
Functional Delineation of a Protein–Membrane Interaction Hotspot Site on the HIV-1 Neutralizing Antibody 10E8

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
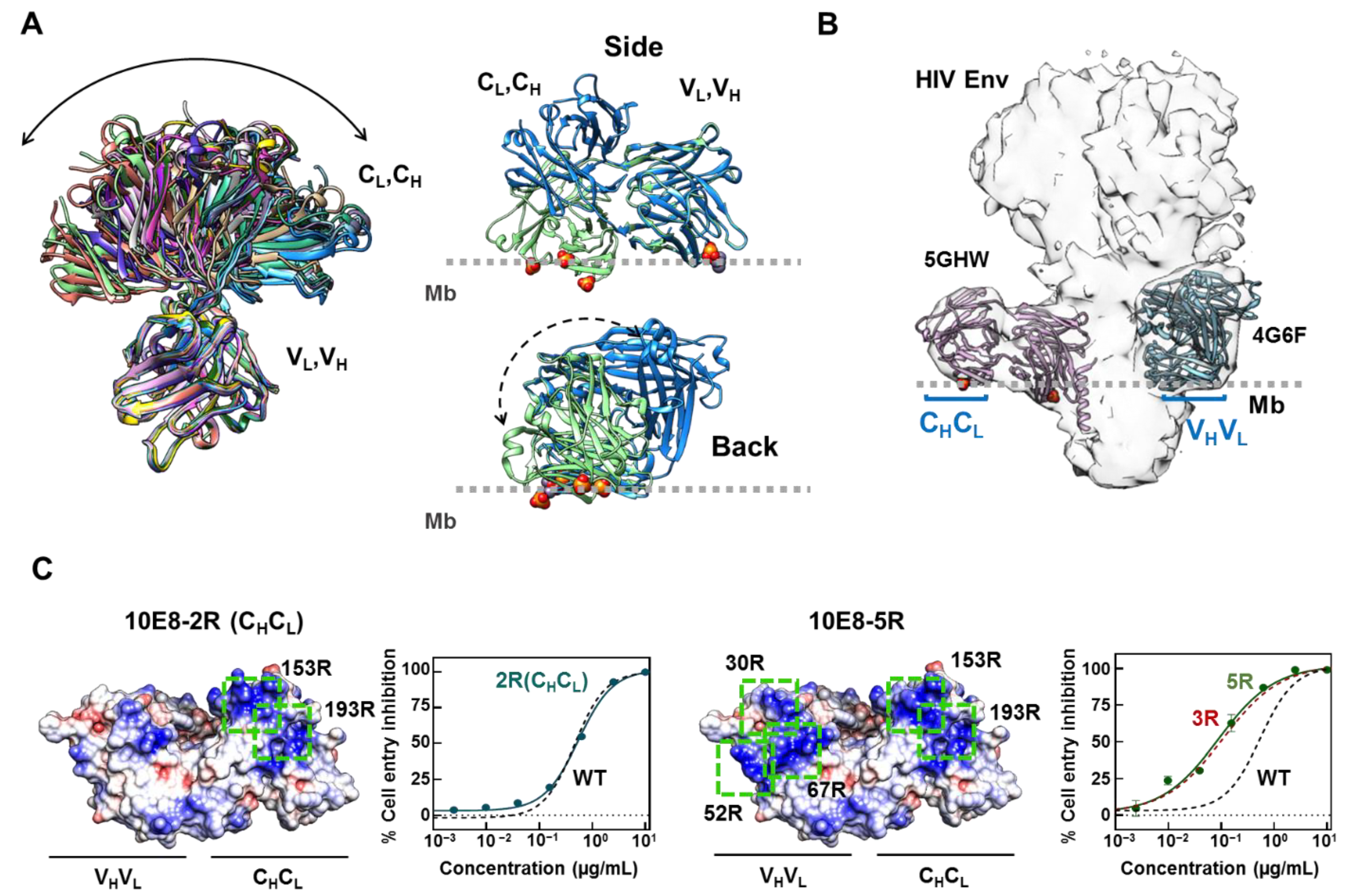
2.1. Inclusion of Arg Residues in the Surface of the Constant Domain That Can Accommodate the Membrane Does Not Improve Antibody Potency
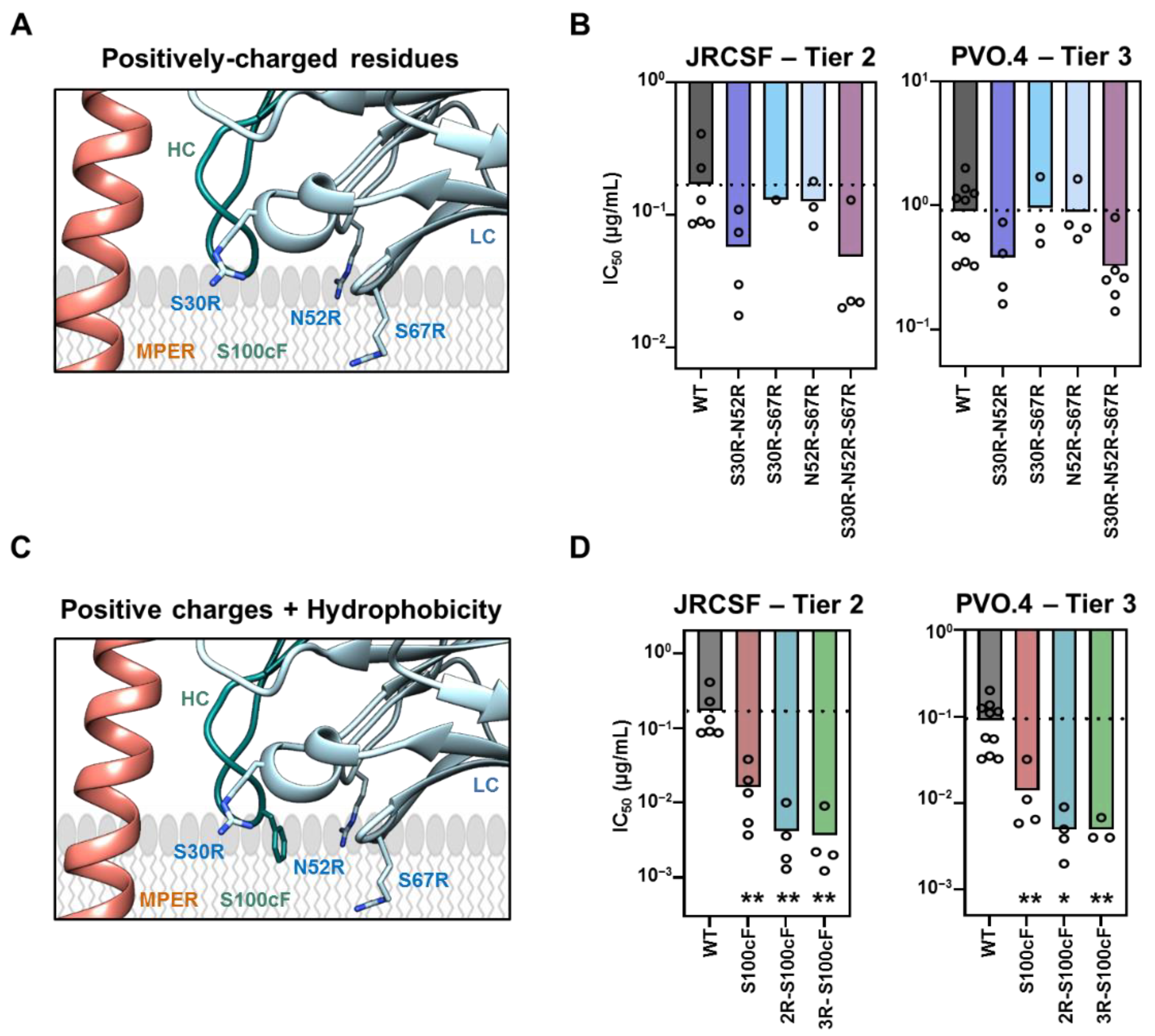
2.2. Requirements for Arg-Mediated 10E8 Potentiation and Complementation with HC-Ser100cPhe Mutation
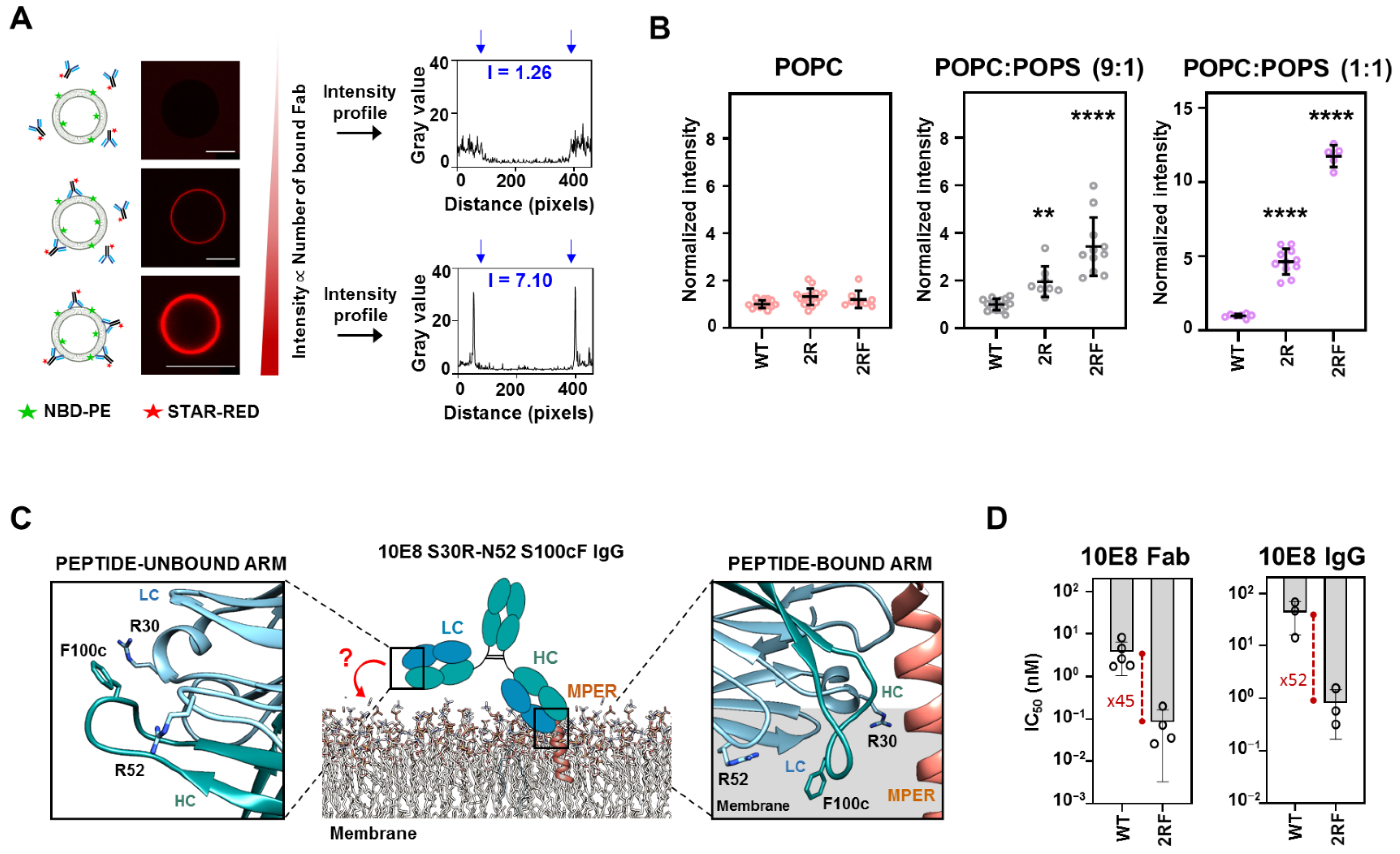
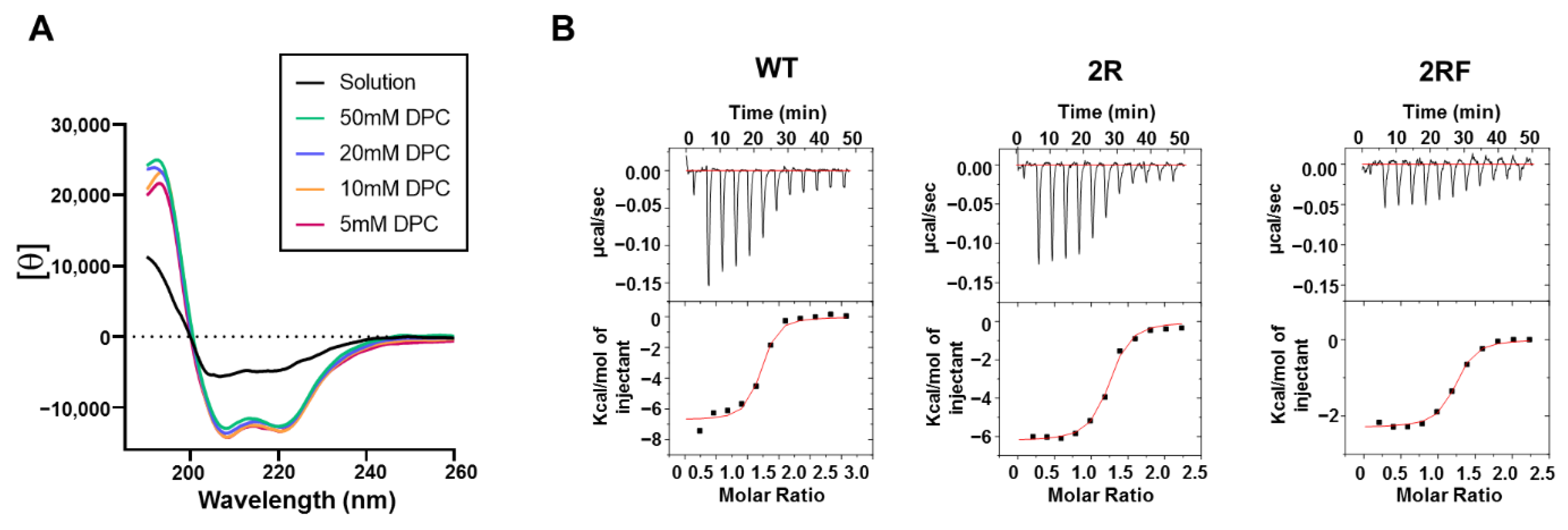
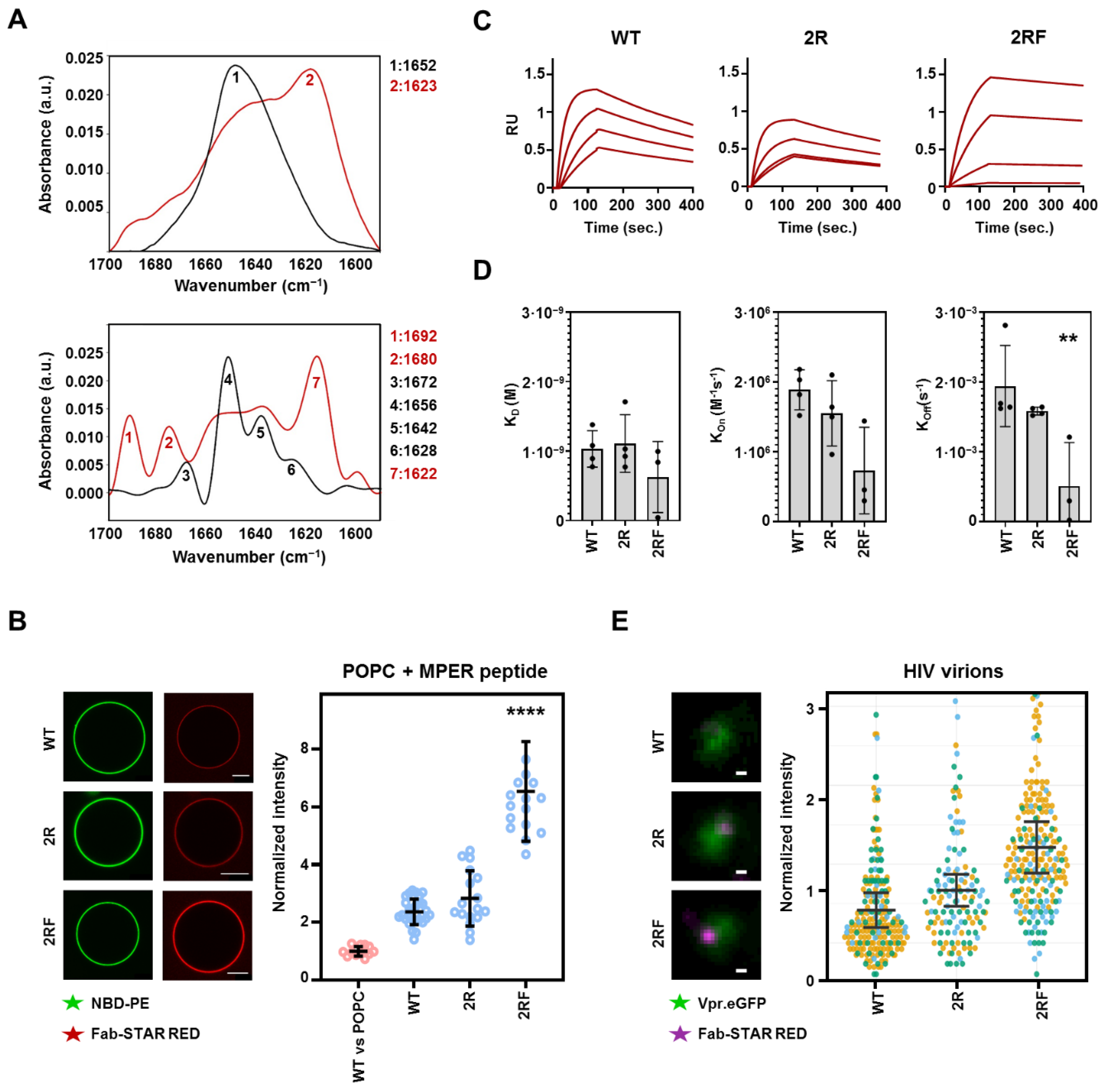
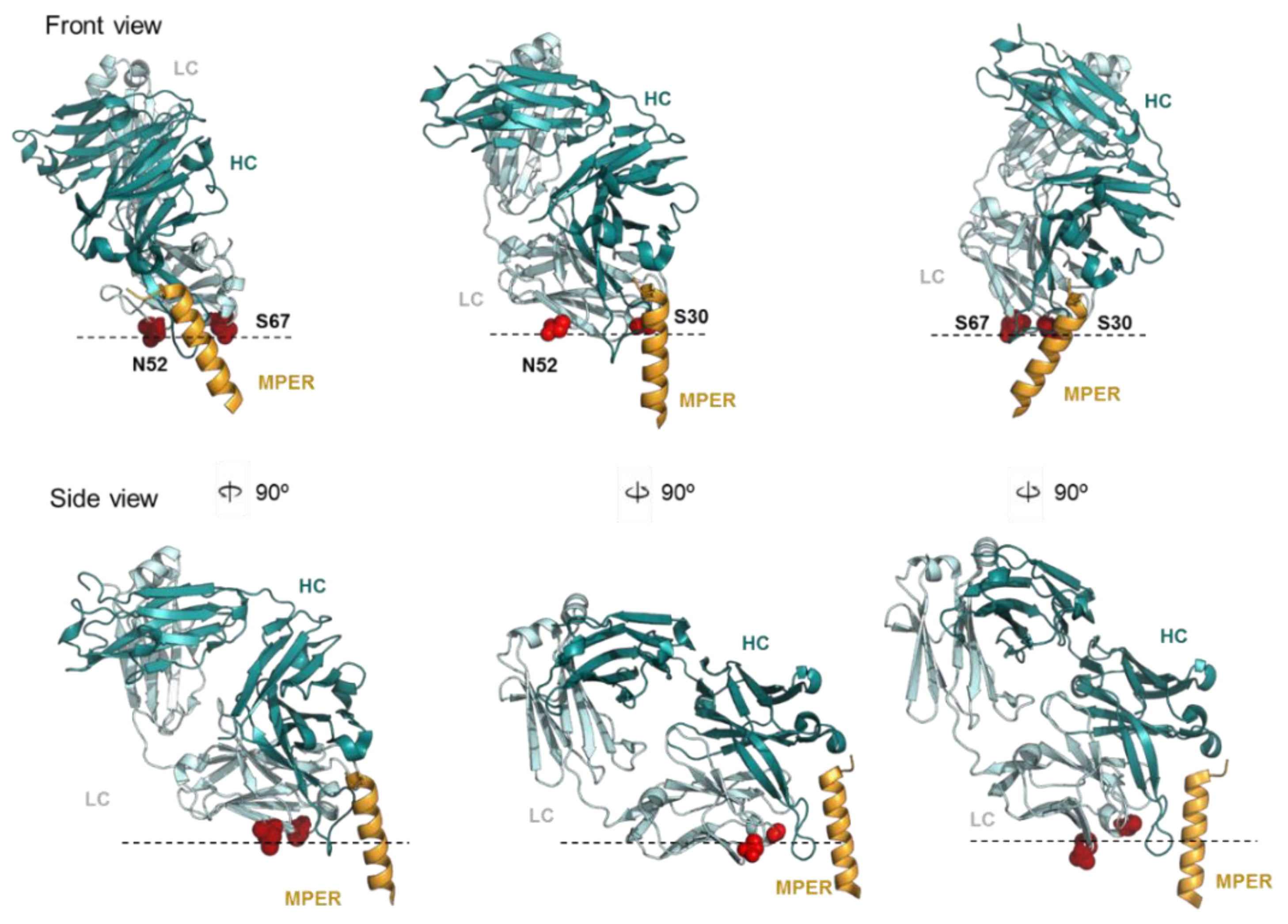
2.3. LC Arg and HC S100cF Substitutions Promote Antibody–Membrane Interaction
2.4. 10E8 Substitutions Promote MPER-Binding in Model Membranes and Native Environments
3. Discussion
Concluding Remarks
4. Materials and Methods
4.1. Reagents
4.2. Antibody Production, Characterization and Labeling
4.3. Circular Dichroism
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Pseudovirus Production
4.6. Cell-Entry Inhibition Assay
4.7. Transmission Infrared Spectroscopy
4.8. Single-Vesicle-Binding Assay
4.9. Isothermal Titration Calorimetry (ITC)
4.10. Surface Plasmon Resonance (SPR) Measurements
4.11. STED Imaging
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blattner, C.; Lee, J.H.; Sliepen, K.; Derking, R.; Falkowska, E.; de la Peña, A.T.; Cupo, A.; Julien, J.-P.; van Gils, M.; Lee, P.S.; et al. Structural Delineation of a Quaternary, Cleavage-Dependent Epitope at the gp41-gp120 Interface on Intact HIV-1 Env Trimers. Immunity 2014, 40, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Sok, D.; Burton, D.R. Recent progress in broadly neutralizing antibodies to HIV. Nat. Immunol. 2018, 19, 1179–1188. [Google Scholar] [CrossRef]
- Burton, D.R. Advancing an HIV vaccine; advancing vaccinology. Nat. Rev. Immunol. 2018, 19, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 2019, 568, 415–419. [Google Scholar] [CrossRef]
- Carravilla, P.; Chojnacki, J.; Rujas, E.; Insausti, S.; Largo, E.; Waithe, D.; Apellaniz, B.; Sicard, T.; Julien, J.-P.; Eggeling, C.; et al. Molecular recognition of the native HIV-1 MPER revealed by STED microscopy of single virions. Nat. Commun. 2019, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.M.; Leaman, D.P.; Lovendahl, K.N.; Croft, J.T.; Benhaim, M.A.; Hodge, E.A.; Zwick, M.B.; Lee, K.K. Cryo-ET of Env on intact HIV virions reveals structural variation and positioning on the Gag lattice. Cell 2022, 185, 641–653. [Google Scholar] [CrossRef]
- Zwick, M.B.; Labrijn, A.F.; Wang, M.; Spenlehauer, C.; Saphire, E.O.; Binley, J.M.; Moore, J.P.; Stiegler, G.; Katinger, H.; Burton, D.R.; et al. Broadly Neutralizing Antibodies Targeted to the Membrane-Proximal External Region of Human Immunodeficiency Virus Type 1 Glycoprotein gp41. J. Virol. 2001, 75, 10892–10905. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef]
- Williams, L.D.; Ofek, G.; Schätzle, S.; McDaniel, J.R.; Lu, X.; Nicely, N.I.; Wu, L.; Lougheed, C.S.; Bradley, T.; Louder, M.K.; et al. Potent and broad HIV-neutralizing antibodies in memory B cells and plasma. Sci. Immunol. 2017, 2, eaal2200. [Google Scholar] [CrossRef]
- Krebs, S.J.; Kwon, Y.D.; Schramm, C.A.; Law, W.H.; Donofrio, G.; Zhou, K.H.; Gift, S.; Dussupt, V.; Georgiev, I.S.; Schätzle, S.; et al. Longitudinal Analysis Reveals Early Development of Three MPER-Directed Neutralizing Antibody Lineages from an HIV-1-Infected Individual. Immunity 2019, 50, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Fenwick, C.; Caillat, C.; Silacci, C.; Guseva, S.; Dehez, F.; Chipot, C.; Barbieri, S.; Minola, A.; Jarrossay, D.; et al. Structural Basis for Broad HIV-1 Neutralization by the MPER-Specific Human Broadly Neutralizing Antibody LN01. Cell Host Microbe 2019, 26, 623–637. [Google Scholar] [CrossRef]
- Zhang, L.; IAVI Protocol G Investigators; Irimia, A.; He, L.; Landais, E.; Rantalainen, K.; Leaman, D.P.; Vollbrecht, T.; Stano, A.; Sands, D.I.; et al. An MPER antibody neutralizes HIV-1 using germline features shared among donors. Nat. Commun. 2019, 10, 5389. [Google Scholar] [CrossRef] [PubMed]
- Caillat, C.; Guilligay, D.; Sulbaran, G.; Weissenhorn, W. Neutralizing Antibodies Targeting HIV-1 gp41. Viruses 2020, 12, 1210. [Google Scholar] [CrossRef]
- Kim, A.; Leaman, D.P.; Zwick, M.B. Antibody to gp41 MPER Alters Functional Properties of HIV-1 Env without Complete Neutralization. PLoS Pathog. 2014, 10, e1004271. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.D.; Georgiev, I.S.; Ofek, G.; Zhang, B.; Asokan, M.; Bailer, R.T.; Bao, A.; Caruso, W.; Chen, X.; Choe, M.; et al. Optimization of the Solubility of HIV-1-Neutralizing Antibody 10E8 through Somatic Variation and Structure-Based Design. J. Virol. 2016, 90, 5899–5914. [Google Scholar] [CrossRef]
- Rujas, E.; Caaveiro, J.M.M.; Partida-Hanon, A.; Gulzar, N.; Morante, K.; Apellániz, B.; García-Porras, M.; Bruix, M.; Tsumoto, K.; Scott, J.K.; et al. Structural basis for broad neutralization of HIV-1 through the molecular recognition of 10E8 helical epitope at the membrane interface. Sci. Rep. 2016, 6, 38177. [Google Scholar] [CrossRef] [PubMed]
- Irimia, A.; Serra, A.M.; Sarkar, A.; Jacak, R.; Kalyuzhniy, O.; Sok, D.; Saye-Francisco, K.L.; Schiffner, T.; Tingle, R.; Kubitz, M.; et al. Lipid interactions and angle of approach to the HIV-1 viral membrane of broadly neutralizing antibody 10E8: Insights for vaccine and therapeutic design. PLoS Pathog. 2017, 13, e1006212. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.D.; Chuang, G.-Y.; Zhang, B.; Bailer, R.T.; Doria-Rose, N.A.; Gindin, T.S.; Lin, B.; Louder, M.K.; McKee, K.; O’Dell, S.; et al. Surface-Matrix Screening Identifies Semi-specific Interactions that Improve Potency of a Near Pan-reactive HIV-1-Neutralizing Antibody. Cell Rep. 2018, 22, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Asokan, M.; Rudicell, R.S.; Louder, M.; McKee, K.; O'Dell, S.; Stewart-Jones, G.; Wang, K.; Xu, L.; Chen, X.; Choe, M.; et al. Bispecific Antibodies Targeting Different Epitopes on the HIV-1 Envelope Exhibit Broad and Potent Neutralization. J. Virol. 2015, 89, 12501–12512. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pegu, A.; Rao, E.; Doria-Rose, N.; Beninga, J.; McKee, K.; Lord, D.M.; Wei, R.R.; Deng, G.; Louder, M.; et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 2017, 358, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yu, J.; Lanzi, A.; Yao, X.; Andrews, C.D.; Tsai, L.; Gajjar, M.R.; Sun, M.; Seaman, M.S.; Padte, N.N.; et al. Engineered Bispecific Antibodies with Exquisite HIV-1-Neutralizing Activity. Cell 2016, 165, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, J.; Guenaga, J.; Turner, H.L.; McKee, K.; Louder, M.K.; O’Dell, S.; Chiang, C.-I.; Lei, L.; Galkin, A.; Andrianov, A.K.; et al. Rational design of a trispecific antibody targeting the HIV-1 Env with elevated anti-viral activity. Nat. Commun. 2018, 9, 877. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.N.; Sok, D.; Tran, K.; Movsesyan, A.; Dubrovskaya, V.; Burton, D.R.; Wyatt, R.T. Targeting the HIV-1 Spike and Coreceptor with Bi- and Trispecific Antibodies for Single-Component Broad Inhibition of Entry. J. Virol. 2018, 92, e00384-18. [Google Scholar] [CrossRef]
- Wagh, K.; Seaman, M.S.; Zingg, M.; Fitzsimons, T.; Barouch, D.H.; Burton, D.R.; Connors, M.; Ho, D.D.; Mascola, J.R.; Nussenzweig, M.C.; et al. Potential of conventional & bispecific broadly neutralizing antibodies for prevention of HIV-1 subtype A, C & D infections. PLoS Pathog. 2018, 14, e1006860. [Google Scholar] [CrossRef]
- Rujas, E.; Cui, H.; Burnie, J.; Aschner, C.B.; Zhao, T.; Insausti, S.; Muthuraman, K.; Semesi, A.; Ophel, J.; Nieva, J.L.; et al. Engineering pan–HIV-1 neutralization potency through multispecific antibody avidity. Proc. Natl. Acad. Sci. USA 2022, 119, e2112887119. [Google Scholar] [CrossRef]
- Pegu, A.; Yang, Z.-Y.; Boyington, J.C.; Wu, L.; Ko, S.-Y.; Schmidt, S.D.; McKee, K.; Kong, W.-P.; Shi, W.; Chen, X.; et al. Neutralizing antibodies to HIV-1 envelope protect more effectively in vivo than those to the CD4 receptor. Sci. Transl. Med. 2014, 6, 243ra88. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, H.; Tang, X.; Li, L.; Geng, X.; Zhu, Y.; Chong, H.; He, Y. Cell membrane-anchored anti-HIV single-chain antibodies and bifunctional inhibitors targeting the gp41 fusion protein: New strategies for HIV gene therapy. Emerg. Microbes Infect. 2022, 11, 30–49. [Google Scholar] [CrossRef]
- Lee, J.H.; Ozorowski, G.; Ward, A.B. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 2016, 351, 1043–1048. [Google Scholar] [CrossRef]
- Rantalainen, K.; Berndsen, Z.T.; Antanasijevic, A.; Schiffner, T.; Zhang, X.; Lee, W.-H.; Torres, J.L.; Zhang, L.; Irimia, A.; Copps, J.; et al. HIV-1 Envelope and MPER Antibody Structures in Lipid Assemblies. Cell Rep. 2020, 31, 107583. [Google Scholar] [CrossRef]
- Rujas, E.; Leaman, D.P.; Insausti, S.; Ortigosa-Pascual, L.; Zhang, L.; Zwick, M.B.; Nieva, J.L. Functional Optimization of Broadly Neutralizing HIV-1 Antibody 10E8 by Promotion of Membrane Interactions. J. Virol. 2018, 92, e02249-17. [Google Scholar] [CrossRef] [Green Version]
- Rujas, E.; Insausti, S.; Leaman, D.P.; Carravilla, P.; González-Resines, S.; Monceaux, V.; Sánchez-Eugenia, R.; García-Porras, M.; Iloro, I.; Zhang, L.; et al. Affinity for the Interface Underpins Potency of Antibodies Operating in Membrane Environments. Cell Rep. 2020, 32, 108037. [Google Scholar] [CrossRef] [PubMed]
- Torralba, J.; de la Arada, I.; Carravilla, P.; Insausti, S.; Rujas, E.; Largo, E.; Eggeling, C.; Arrondo, J.L.R.; Apellaniz, B.; Nieva, J.L. Cholesterol Constrains the Antigenic Configuration of the Membrane-Proximal Neutralizing HIV-1 Epitope. ACS Infect. Dis. 2020, 6, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Caillat, C.; Guilligay, D.; Torralba, J.; Friedrich, N.; Nieva, J.L.; Trkola, A.; Chipot, C.J.; Dehez, F.L.; Weissenhorn, W. Structure of HIV-1 gp41 with its membrane anchors targeted by neutralizing antibodies. eLife 2021, 10, e65005. [Google Scholar] [CrossRef] [PubMed]
- Leaman, D.P.; Stano, A.; Chen, Y.; Zhang, L.; Zwick, M.B. Membrane Env Liposomes Facilitate Immunization with Multivalent Full-Length HIV Spikes. J. Virol. 2021, 95, e00005-21. [Google Scholar] [CrossRef]
- Wang, Y.; Kaur, P.; Sun, Z.-Y.J.; Elbahnasawy, M.A.; Hayati, Z.; Qiao, Z.-S.; Bui, N.N.; Chile, C.; Nasr, M.L.; Wagner, G.; et al. Topological analysis of the gp41 MPER on lipid bilayers relevant to the metastable HIV-1 envelope prefusion state. Proc. Natl. Acad. Sci. USA 2019, 116, 22556. [Google Scholar] [CrossRef]
- Kwon, B.; Lee, M.; Waring, A.J.; Hong, M. Oligomeric Structure and Three-Dimensional Fold of the HIV gp41 Membrane-Proximal External Region and Transmembrane Domain in Phospholipid Bilayers. J. Am. Chem. Soc. 2018, 140, 8246–8259. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Zemla, A.; Wilson, I.A.; Rupp, B. Antibody Elbow Angles are Influenced by their Light Chain Class. J. Mol. Biol. 2006, 357, 1566–1574. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- White, S.H.; Wimley, W.C. Membrane protein folding and stability: Physical Principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef]
- Vajda, S.; Porter, K.A.; Kozakov, D. Progress toward improved understanding of antibody maturation. Curr. Opin. Struct. Biol. 2021, 67, 226–231. [Google Scholar] [CrossRef]
- Arrondo, J.L.R.; Muga, A.; Castresana, J.; Goñi, F.M. Quantitative studies of the structure of proteins in solution by fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 1993, 59, 23–56. [Google Scholar] [CrossRef]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. STED super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Mulgrewnesbitt, A.; Diraviyam, K.; Wang, J.; Singh, S.; Murray, P.; Li, Z.; Rogers, L.; Mirkovic, N.; Murray, D. The role of electrostatics in protein–membrane interactions. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2006, 1761, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.M.; He, T.; Fuglebakk, E.; Grauffel, C.; Yang, B.; Roberts, M.F.; Gershenson, A.; Reuter, N. A Role for Weak Electrostatic Interactions in Peripheral Membrane Protein Binding. Biophys. J. 2016, 110, 1367–1378. [Google Scholar] [CrossRef]
- Alam, S.M.; Morelli, M.; Dennison, S.M.; Liao, H.-X.; Zhang, R.; Xia, S.-M.; Rits-Volloch, S.; Sun, L.; Harrison, S.C.; Haynes, B.F.; et al. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2009, 106, 20234–20239. [Google Scholar] [CrossRef]
- Bobardt, M.D.; Cheng, G.; de Witte, L.; Selvarajah, S.; Chatterji, U.; Sanders-Beer, B.E.; Geijtenbeek, T.B.H.; Chisari, F.V.; Gallay, P.A. Hepatitis C virus NS5A anchor peptide disrupts human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 2008, 105, 5525–5530. [Google Scholar] [CrossRef]
- Granqvist, N.; Yliperttula, M.; Välimäki, S.; Pulkkinen, P.; Tenhu, H.; Viitala, T. Control of the Morphology of Lipid Layers by Substrate Surface Chemistry. Langmuir 2014, 30, 2799–2809. [Google Scholar] [CrossRef]
- Galiani, S.; Waithe, D.; Reglinski, K.; Cruz-Zaragoza, L.D.; Garcia, E.; Clausen, M.P.; Schliebs, W.; Erdmann, R.; Eggeling, C. Super-resolution Microscopy Reveals Compartmentalization of Peroxisomal Membrane Proteins. J. Biol. Chem. 2016, 291, 16948–16962. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fab | KD (nM) | n | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|
| WT | 32.39 | 1.12 | −6.73 | −3.49 |
| 2R | 46.29 | 1.17 | −6.24 | −3.75 |
| 2RF | 41.3 | 1.16 | −2.3 | −7.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Insausti, S.; Garcia-Porras, M.; Torralba, J.; Morillo, I.; Ramos-Caballero, A.; de la Arada, I.; Apellaniz, B.; Caaveiro, J.M.M.; Carravilla, P.; Eggeling, C.; et al. Functional Delineation of a Protein–Membrane Interaction Hotspot Site on the HIV-1 Neutralizing Antibody 10E8. Int. J. Mol. Sci. 2022, 23, 10767. https://doi.org/10.3390/ijms231810767
Insausti S, Garcia-Porras M, Torralba J, Morillo I, Ramos-Caballero A, de la Arada I, Apellaniz B, Caaveiro JMM, Carravilla P, Eggeling C, et al. Functional Delineation of a Protein–Membrane Interaction Hotspot Site on the HIV-1 Neutralizing Antibody 10E8. International Journal of Molecular Sciences. 2022; 23(18):10767. https://doi.org/10.3390/ijms231810767
Chicago/Turabian StyleInsausti, Sara, Miguel Garcia-Porras, Johana Torralba, Izaskun Morillo, Ander Ramos-Caballero, Igor de la Arada, Beatriz Apellaniz, Jose M. M. Caaveiro, Pablo Carravilla, Christian Eggeling, and et al. 2022. "Functional Delineation of a Protein–Membrane Interaction Hotspot Site on the HIV-1 Neutralizing Antibody 10E8" International Journal of Molecular Sciences 23, no. 18: 10767. https://doi.org/10.3390/ijms231810767