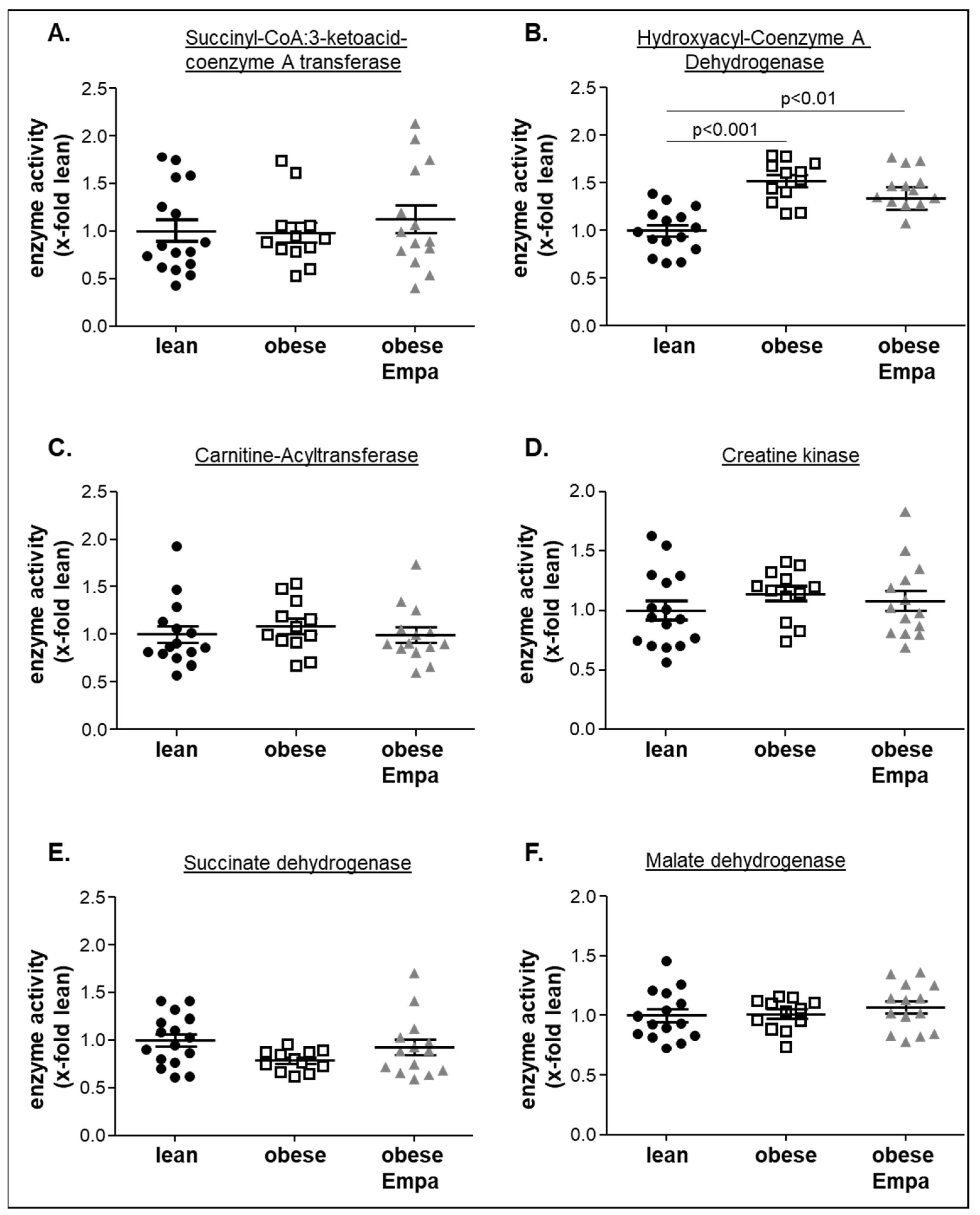
2.5. Impact of Empaliflozin on Metabolic Key Enzymes
3. Discussion
- Improved skeletal muscle contractility and reduced lipid content with some alterations of atrophy-related proteins;
- Improved mitochondrial function especially in complex IV without modulating the protein expression of mitochondrial complex proteins;
- Had no effect on key enzymes of different metabolic pathways such as ketone body utilization, ß-oxidation or Krebs cycle.
3.1. Empagliflozin and Skeletal Muscle Mass and Function
3.2. Empagliflozin and Mitochondrial Function
3.3. Empagliflozin and Metabolic Alterations
3.4. Study Limitations
4. Materials and Methods
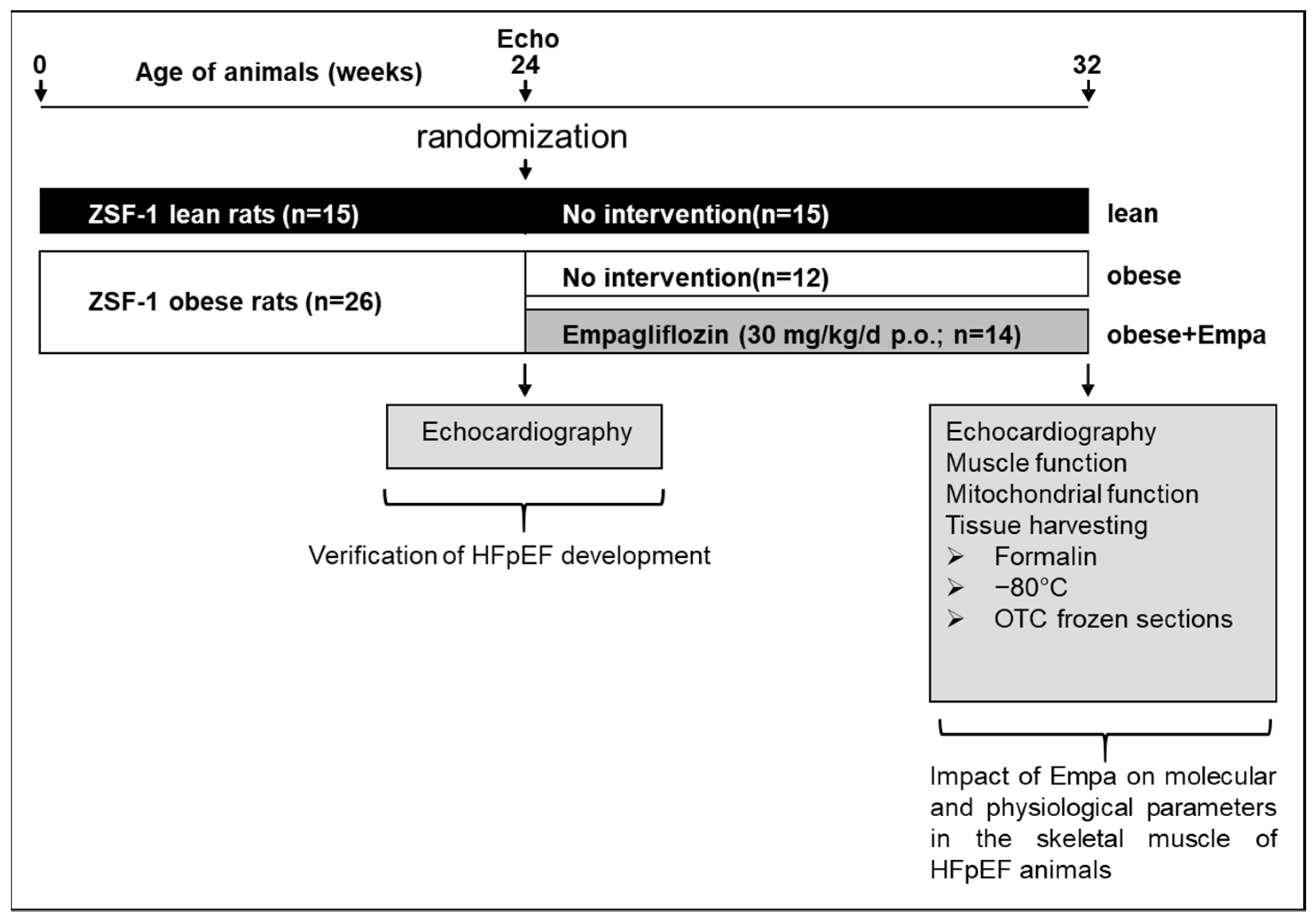
4.1. Study Design
4.2. Echocardiography and Invasive Hemodynamic Measurements
4.3. Skeletal Muscle Function and Cross Sectional Area
4.4. Muscle Mitochondrial Respiration
4.5. Western Blot Analysis
4.6. Enzyme Activity Measurements
4.7. Histological Analysis
4.8. Triglyceride and Glucose Content
4.9. Statistical Analyses
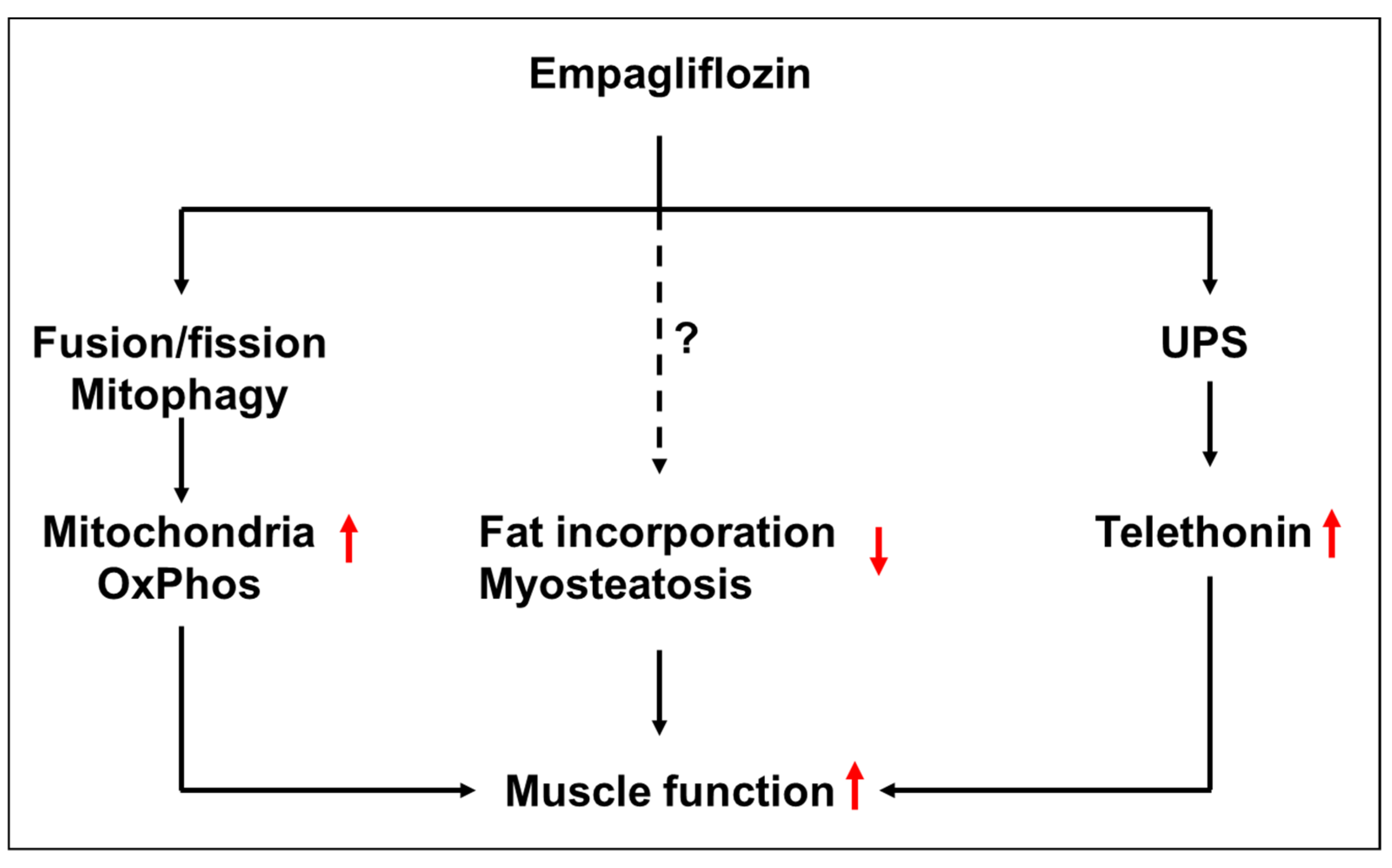
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatia, R.S.; Tu, J.V.; Lee, D.S.; Austin, P.C.; Fang, J.; Haouzi, A.; Gong, Y.; Liu, P.P. Outcome of Heart Failure with Preserved Ejection Fraction in a Population-Based Study. N. Engl. J. Med. 2006, 355, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, D.M.; Oduah, M.; Kiebalo, T.; Nartowicz, S.; Bęben, M.; Pochylski, M.; Ciepłucha, A.; Gwizdała, A.; Lesiak, M.; Straburzyńska-Migaj, E. Heart Failure with Preserved Ejection Fraction-a Concise Review. Curr. Cardiol. Rep. 2020, 22, 82–892. [Google Scholar] [CrossRef] [PubMed]
- Schmederer, Z.; Rolim, N.; Bowen, T.S.; Linke, A.; Wisloff, U.; Adams, V. Endothelial function is disturbed in a hypertensive diabetic animal model of HFpEF: Moderate continuous vs. high intensity interval training. Int. J. Cardiol. 2018, 273, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, E.; Sugiyama, S.; Matsuzawa, Y.; Konishi, M.; Suzuki, H.; Nozaki, T.; Ohba, K.; Matsubara, J.; Maeda, H.; Horibata, Y.; et al. Incremental Prognostic Significance of Peripheral Endothelial Dysfunction in Patients with Heart Failure with Normal Left Ventricular Ejection Fraction. J. Am. Coll. Cardiol. 2012, 60, 1778–1786. [Google Scholar] [CrossRef]
- Haykowsky, M.J.; Kouba, E.J.; Brubaker, P.H.; Nicklas, B.J.; Eggebeen, J.; Kitzman, D.W. Skeletal Muscle Composition and Its Relation to Exercise Intolerance in Older Patients with Heart Failure and Preserved Ejection Fraction. Am. J. Cardiol. 2014, 113, 1211–1216. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure with Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef]
- Bowen, T.S.; Rolim, N.P.L.; Fischer, T.; Baekkerud, F.H.; Medeiros, A.; Werner, S.; Brønstad, E.; Rognmo, O.; Mangner, N.; Linke, A.; et al. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur. J. Heart Fail. 2015, 17, 263–272. [Google Scholar] [CrossRef]
- Seiler, M.; Bowen, T.S.; Rolim, N.; Dieterlen, M.; Werner, S.; Hoshi, T.; Fischer, T.; Mangner, N.; Linke, A.; Schuler, G.; et al. Skeletal muscle alterations are exacerbated in heart failure with reduced compared with preserved ejection fraction: Mediated by circulating cytokines? Circ. Heart Fail. 2016, 9, e003027. [Google Scholar] [CrossRef]
- Bowen, T.S.; Herz, C.; Rolim, N.P.; Berre, A.-M.O.; Halle, M.; Kricke, A.; Linke, A.; da Silva, G.J.; Wisloff, U.; Adams, V. Effects of Endurance Training on Detrimental Structural, Cellular, and Functional Alterations in Skeletal Muscles of Heart Failure with Preserved Ejection Fraction. J. Card. Fail. 2018, 24, 603–613. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2003, 18, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Wunderlich, S.; Mangner, N.; Hommel, J.; Esefeld, K.; Gielen, S.; Halle, M.; Ellingsen, P.; Van Craenenbroeck, E.M.; Wisløff, U.; et al. Ubiquitin-proteasome-system and enzymes of energy metabolism in skeletal muscle of patients with HFpEF and HFrEF. ESC Heart Fail. 2021, 8, 2556–2568. [Google Scholar] [CrossRef] [PubMed]
- Wintrich, J.; Kindermann, I.; Ukena, C.; Selejan, S.; Werner, C.; Maack, C.; Laufs, U.; Tschöpe, C.; Anker, S.D.; Lam, C.S.P.; et al. Therapeutic approaches in heart failure with preserved ejection fraction: Past, present and future. Clin. Res. Cardiol. 2020, 109, 1079–1098. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Okamura, T.; Hashimoto, Y.; Osaka, T.; Fukuda, T.; Hamaguchi, M.; Fukui, M. The sodium-glucose cotransporter 2 inhibitor luseogliflozin can suppress muscle atrophy in Db/Db mice by suppressing the expression of foxo1. J. Clin. Biochem. Nutr. 2019, 65, 23–28. [Google Scholar] [CrossRef]
- Nambu, H.; Takada, S.; Fukushima, A.; Matsumoto, J.; Kakutani, N.; Maekawa, S.; Shirakawa, R.; Nakano, I.; Furihata, T.; Katayama, T.; et al. Empagliflozin restores lowered exercise endurance capacity via the activation of skeletal muscle fatty acid oxidation in a murine model of heart failure. Eur. J. Pharmacol. 2019, 866, 172810. [Google Scholar]
- Afsar, B.; Hornum, M.; Afsar, R.E.; Ertuglu, L.A.; Ortiz, A.; Covic, A.; van Raalte, D.H.; Cherney, D.Z.; Kanbay, M. Mitochondrion-driven nephroprotective mechanisms of novel glucose lowering medications. Mitochondrion 2021, 58, 72–82. [Google Scholar] [CrossRef]
- Croteau, D.; Luptak, I.; Chambers, J.M.; Hobai, I.; Panagia, M.; Pimentel, D.R.; Siwik, D.A.; Qin, F.; Colucci, W.S. Effects of Sodium-Glucose Linked Transporter 2 Inhibition with Ertugliflozin on Mitochondrial Function, Energetics, and Metabolic Gene Expression in the Presence and Absence of Diabetes Mellitus in Mice. J. Am. Heart Assoc. 2021, 10, e019995. [Google Scholar] [CrossRef]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2020, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Murray, E.K.; Eaton, D.M.; Huynh, A.T.; Tomar, D.; Garbincius, J.F.; Kolmetzky, D.W.; Berretta, R.M.; Wallner, M.; Houser, S.R.; et al. Molecular Signature of HFpEF: Systems Biology in a Cardiac-Centric Large Animal Model. JACC Basic Transl. Sci. 2021, 6, 650–672. [Google Scholar] [CrossRef] [PubMed]
- Schauer, A.; Draskowski, R.; Jannasch, A.; Kirchhoff, V.; Goto, K.; Männel, A.; Barthel, P.; Augstein, A.; Winzer, E.; Tugtekin, M.; et al. ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail. 2020, 7, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Schauer, A.; Adams, V.; Augstein, A.; Jannasch, A.; Draskowski, R.; Kirchhoff, V.; Goto, K.; Mittag, J.; Galli, R.; Männel, A.; et al. Sacubitril/Valsartan Improves Diastolic Function but Not Skeletal Muscle Function in a Rat Model of HFpEF. Int. J. Mol. Sci. 2021, 22, 3570. [Google Scholar] [CrossRef]
- Adams, V.; Schauer, A.; Augstein, A.; Kirchhoff, V.; Draskowski, R.; Jannasch, A.; Goto, K.; Lyall, G.; Männel, A.; Barthel, P.; et al. Targeting MuRF1 by small molecules in a HFpEF rat model improves myocardial diastolic function and skeletal muscle contractility. J. Cachex Sarcopenia Muscle 2022, 13, 1565–1581. [Google Scholar] [CrossRef] [PubMed]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Heng, A.E.; Ventadour, S.; Jarzaguet, M.; Pouch-Pélissier, M.N.; Guezennec, C.Y.; Bigard, X.; Attaix, D.; Taillandier, D. Coordinate expression of the 19S regulatory complex and evidence for ubiquitin-dependent telethonin degradation in the unloaded soleus muscle. Int. J. Biochem. Cell Biol. 2008, 40, 2544–2552. [Google Scholar] [CrossRef]
- Nassif, M.E.; Windsor, S.L.; Borlaug, B.A.; Kitzman, D.W.; Shah, S.J.; Tang, F.; Khariton, Y.; Malik, A.O.; Khumri, T.; Umpierrez, G.; et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: A multicenter randomized trial. Nat. Med. 2021, 27, 1954–1960. [Google Scholar] [CrossRef]
- Yamakage, H.; Tanaka, M.; Inoue, T.; Odori, S.; Kusakabe, T.; Satoh-Asahara, N. Effects of dapagliflozin on the serum levels of fibroblast growth factor 21 and myokines and muscle mass in Japanese patients with type 2 diabetes: A randomized, controlled trial. J. Diabetes Investig. 2020, 11, 653–661. [Google Scholar] [CrossRef]
- Sugiyama, S.; Jinnouchi, H.; Kurinami, N.; Hieshima, K.; Yoshida, A.; Jinnouchi, K.; Nishimura, H.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; et al. Dapagliflozin Reduces Fat Mass without Affecting Muscle Mass in Type 2 Diabetes. J. Atheroscler. Thromb. 2018, 25, 467–476. [Google Scholar] [CrossRef]
- Ohta, A.; Kato, H.; Ishii, S.; Sasaki, Y.; Nakamura, Y.; Nakagawa, T.; Nagai, Y.; Tanaka, Y. Ipragliflozin, a sodium glucose co-transporter 2 inhibitor, reduces intrahepatic lipid content and abdominal visceral fat volume in patients with type 2 diabetes. Expert Opin. Pharmacother. 2017, 18, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Sugawara, M.; Fukuda, M. Sodium-glucose cotransporter 2 inhibitor-induced changes in body composition and simultaneous changes in metabolic profile: 52-week prospective LIGHT (Luseogliflozin: The Components of Weight Loss in Japanese Patients with Type 2 Diabetes Mellitus) Study. J. Diabetes Investig. 2018, 10, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Q.; Li, G.-Q.; Zhong, Y.; Wang, J.; Wang, A.-N.; Zhou, X.; Mao, X.-M. Empagliflozin improves chronic hypercortisolism-induced abnormal myocardial structure and cardiac function in mice. Ther. Adv. Chronic Dis. 2020, 11, 2040622320974833. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar]
- Goto, K.; Schauer, A.; Augstein, A.; Methawasin, M.; Granzier, H.; Halle, M.; van Craenenbroeck, E.M.; Rolim, N.; Gielen, S.; Pieske, B.; et al. Muscular changes in animal models of heart failure with preserved ejection fraction: What comes closest to the patient? ESC Heart Fail. 2021, 8, 139–150. [Google Scholar] [CrossRef]
- Otsuka, H.; Yokomizo, H.; Nakamura, S.; Izumi, Y.; Takahashi, M.; Obara, S.; Nakao, M.; Ikeda, Y.; Sato, N.; Sakamoto, R.; et al. Differential effect of canagliflozin, a sodium–glucose cotransporter 2 (SGLT2) inhibitor, on slow and fast skeletal muscles from nondiabetic mice. Biochem. J. 2022, 479, 425–444. [Google Scholar] [CrossRef]
- Bodmer, N.K.; Theisen, K.E.; Dima, R.I. Molecular Investigations into the Mechanics of a Muscle Anchoring Complex. Biophys. J. 2015, 108, 2322–2332. [Google Scholar] [CrossRef]
- Furukawa, T.; Ono, Y.; Tsuchiya, H.; Katayama, Y.; Bang, M.; Labeit, D.; Labeit, S.; Inagaki, N.; Gregorio, C.C. Specific interaction of the potassium channel ß-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol 2001, 313, 775–784. [Google Scholar] [CrossRef]
- Sadikot, T.; Hammond, C.; Ferrari, M.B. Distinct roles for telethonin N-versus C-terminus in sarcomere assembly and maintenance. Dev. Dyn. 2010, 239, 1124–1135. [Google Scholar] [CrossRef]
- Correa-De-Araujo, R.; Addison, O.; Miljkovic, I.; Goodpaster, B.H.; Bergman, B.C.; Clark, R.V.; Elena, J.W.; Esser, K.A.; Ferrucci, L.; Harris-Love, M.O.; et al. Myosteatosis in the Context of Skeletal Muscle Function Deficit: An Interdisciplinary Workshop at the National Institute on Aging. Front. Physiol. 2020, 11, 963. [Google Scholar] [CrossRef]
- Anderson, M.; Parrott, C.F.; Mark, J.H.; Peter, H.B. Skeletal muscle abnormalities in heart failure with preserved ejection fraction. Heart Fail. Rev. 2022; in press. [Google Scholar]
- Molina, A.J.; Bharadwaj, M.S.; Van Horn, C.; Nicklas, B.J.; Lyles, M.F.; Eggebeen, J.; Haykowsky, M.J.; Brubaker, P.H.; Kitzman, D.W. Skeletal Muscle Mitochondrial Content, Oxidative Capacity, and Mfn2 Expression Are Reduced in Older Patients with Heart Failure and Preserved Ejection Fraction and Are Related to Exercise Intolerance. JACC Heart Fail. 2016, 4, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Schär, M.; Panjrath, G.S.; Zhang, Y.; Sharma, K.; Bottomley, P.A.; Golozar, A.; Steinberg, A.; Gerstenblith, G.; Russell, S.D.; et al. Fatigability, Exercise Intolerance, and Abnormal Skeletal Muscle Energetics in Heart Failure. Circ. Heart Fail. 2017, 10, e004129. [Google Scholar] [CrossRef] [PubMed]
- Zamani, P.; Proto, E.A.; Wilson, N.; Fazelinia, H.; Ding, H.; Spruce, L.A.; Davilia, A., Jr.; Hanff, T.C.; Mazurek, J.A.; Prenner, S.B.; et al. Multimodality assessment of heart failure with preserved ejection fraction skeletal muscle reveals differences in the machinery of energy fuel metabolism. ESC Heart Fail. 2021, 8, 2698–2712. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Guo, Z.; Chang, X.; Li, Z.; Wu, F.; He, J.; Cao, T.; Wang, K.; Shi, N.; Zhou, H.; et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKa1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 2022, 52, 102288. [Google Scholar] [CrossRef]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Mangner, N.; Garbade, J.; Heyne, E.; van den Berg, M.; Winzer, E.B.; Hommel, J.; Sandri, M.; Jozwiak-Nozdrzykowska, J.; Meyer, A.L.; Lehmann, S.; et al. Molecular Mechanisms of Diaphragm Myopathy in Humans with Severe Heart Failure. Circ. Res. 2021, 128, 706–719. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Empagliflozin’s Fuel Hypothesis: Not so Soon. Cell Metab. 2016, 24, 200–202. [Google Scholar] [CrossRef]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin Increases Cardiac Energy-áProduction-áin Diabetes: Novel Translational Insights into the Heart Failure Benefits-áof-áSGLT2 Inhibitors. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Haykowsky, M.J.; Tomczak, C.R.; Scott, J.; Paterson, D.I.; Kitzman, D.W. Determinants of exercise intolerance in patients with heart failure and reduced or preserved ejection fraction. J. Appl. Physiol. 2015, 119, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Kitzman, D.W.; Nicklas, B.; Kraus, W.E.; Lyles, M.F.; Eggebeen, J.; Morgan, T.M.; Haykowsky, M. Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Am. J. Physiol. Circ. Physiol. 2014, 306, H1364–H1370. [Google Scholar] [CrossRef] [PubMed]
- Espino-Gonzalez, E.; Tickle, P.G.; Benson, A.P.; Kissane, R.W.P.; Askew, G.N.; Egginton, S.; Bowen, T.S. Abnormal skeletal muscle blood flow, contractile mechanics and fibre morphology in a rat model of obese-HFpEF. J. Physiol. 2021, 599, 981–1001. [Google Scholar] [CrossRef]
- Fonarow, G.C.; Stough, W.G.; Abraham, W.T.; Albert, N.M.; Gheorghiade, M.; Greenberg, B.H.; O’Connor, C.M.; Sun, J.L.; Yancy, C.W.; Young, J.B.; et al. Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure: A report from the OPTIMIZE-HF Registry. J. Am. Coll. Cardiol. 2007, 50, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.; Cox, M.; Neely, M.L.; Heidenreich, P.A.; Bhatt, D.L.; Eapen, Z.J.; Hernandez, A.F.; Butler, J.; Yancy, C.W.; Fonarow, G.C. Outcomes in patients with heart failure with preserved, borderline, and reduced ejection fraction in the Medicare population. Am. Heart J. 2014, 168, 721–730.e3. [Google Scholar] [CrossRef]
- Takashi, O.; Takashi, H. Occurrence of two 3-Hydroxyacyl-CoA dehydrogenases in rat liver. Biochim. Biohphys. Acta 1979, 574, 258–267. [Google Scholar] [CrossRef]
- Dzeja, P.P.; Pucar, D.; Redfield, M.M.; Burnett, J.C.; Terzic, A. Reduced activity of enzymes coupling ATP-generating with ATP-consuming processes in the failing myocardium. Mol. Cell. Biochem. 1999, 201, 33–40. [Google Scholar] [CrossRef]
- Hiltunen, J.K.; Saukko, P.; Hirvonen, J. Correlations between enzyme histochemical reactions and respective enzyme activities in global ischaemic rat hearts. Br. J. Exp. Pathol. 1985, 66, 743–752. [Google Scholar]
- Turko, I.V.; Marcondes, S.; Murad, F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2289–H2294. [Google Scholar] [CrossRef]
- Zipfel, W.R.; Williams, R.M.; Webb, W.W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003, 21, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Büttner, P.; Galli, R.; Husser, D.; Bollmann, A. Label-free Imaging of Myocardial Remodeling in Atrial Fibrillation Using Nonlinear Optical Microscopy: A Feasibility Study. J. Atr. Fibrillation 2018, 10, 1644. [Google Scholar] [PubMed]
- Pezacki, J.P.; Blake, J.A.; Danielson, D.C.; Kennedy, D.C.; Lyn, R.K.; Singaravelu, R. Chemical contrast for imaging living systems: Molecular vibrations drive CARS microscopy. Nat. Chem. Biol. 2011, 7, 137–145. [Google Scholar] [CrossRef]
- Vogler, N.; Heuke, S.; Bocklitz, T.W.; Schmitt, M.; Popp, J. Multimodal Imaging Spectroscopy of Tissue. Annu. Rev. Anal. Chem. 2015, 8, 359–387. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Lean (n = 15) | Obese (n = 12) | Obese + Empa (n = 14) |
|---|---|---|---|
| Body weight (g) | 255 ± 4 | 532 ± 9 *** | 488 ± 6 ***,§§§ |
| Tibia length (TL, mm) | 35.6 ± 0.1 | 35.6 ± 0.1 | 35.3 ± 0.1 |
| Heart weight/TL (mg/mm) | 27.75 ± 0.65 | 43.82 ± 2.02 *** | 36.70 ± 1.05 ***,§§§ |
| Lung weight (wet/dry) | 4.56 ± 0.02 | 4.22 ± 0.04 *** | 4.22 ± 0.06 *** |
| Lung wet weight/TL (mg/mm) | 10.89 ± 0.11 | 11.91 ± 0.21 *** | 11.04 ± 0.18 §§ |
| Kidney weight/TL (mg/mm) | 28.2 ± 0.5 | 46.6 ± 1.4 *** | 49.4 ± 1.0 *** |
| blood glucose (mmol/L) | 19.9 ± 1.8 | 37.2 ± 1.5 *** | 25.6 ± 0.6 §§§ |
| Urinary glucose (mmol/L) | 2.3 ± 0.6 | 15.2 ± 3.8 | 142.5 ± 21.6 ***,§§§ |
| Echocardiography/invasive hemodynamic | |||
| LVEF (%) | 68.0 ± 1.4 | 67.7 ± 1.7 | 68.0 ± 1.2 |
| LVFS (%) | 23.6 ± 1.1 | 24.9 ± 1.1 | 22.9 ± 0.8 |
| Aortic blood pressure (mmHg) | 97 ± 3 | 129 ± 3 *** | 117 ± 5 *** |
| E/é | 17.5 ± 0.7 | 22.8 ± 1.1 *** | 19.5 ± 0.8 § |
| LVEDP (mmHg) | 5.96 ± 0.35 | 8.02 ± 0.61 *** | 7.05 ± 0.37 § |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winzer, E.B.; Schauer, A.; Langner, E.; Augstein, A.; Goto, K.; Männel, A.; Barthel, P.; Jannasch, A.; Labeit, S.; Mangner, N.; et al. Empagliflozin Preserves Skeletal Muscle Function in a HFpEF Rat Model. Int. J. Mol. Sci. 2022, 23, 10989. https://doi.org/10.3390/ijms231910989
Winzer EB, Schauer A, Langner E, Augstein A, Goto K, Männel A, Barthel P, Jannasch A, Labeit S, Mangner N, et al. Empagliflozin Preserves Skeletal Muscle Function in a HFpEF Rat Model. International Journal of Molecular Sciences. 2022; 23(19):10989. https://doi.org/10.3390/ijms231910989
Chicago/Turabian StyleWinzer, Ephraim B., Antje Schauer, Erik Langner, Antje Augstein, Keita Goto, Anita Männel, Peggy Barthel, Anett Jannasch, Siegfried Labeit, Norman Mangner, and et al. 2022. "Empagliflozin Preserves Skeletal Muscle Function in a HFpEF Rat Model" International Journal of Molecular Sciences 23, no. 19: 10989. https://doi.org/10.3390/ijms231910989