
Microsatellite Variation in the Most Devastating Beetle Pests (Coleoptera: Curculionidae) of Agricultural and Forest Crops

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Insect Samples
2.2. Sample Preparation and DNA Extraction
2.3. Next-Generation Sequencing and Genome Assembly
2.4. Genome Sequences
2.5. Identification of Microsatellites
2.6. Assigning Microsatellites to Genomic Regions
2.7. Statistical Analysis
3. Results
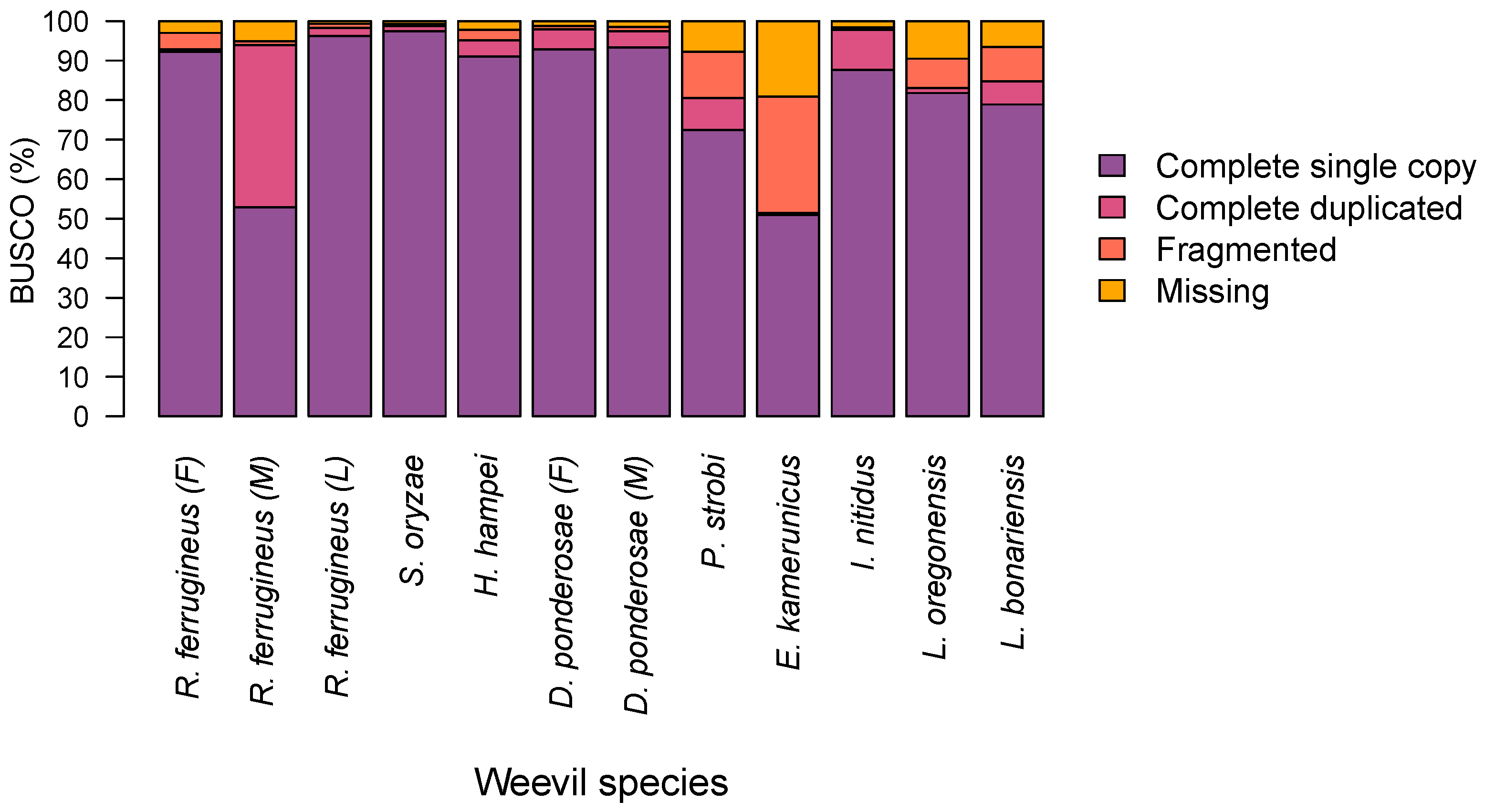
3.1. Genome Assembly and Assessing of Draft Genome Completeness
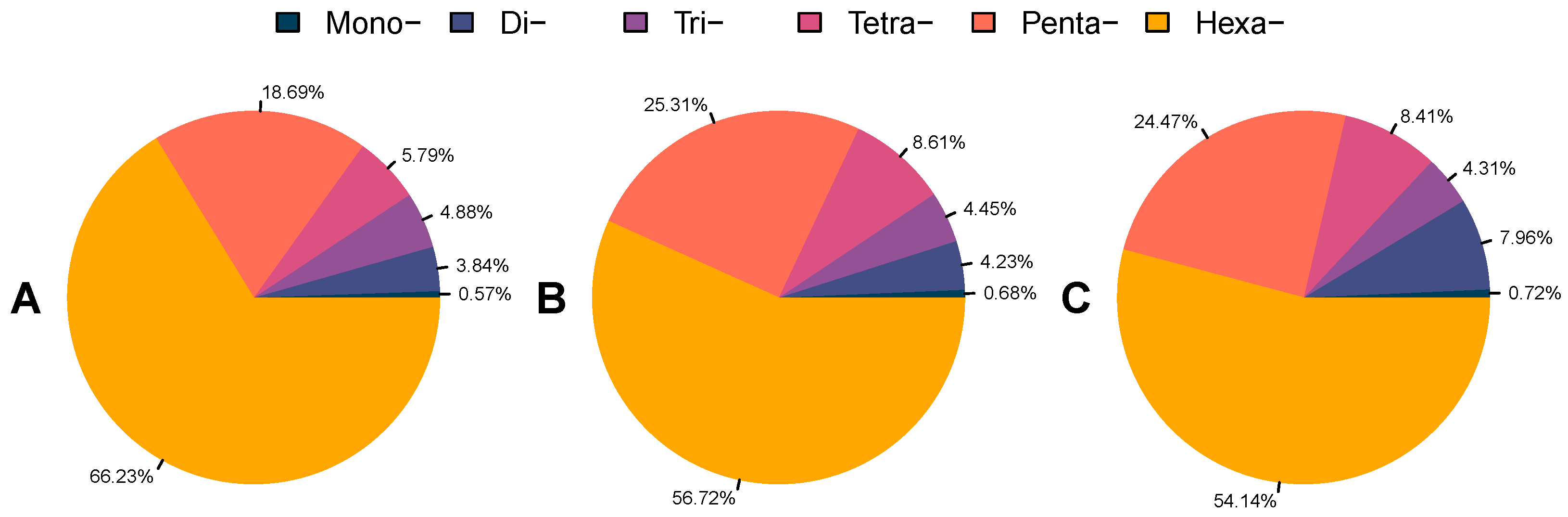
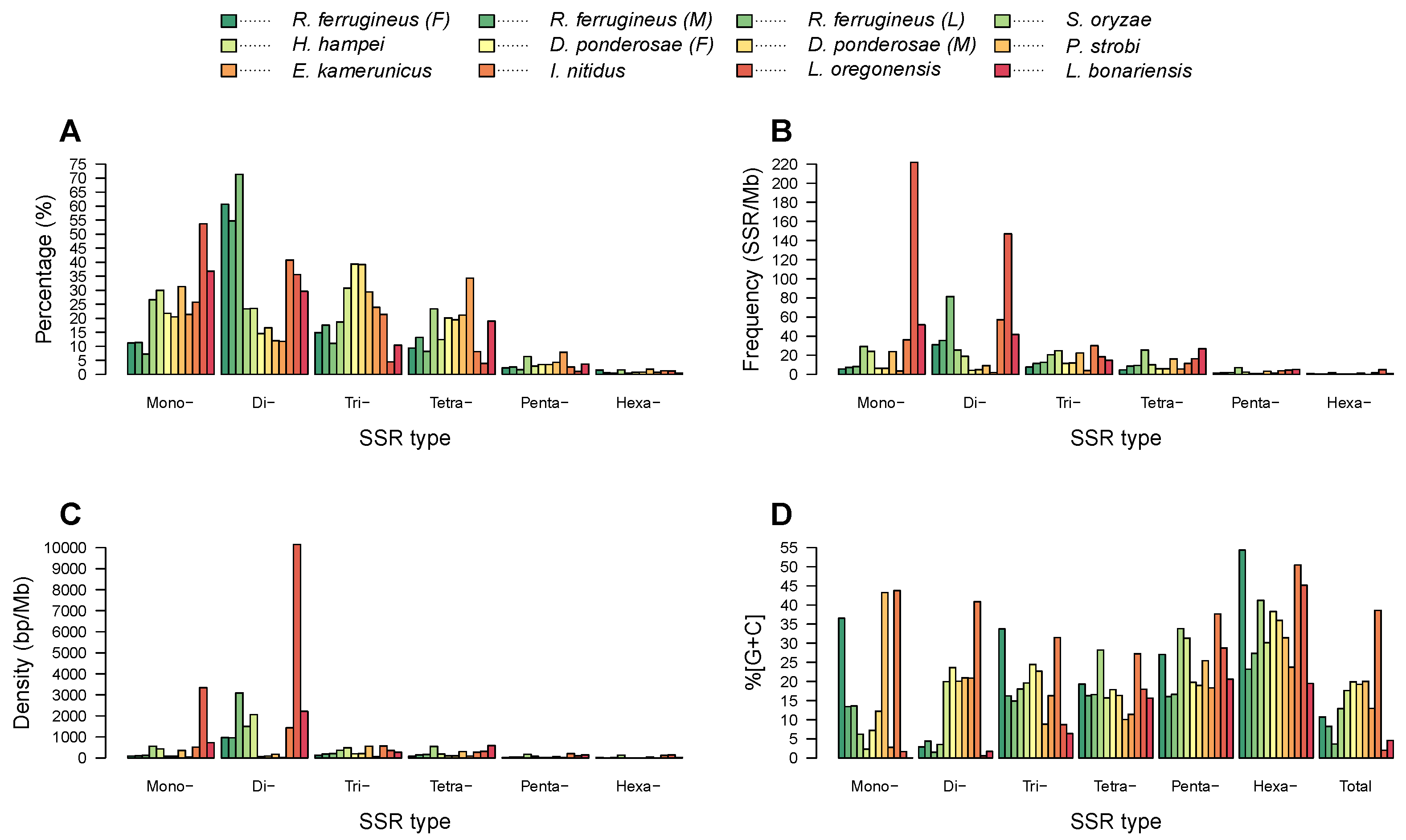
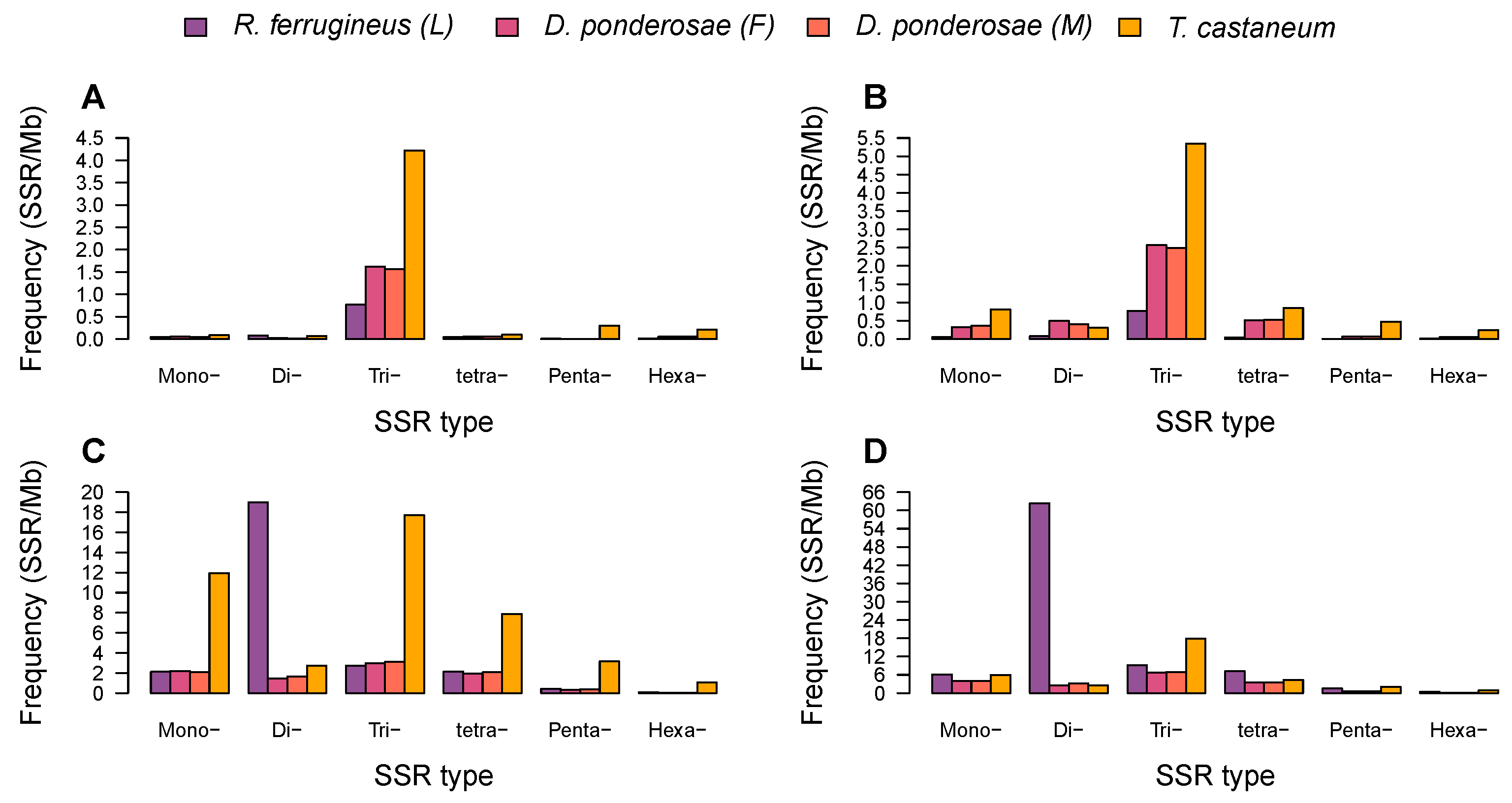
3.2. Identification and Characterization of Microsatellites in Beetle Genomes
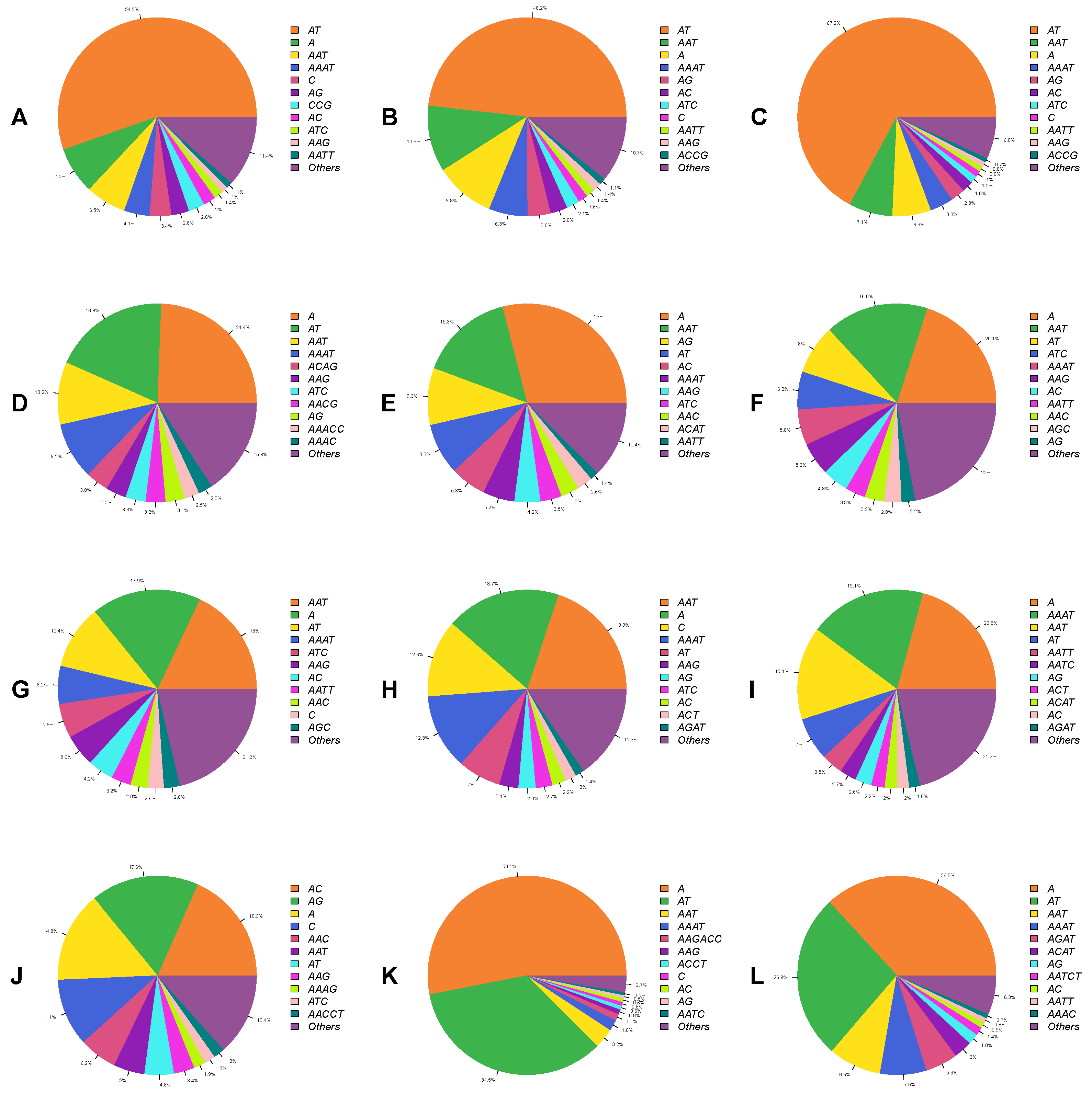
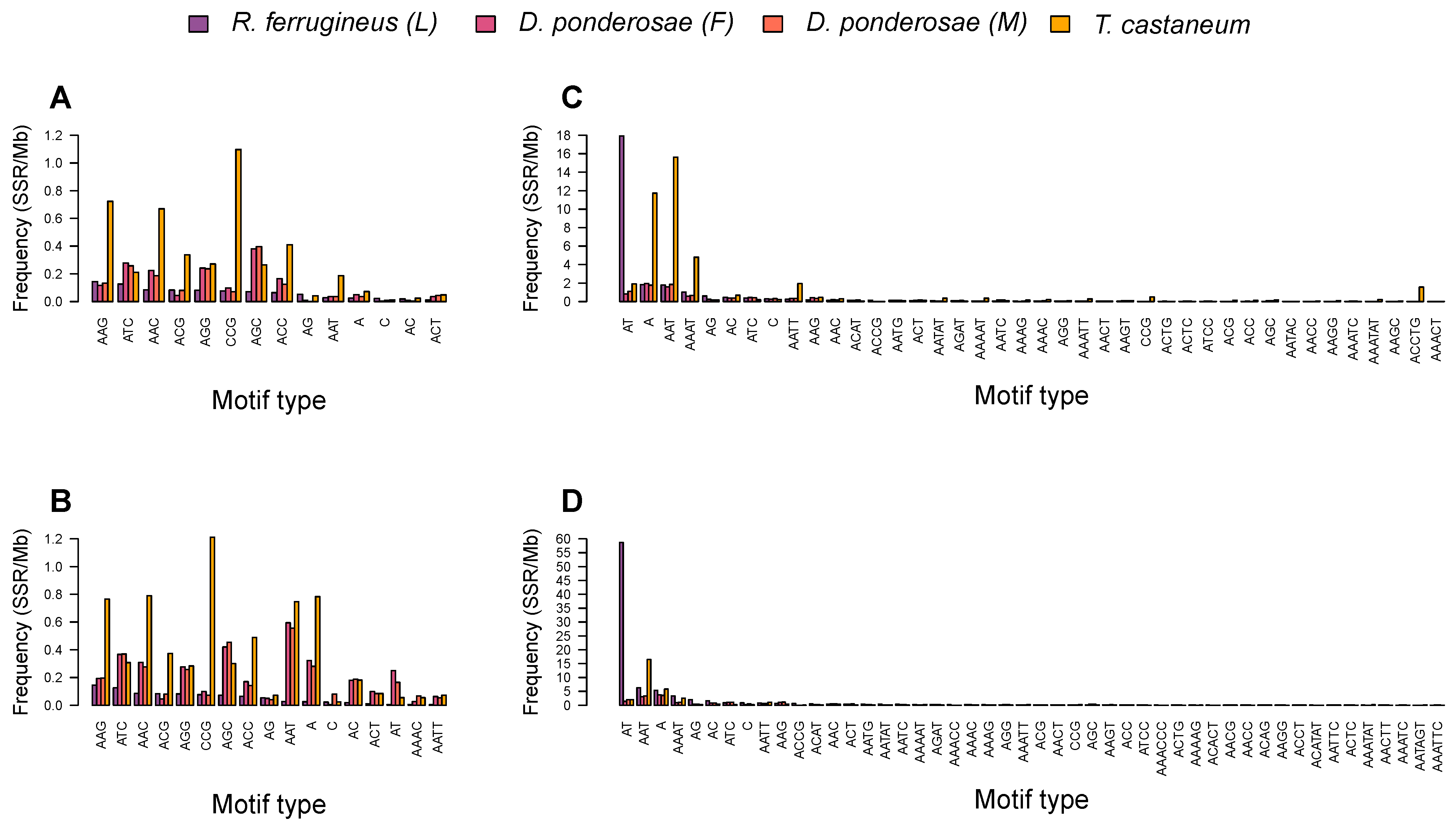
3.3. Diversity of Microsatellite Motifs in Beetle Genomes
3.4. Microsatellite Distribution and Motif Diversity According to Genomic Region
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bozdoğan, H.; Erbey, M.; Aksoy, H.A. Total amount of protein, lipid and carbohydrate of some adult species belong to curculionidae family (Coleoptera: Curculionidae). J. Entomol. Zool. Stud 2016, 4, 242–248. [Google Scholar]
- Bhatti, A.R.; Zia, A.; Mastoi, M.I.; Shehzad, M.I.A.; Iqbal, J. Tanymecus xanthuruschevrolat, 1880 (curculionidae: Entiminae), a new addition to curculionid fauna of pakistan. Pak. Entomol 2018, 40, 91–94. [Google Scholar]
- Rugman-Jones, P.F.; Hoddle, C.D.; Hoddle, M.S.; Stouthamer, R. The lesser of two weevils: Molecular-genetics of pest palm weevil populations confirm Rhynchophorus vulneratus (Panzer 1798) as a valid species distinct from R. ferrugineus (Olivier 1790), and reveal the global extent of both. PLoS ONE 2013, 8, e78379. [Google Scholar] [CrossRef]
- Aguirre, C.; Olivares, N.; Luppichini, P.; Hinrichsen, P. A PCR-based diagnostic system for differentiating two weevil species (Coleoptera: Curculionidae) of economic importance to the chilean citrus industry. J. Econ. Entomol. 2015, 108, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Milosavljević, I.; El-Shafie, H.A.; Faleiro, J.R.; Hoddle, C.D.; Lewis, M.; Hoddle, M.S. Palmageddon: The wasting of ornamental palms by invasive palm weevils, Rhynchophorus spp. J. Pest Sci. 2019, 92, 143–156. [Google Scholar] [CrossRef]
- Chen, H.; Chen, Z.; Zhou, Y. Rice water weevil (Coleoptera: Curculionidae) in mainland China: Invasion, spread and control. Crop Prot. 2005, 24, 695–702. [Google Scholar] [CrossRef]
- Bentz, B.J.; Jönsson, A.M.; Schroeder, M.; Weed, A.; Wilcke, R.A.I.; Larsson, K. Ips typographus and Dendroctonus ponderosae models project thermal suitability for intra-and inter-continental establishment in a changing climate. Front. For. Glob. Chang. 2019, 2, 1. [Google Scholar]
- Hansen, E.M.; Amacher, M.C.; Van Miegroet, H.; Long, J.N.; Ryan, M.G. Carbon dynamics in central US Rockies lodgepole pine type after mountain pine beetle outbreaks. For. Sci. 2015, 61, 665–679. [Google Scholar] [CrossRef]
- Griffith, R.; Koshy, P. Chapter Il Nematode Parasites of Coconut and Other Paims. Plant Parasit. Nematodes Subtrop. Trop. Agric. 1990, 363. [Google Scholar] [CrossRef]
- Cruz, L.F.; Menocal, O.; Mantilla, J.; Ibarra-Juarez, L.A.; Carrillo, D. Xyleborus volvulus (Coleoptera: Curculionidae): Biology and fungal associates. Appl. Environ. Microbiol. 2019, 85, e01190-19. [Google Scholar]
- Faleiro, J. A review of the issues and management of the red palm weevil Rhynchophorus ferrugineus (Coleoptera: Rhynchophoridae) in coconut and date palm during the last one hundred years. Int. J. Trop. Insect Sci. 2006, 26, 135–154. [Google Scholar]
- Chamorro, M.L.; de Medeiros, B.A.; Farrell, B.D. First phylogenetic analysis of Dryophthorinae (Coleoptera, Curculionidae) based on structural alignment of ribosomal DNA reveals Cenozoic diversification. Ecol. Evol. 2021, 11, 1984–1998. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Cao, L.J.; Hoffmann, A.A.; Gong, Y.J.; Chen, J.C.; Chen, H.S.; Wang, X.B.; Zeng, A.P.; Wei, S.J.; Zhou, Z.S. Rapid and strong population genetic differentiation and genomic signatures of climatic adaptation in an invasive mealybug. Divers. Distrib. 2020, 26, 610–622. [Google Scholar] [CrossRef]
- Katti, M.V.; Ranjekar, P.K.; Gupta, V.S. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 2001, 18, 1161–1167. [Google Scholar] [CrossRef]
- Bagshaw, A.T. Functional mechanisms of microsatellite DNA in eukaryotic genomes. Genome Biol. Evol. 2017, 9, 2428–2443. [Google Scholar] [CrossRef] [Green Version]
- Kashi, Y.; King, D.G. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006, 22, 253–259. [Google Scholar] [CrossRef]
- Kaakeh, W. Longevity, fecundity, and fertility of the red palm weevil, Rynchophorus ferrugineus Olivier (Coleoptera: Curculionidae) on natural and artificial diets. Emir. J. Food Agric. 2005, 23–33. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Avvaru, A.K.; Sowpati, D.T.; Mishra, R.K. PERF: An exhaustive algorithm for ultra-fast and efficient identification of microsatellites from large DNA sequences. Bioinformatics 2017, 27, 573. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hou, W.; Sun, T.; Xu, Y.; Li, P.; Yue, B.; Fan, Z.; Li, J. Genome-wide mining and comparative analysis of microsatellites in three macaque species. Mol. Genet. Genom. 2017, 292, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.H.; Jiang, X.M.; Yan, C.C.; Zhang, W.Q.; Xiao, G.S.; Yue, B.S.; Zhou, C.Q. Distribution patterns and variation analysis of simple sequence repeats in different genomic regions of bovid genomes. Sci. Rep. 2018, 8, 14407. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Pethiyagoda, C. Simple repetitive DNA sequences from primates: Compilation and analysis. J. Mol. Evol. 1995, 40, 120–126. [Google Scholar] [CrossRef]
- Li, C.Y.; Liu, L.; Yang, J.; Li, J.B.; Su, Y.; Zhang, Y.; Wang, Y.Y.; Zhu, Y.Y. Genome-wide analysis of microsatellite sequence in seven filamentous fungi. Interdiscip. Sci. Comput. Life Sci. 2009, 1, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Hazzouri, K.M.; Sudalaimuthuasari, N.; Kundu, B.; Nelson, D.; Al-Deeb, M.A.; Le Mansour, A.; Spencer, J.J.; Desplan, C.; Amiri, K. The genome of pest Rhynchophorus ferrugineus reveals gene families important at the plant-beetle interface. Commun. Biol. 2020, 3, 323. [Google Scholar] [CrossRef]
- Dias, G.B.; Altammami, M.A.; El-Shafie, H.A.; Alhoshani, F.M.; Al-Fageeh, M.B.; Bergman, C.M.; Manee, M.M. Haplotype-resolved genome assembly enables gene discovery in the red palm weevil Rhynchophorus ferrugineus. Sci. Rep. 2021, 11, 9987. [Google Scholar] [CrossRef]
- Parisot, N.; Vargas-Chávez, C.; Goubert, C.; Baa-Puyoulet, P.; Balmand, S.; Beranger, L.; Blanc, C.; Bonnamour, A.; Boulesteix, M.; Burlet, N.; et al. The transposable element-rich genome of the cereal pest Sitophilus oryzae. BMC Biol. 2021, 19, 241. [Google Scholar] [CrossRef]
- Vega, F.E.; Brown, S.M.; Chen, H.; Shen, E.; Nair, M.B.; Ceja-Navarro, J.A.; Brodie, E.L.; Infante, F.; Dowd, P.F.; Pain, A. Draft genome of the most devastating insect pest of coffee worldwide: The coffee berry borer, Hypothenemus hampei. Sci. Rep. 2015, 5, 12525. [Google Scholar] [CrossRef]
- Apriyanto, A. Draft genome sequence, annotation, and SSR mining data of Elaeidobius kamerunicus Faust., an essential oil palm pollinating weevil. Data Brief 2021, 34, 106745. [Google Scholar] [CrossRef] [PubMed]
- Harrop, T.W.; Le Lec, M.F.; Jauregui, R.; Taylor, S.E.; Inwood, S.N.; van Stijn, T.; Henry, H.; Skelly, J.; Ganesh, S.; Ashby, R.L.; et al. Genetic diversity in invasive populations of argentine stem weevil associated with adaptation to biocontrol. Insects 2020, 11, 441. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Mishra, R.K.; Singh, L. Genome-wide analysis of microsatellite repeats in humans: Their abundance and density in specific genomic regions. Genome Biol. 2003, 4, R13. [Google Scholar] [CrossRef]
- Manee, M.M.; Al-Shomrani, B.M.; Al-Fageeh, M.B. Genome-wide characterization of simple sequence repeats in Palmae genomes. Genes Genom. 2020, 42, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Xu, H.; Song, J.; Xu, J.; Zhu, Y.; Chen, S. Genome-wide analysis of simple sequence repeats in the model medicinal mushroom Ganoderma lucidum. Gene 2013, 512, 331–336. [Google Scholar] [CrossRef]
- Karaoglu, H.; Lee, C.M.Y.; Meyer, W. Survey of simple sequence repeats in completed fungal genomes. Mol. Biol. Evol. 2005, 22, 639–649. [Google Scholar] [CrossRef]
- Xu, Y.; Li, W.; Hu, Z.; Zeng, T.; Shen, Y.; Liu, S.; Zhang, X.; Li, J.; Yue, B. Genome-wide mining of perfect microsatellites and tetranucleotide orthologous microsatellites estimates in six primate species. Gene 2018, 643, 124–132. [Google Scholar] [CrossRef]
- Sharma, P.C.; Grover, A.; Kahl, G. Mining microsatellites in eukaryotic genomes. Trends Biotechnol. 2007, 25, 490–498. [Google Scholar] [CrossRef]
- Webster, M.T.; Smith, N.G.; Ellegren, H. Microsatellite evolution inferred from human–chimpanzee genomic sequence alignments. Proc. Natl. Acad. Sci. USA 2002, 99, 8748–8753. [Google Scholar] [CrossRef]
- Pascual, M.; Schug, M.D.; Aquadro, C.F. High density of long dinucleotide microsatellites in Drosophila subobscura. Mol. Biol. Evol. 2000, 17, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A. Optimizing depth and type of high-throughput sequencing data for microsatellite discovery. Appl. Plant Sci. 2019, 7, e11298. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Wang, S.; He, K.; Jiang, M.; Li, F. Large-scale analysis reveals that the genome features of simple sequence repeats are generally conserved at the family level in insects. BMC Genom. 2017, 18, 848. [Google Scholar] [CrossRef]
- Wang, X.T.; Zhang, Y.J.; Qiao, L.; Chen, B. Comparative analyses of simple sequence repeats (SSRs) in 23 mosquito species genomes: Identification, characterization and distribution (Diptera: Culicidae). Insect Sci. 2019, 26, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Peng, Z.; Sun, H.; Yue, X.; Lou, Y.; Dong, L.; Wang, L.; Gao, Z. Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci. Rep. 2015, 5, 8018. [Google Scholar] [CrossRef]
- Wang, Q.; Fang, L.; Chen, J.; Hu, Y.; Si, Z.; Wang, S.; Chang, L.; Guo, W.; Zhang, T. Genome-wide mining, characterization and development of microsatellite markers in Gossypium species. Sci. Rep. 2015, 5, 10638. [Google Scholar] [CrossRef]
- Kumpatla, S.P.; Mukhopadhyay, S. Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 2005, 48, 985–998. [Google Scholar] [CrossRef]
- Peng, X.; Yang, Z.; Xu, L.; Wang, H.; Guo, C.; Hu, P. Genome Survey Sequencing and Identification of Genomic SSR Markers for Batocera Horsfieldi (Coleoptera: Cerambycidae). 2021. Available online: https://www.researchsquare.com/article/rs-498077/v1 (accessed on 21 July 2022).
- Manee, M.M.; Algarni, A.T.; Alharbi, S.N.; Al-Shomrani, B.M.; Ibrahim, M.A.; Binghadir, S.A.; Al-Fageeh, M.B. Genome-wide characterization and analysis of microsatellite sequences in camelid species. Mammal Res. 2020, 65, 359–373. [Google Scholar] [CrossRef]
- Kim, T.S.; Booth, J.G.; Gauch, H.G.; Sun, Q.; Park, J.; Lee, Y.H.; Lee, K. Simple sequence repeats in Neurospora crassa: Distribution, polymorphism and evolutionary inference. BMC Genom. 2008, 9, 31. [Google Scholar] [CrossRef]
- Bacolla, A.; Larson, J.E.; Collins, J.R.; Li, J.; Milosavljevic, A.; Stenson, P.D.; Cooper, D.N.; Wells, R.D. Abundance and length of simple repeats in vertebrate genomes are determined by their structural properties. Genome Res. 2008, 18, 1545–1553. [Google Scholar] [CrossRef]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, G.; Li, X.; Wang, L.; Yuan, J.; Deng, C.; Gao, W. Genome-wide identification and validation of simple sequence repeats (SSRs) from Asparagus officinalis. Mol. Cell. Probes 2016, 30, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhao, J.; Liu, M.; Liu, P.; Dai, L.; Zhao, Z. Genome-wide characterization of simple sequence repeat (SSR) loci in Chinese jujube and jujube SSR primer transferability. PLoS ONE 2015, 10, e0127812. [Google Scholar]
- Sureshkumar, S.; Todesco, M.; Schneeberger, K.; Harilal, R.; Balasubramanian, S.; Weigel, D. A genetic defect caused by a triplet repeat expansion in Arabidopsis thaliana. Science 2009, 323, 1060–1063. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within genes: Structure, function, and evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef]
- Qi, W.H.; Yan, C.c.; Li, W.J.; Jiang, X.M.; Li, G.Z.; Zhang, X.Y.; Hu, T.Z.; Li, J.; Yue, B.S. Distinct patterns of simple sequence repeats and GC distribution in intragenic and intergenic regions of primate genomes. Aging (Albany NY) 2016, 8, 2635. [Google Scholar] [CrossRef]
- Hong, C.P.; Piao, Z.Y.; Kang, T.W.; Batley, J.; Yang, T.; Hur, Y.; Bhak, J.; Park, B.; Edwards, D.; Lim, Y.P.; et al. Genomic distribution of simple sequence repeats in Brassica rapa. Mol. Cells 2007, 23, 349. [Google Scholar]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar]
- Vinogradov, A.E. DNA helix: The importance of being GC-rich. Nucleic Acids Res. 2003, 31, 1838–1844. [Google Scholar] [CrossRef]
- Kudla, G.; Lipinski, L.; Caffin, F.; Helwak, A.; Zylicz, M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006, 4, e180. [Google Scholar] [CrossRef]
- Zhang, Z.Q. Animal Biodiversity: An Outline of Higher-Level Classification and Survey of Taxonomic Richness; Magnolia Press: Auckland, New Zealand, 2011. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insect Name | Common Name | Genome Size (Mb) | Number of SSRs | Frequency (SSR/Mb) | Density (bp/Mb) | SSRs Content (%) | Reference |
|---|---|---|---|---|---|---|---|
| R. ferrugineus (F) | Female red palm weevil | 1121.36 | 57,175 | 50.99 | 1320.92 | 0.13 | This study |
| R. ferrugineus (M) | Male red palm weevil | 782.10 | 50,723 | 64.86 | 1445.93 | 0.14 | [27] |
| R. ferrugineus (L) | Red palm weevil larva | 589.40 | 67,261 | 114.11 | 3649.45 | 0.36 | [28] |
| S. oryzae | Rice weevil | 770.57 | 84,391 | 109.52 | 3287.11 | 0.33 | [29] |
| H. hampei | Coffee berry borer | 162.57 | 13,092 | 80.53 | 3260.24 | 0.33 | [30] |
| D. ponderosae (F) | Female mountain pine beetle | 223.74 | 6505 | 29.07 | 481.68 | 0.05 | Unpublished |
| D. ponderosae (M) | Male mountain pine beetle | 224.79 | 6803 | 30.26 | 511.44 | 0.05 | Unpublished |
| P. strobi | White pine weevil | 2025.02 | 154,511 | 76.30 | 1516.87 | 0.15 | Unpublished |
| E. kamerunicus | African oil palm weevil | 269.64 | 4397 | 16.31 | 249.98 | 0.02 | [31] |
| I. nitidus | Qinghai spruce bark beetle | 345.00 | 48372 | 140.21 | 3127.27 | 0.31 | Unpublished |
| L. oregonensis | Carrot weevil | 1293.28 | 534,123 | 412.99 | 14,406.26 | 1.44 | Unpublished |
| L. bonariensis | Argentine stem weevil | 1112.44 | 156,716 | 140.88 | 3976.45 | 0.40 | [32] |
| Repeat Type | Parameter | R. ferrugineus (F) | R. ferrugineus (M) | R. ferrugineus (L) | S. oryzae | H. hampei | D. ponderosae (F) | D. ponderosae (M) | P. strobi | E. kamerunicus | I. nitidus | L. oregonensis | L. bonariensis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mono- | Number of SSRs | 6393 | 5748 | 4906 | 22,484 | 3924 | 1415 | 1395 | 48,345 | 939 | 12,459 | 286,859 | 57,721 |
| Total length (bp) | 98,929 | 79,577 | 69,520 | 429,331 | 70,509 | 18,625 | 18,802 | 725,181 | 11,752 | 178,828 | 4,316,826 | 809,526 | |
| Average length (bp) | 15.47 | 13.84 | 14.17 | 19.09 | 17.97 | 13.16 | 13.48 | 15.00 | 12.52 | 14.35 | 15.05 | 14.02 | |
| Frequency (SSR/Mb) | 5.70 | 7.35 | 8.32 | 29.18 | 24.14 | 6.32 | 6.21 | 23.87 | 3.48 | 36.11 | 221.81 | 51.89 | |
| Density (bp/Mb) | 88.22 | 101.75 | 117.95 | 557.16 | 433.71 | 83.24 | 83.64 | 358.11 | 43.58 | 518.33 | 3337.89 | 727.70 | |
| Di- | Number of SSRs | 34,681 | 27,781 | 47,993 | 196,96 | 3077 | 944 | 1128 | 18,612 | 517 | 19,728 | 190,064 | 46,373 |
| Total length (bp) | 1,084,748 | 748,002 | 1,823,936 | 1,157,184 | 335,416 | 15,834 | 19,970 | 360,806 | 7412 | 493,798 | 13,130,600 | 2,463,404 | |
| Average length (bp) | 31.28 | 26.92 | 38.00 | 58.75 | 109.00 | 16.77 | 17.70 | 19.39 | 14.34 | 25.03 | 69.09 | 53.12 | |
| Frequency (SSR/Mb) | 30.93 | 35.52 | 81.43 | 25.56 | 18.93 | 4.22 | 5.02 | 9.19 | 1.92 | 57.18 | 146.96 | 41.69 | |
| Density (bp/Mb) | 967.35 | 956.40 | 3094.55 | 1501.73 | 2063.20 | 70.77 | 88.84 | 178.17 | 27.49 | 1431.28 | 10,152.94 | 2214.42 | |
| Tri- | Number of SSRs | 8517 | 8897 | 7407 | 15,786 | 4028 | 2558 | 2663 | 45,395 | 1050 | 10,357 | 23,752 | 16,301 |
| Total length (bp) | 142,764 | 149,838 | 126,207 | 284,631 | 79,143 | 44,475 | 46,941 | 1,137,378 | 15,882 | 196,965 | 457,914 | 304,446 | |
| Average length (bp) | 16.76 | 16.84 | 17.04 | 18.03 | 19.64821 | 17.39 | 17.63 | 25.06 | 15.13 | 19.02 | 19.28 | 18.68 | |
| Frequency (SSR/Mb) | 7.60 | 11.38 | 12.57 | 20.49 | 24.78 | 11.43 | 11.85 | 22.42 | 3.89 | 30.02 | 18.37 | 14.65 | |
| Density (bp/Mb) | 127.31 | 191.58 | 214.13 | 369.38 | 486.82 | 198.78 | 208.82 | 561.66 | 58.90 | 570.90 | 354.07 | 273.67 | |
| Tetra- | Number of SSRs | 5368 | 6700 | 5515 | 19,698 | 1627 | 1310 | 1323 | 32,698 | 1510 | 3915 | 21,085 | 29,736 |
| Total length (bp) | 94,688 | 116,572 | 97,284 | 422,340 | 30,380 | 22,688 | 22,752 | 621,160 | 24,604 | 95,912 | 406,368 | 654,160 | |
| Average length (bp) | 17.64 | 17.40 | 17.64 | 21.44 | 18.67 | 17.32 | 17.20 | 18.99 | 16.29 | 24.50 | 19.27 | 21.99 | |
| Frequency (SSR/Mb) | 4.79 | 8.57 | 9.36 | 25.56 | 10.01 | 5.86 | 5.89 | 16.15 | 5.60 | 11.35 | 16.30 | 26.73 | |
| Density (bp/Mb) | 84.44 | 149.05 | 165.06 | 548.09 | 186.87 | 101.40 | 101.21 | 306.74 | 91.25 | 278.00 | 314.21 | 588.04 | |
| Penta- | Number of SSRs | 1318 | 1324 | 1151 | 5375 | 382 | 230 | 239 | 6630 | 348 | 1289 | 5786 | 5784 |
| Total length (bp) | 30,555 | 28,995 | 25,330 | 135,845 | 13,175 | 4900 | 5145 | 147,940 | 6960 | 70,845 | 136,620 | 158,590 | |
| Average length (bp) | 23.18 | 21.90 | 22.01 | 25.27 | 34.49 | 21.30 | 21.52 | 22.31 | 20.00 | 54.96 | 23.61 | 27.42 | |
| Frequency (SSR/Mb) | 1.18 | 1.69 | 1.95 | 6.98 | 2.35 | 1.03 | 1.06 | 3.27 | 1.29 | 3.74 | 4.47 | 5.20 | |
| Density (bp/Mb) | 27.25 | 37.07 | 42.98 | 176.29 | 81.04 | 21.90 | 22.89 | 73.06 | 25.81 | 205.35 | 105.64 | 142.56 | |
| Hexa- | Number of SSRs | 898 | 273 | 289 | 1352 | 54 | 48 | 55 | 2831 | 33 | 624 | 6577 | 801 |
| Total length (bp) | 29,538 | 7872 | 8718 | 103,614 | 1398 | 1248 | 1356 | 79,230 | 792 | 42,576 | 183,012 | 33,432 | |
| Average length (bp) | 32.89 | 28.84 | 30.17 | 76.64 | 25.89 | 26.00 | 24.65 | 27.99 | 24.00 | 68.23 | 27.83 | 41.74 | |
| Frequency (SSR/Mb) | 0.80 | 0.35 | 0.49 | 1.75 | 0.33 | 0.21 | 0.24 | 1.40 | 0.12 | 1.81 | 5.09 | 0.72 | |
| Density (bp/Mb) | 26.34 | 10.07 | 14.79 | 134.47 | 8.60 | 5.58 | 6.03 | 39.13 | 2.94 | 123.41 | 141.51 | 30.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manee, M.M.; Al-Shomrani, B.M.; Altammami, M.A.; El-Shafie, H.A.F.; Alsayah, A.A.; Alhoshani, F.M.; Alqahtani, F.H. Microsatellite Variation in the Most Devastating Beetle Pests (Coleoptera: Curculionidae) of Agricultural and Forest Crops. Int. J. Mol. Sci. 2022, 23, 9847. https://doi.org/10.3390/ijms23179847
Manee MM, Al-Shomrani BM, Altammami MA, El-Shafie HAF, Alsayah AA, Alhoshani FM, Alqahtani FH. Microsatellite Variation in the Most Devastating Beetle Pests (Coleoptera: Curculionidae) of Agricultural and Forest Crops. International Journal of Molecular Sciences. 2022; 23(17):9847. https://doi.org/10.3390/ijms23179847
Chicago/Turabian StyleManee, Manee M., Badr M. Al-Shomrani, Musaad A. Altammami, Hamadttu A. F. El-Shafie, Atheer A. Alsayah, Fahad M. Alhoshani, and Fahad H. Alqahtani. 2022. "Microsatellite Variation in the Most Devastating Beetle Pests (Coleoptera: Curculionidae) of Agricultural and Forest Crops" International Journal of Molecular Sciences 23, no. 17: 9847. https://doi.org/10.3390/ijms23179847