
Post-COVID-19 Parkinsonism and Parkinson’s Disease Pathogenesis: The Exosomal Cargo Hypothesis
 , , ,
, , ,  , ,
, ,  , ,
, ,  ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Viral Ιnfections as Τriggers for Parkinsonism and PD Development
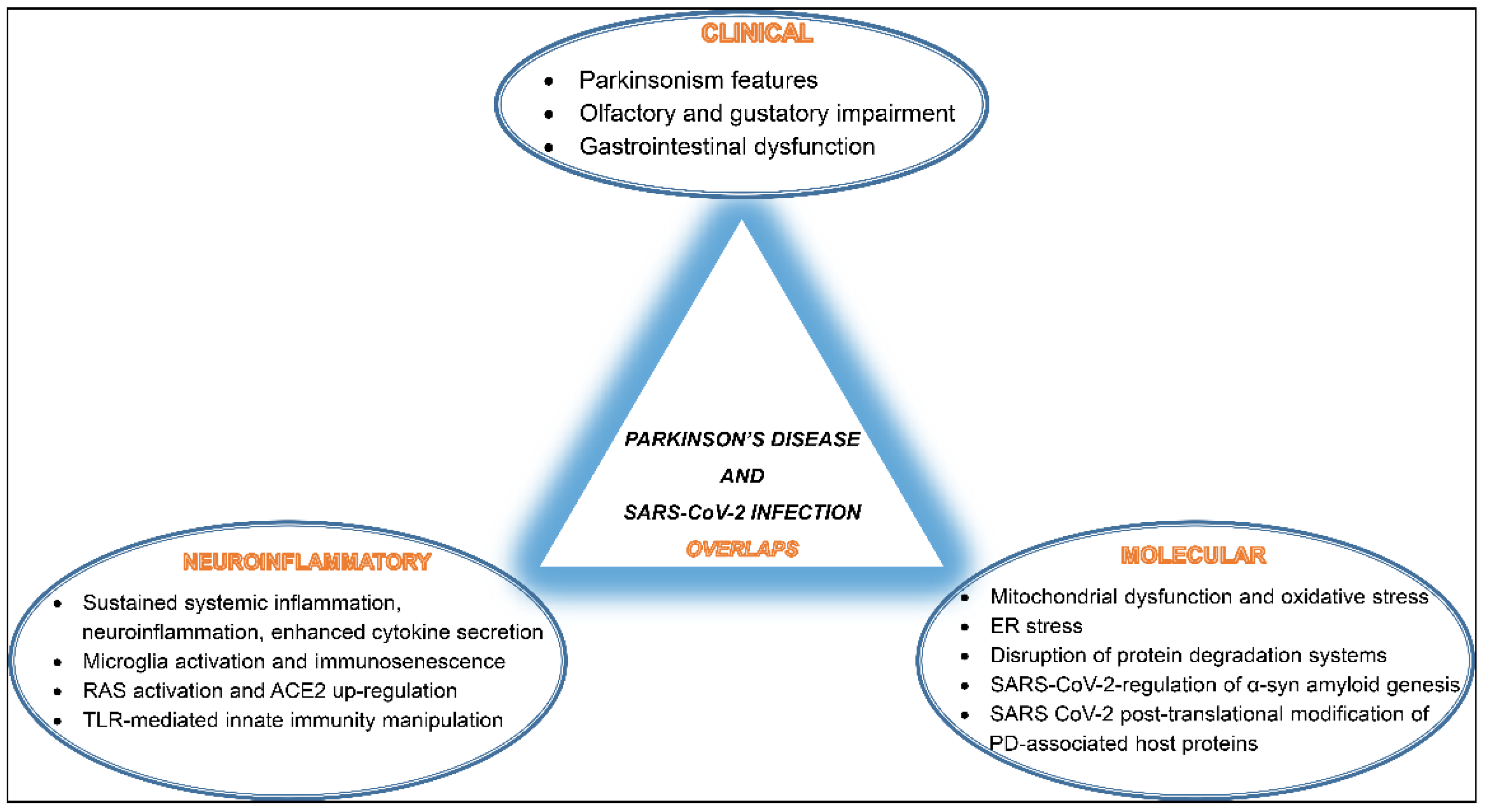
3. SARS-CoV-2 Infection and PD Overlaps
3.1. Clinical Co-Manifestations
3.2. Inflammatory and Molecular Overlapping Pathways
4. The Diverse Roles of Exosomes in Viral Infection and Neurogenerative Disease
4.1. Biogenesis of Exosomes
4.2. Impact of Exosomes in PD Pathogenesis
4.3. Exosomes as Biomarkers in PD Diagnosis
4.4. Exosomes and SARS-CoV-2 Infection
5. SARS-CoV-2-Related Εxosomal Cargo and Its Potential Roles in Post-COVID-19 Parkinsonism and PD Pathogenesis
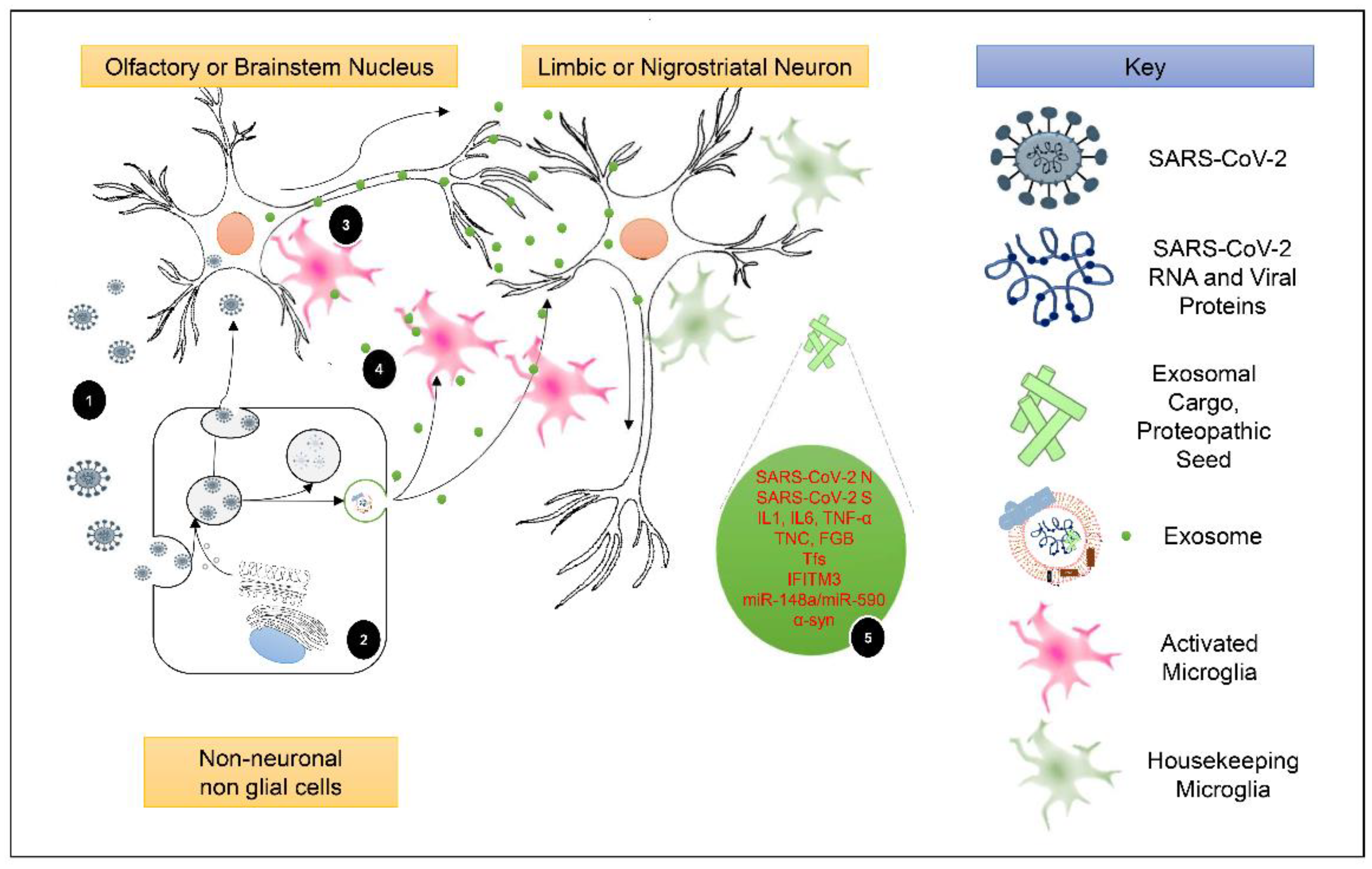
5.1. Exosomal Cargo and Induction of Post-COVID-19 Neuroinflammation
5.2. The Periphery-Exosomes-CNS Axis as a Promoter of Post-COVID-19 Parkinsonism and PD Development
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keener, A.M.; Bordelon, Y.M. Parkinsonism. Proc. Semin. Neurol. 2016, 36, 330–334. [Google Scholar]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Kobylarek, D.; Kozubski, W. Infectious Etiologies of Parkinsonism: Pathomechanisms and Clinical Implications. Front. Neurol. 2019, 10, 652. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Lee, A.; Gilbert, R.M. Epidemiology of Parkinson Disease. Neurol. Clin. 2016, 34, 955–965. [Google Scholar] [CrossRef]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef]
- Twelves, D.; Perkins, K.S.; Counsell, C. Systematic review of incidence studies of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2003, 18, 19–31. [Google Scholar] [CrossRef]
- Karimi-Moghadam, A.; Charsouei, S.; Bell, B.; Jabalameli, M.R. Parkinson Disease from Mendelian Forms to Genetic Susceptibility: New Molecular Insights into the Neurodegeneration Process. Cell. Mol. Neurobiol. 2018, 38, 1153–1178. [Google Scholar] [CrossRef]
- Warner, T.T.; Schapira, A.H. Genetic and environmental factors in the cause of Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S16–S23; discussion S23–S25. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Khoo, T.K.; Yarnall, A.J.; Duncan, G.W.; Coleman, S.; O’Brien, J.T.; Brooks, D.J.; Barker, R.A.; Burn, D.J. The spectrum of nonmotor symptoms in early Parkinson disease. Neurology 2013, 80, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Smeyne, R.J.; Noyce, A.J.; Byrne, M.; Savica, R.; Marras, C. Infection and Risk of Parkinson’s Disease. J. Parkinson’s Dis. 2021, 11, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Baizabal-Carvallo, J.F.; Alonso-Juarez, M. The role of viruses in the pathogenesis of Parkinson’s disease. Neural Regen. Res. 2021, 16, 1200–1201. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T. Viral etiology of Parkinson’s disease: Focus on influenza A virus. Parkinsonism Relat. Disord. 1996, 2, 113–121. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19). Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 9 August 2022).
- Brundin, P.; Nath, A.; Beckham, J.D. Is COVID-19 a Perfect Storm for Parkinson’s Disease? Trends Neurosci. 2020, 43, 931–933. [Google Scholar] [CrossRef]
- Méndez-Guerrero, A.; Laespada-García, M.I.; Gómez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rábano-Suárez, P.; Álvarez-Torres, E.; de Fuenmayor-Fernández de la Hoz, C.P.; Vega Pérez, D.; et al. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef]
- Faber, I.; Brandão, P.R.P.; Menegatti, F.; de Carvalho Bispo, D.D.; Maluf, F.B.; Cardoso, F. Coronavirus Disease 2019 and Parkinsonism: A Non-post-encephalitic Case. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 1721–1722. [Google Scholar] [CrossRef]
- Karabiyik, C.; Lee, M.J.; Rubinsztein, D.C. Autophagy impairment in Parkinson’s disease. Essays Biochem. 2017, 61, 711–720. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S210–S212. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Hansen, C.; Angot, E.; Bergström, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Rastogi, S.; Sharma, V.; Bharti, P.S.; Rani, K.; Modi, G.P.; Nikolajeff, F.; Kumar, S. The Evolving Landscape of Exosomes in Neurodegenerative Diseases: Exosomes Characteristics and a Promising Role in Early Diagnosis. Int. J. Mol. Sci. 2021, 22, 440. [Google Scholar] [CrossRef]
- Gangoda, L.; Boukouris, S.; Liem, M.; Kalra, H.; Mathivanan, S. Extracellular vesicles including exosomes are mediators of signal transduction: Are they protective or pathogenic? Proteomics 2015, 15, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Igarashi, Y. Physiological and pathological roles of exosomes in the nervous system. Biomol. Concepts 2016, 7, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Caobi, A.; Nair, M.; Raymond, A.D. Extracellular Vesicles in the Pathogenesis of Viral Infections in Humans. Viruses 2020, 12, 1200. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Kang, M.H.; Kim, J.H. Diverse Effects of Exosomes on COVID-19: A Perspective of Progress From Transmission to Therapeutic Developments. Front. Immunol. 2021, 12, 716407. [Google Scholar] [CrossRef]
- Karamichali, E.; Foka, P.; Valiakou, V.; Iliadis, P.; Loukaki, D.; Andresaki, K.; Papadopoulou, G.; Georgopoulou, U.; Koskinas, J. Exosomal cargo as a key player of the immune response after direct-acting antiviral treatment in chronic hepatitis C patients. J. Hepatol. 2022, 77, S255. [Google Scholar] [CrossRef]
- Yang, L.; Li, J.; Li, S.; Dang, W.; Xin, S.; Long, S.; Zhang, W.; Cao, P.; Lu, J. Extracellular Vesicles Regulated by Viruses and Antiviral Strategies. Front. Cell Dev. Biol. 2021, 9, 722020. [Google Scholar] [CrossRef]
- Zhang, L.; Ju, Y.; Chen, S.; Ren, L. Recent Progress on Exosomes in RNA Virus Infection. Viruses 2021, 13, 256. [Google Scholar] [CrossRef]
- Rezaie, J.; Aslan, C.; Ahmadi, M.; Zolbanin, N.M.; Kashanchi, F.; Jafari, R. The versatile role of exosomes in human retroviral infections: From immunopathogenesis to clinical application. Cell Biosci. 2021, 11, 19. [Google Scholar] [CrossRef]
- McCall, S.; Vilensky, J.A.; Gilman, S.; Taubenberger, J.K. The relationship between encephalitis lethargica and influenza: A critical analysis. J. Neurovirology 2008, 14, 177–185. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Chesdachai, S.; Jaruvongvanich, V.; Ungprasert, P. Hepatitis C virus infection and risk of Parkinson’s disease: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2018, 30, 9–13. [Google Scholar] [CrossRef]
- Marttila, R.J.; Rinne, U.K. Herpes simplex virus antibodies in patients with Parkinson’s disease. J. Neurol. Sci. 1978, 35, 375–379. [Google Scholar] [CrossRef]
- Mirsattari, S.M.; Power, C.; Nath, A. Parkinsonism with HIV infection. Mov. Disord. Off. J. Mov. Disord. Soc. 1998, 13, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.; Lin, C.H.; Lin, H.F.; Lin, C.L.; Lin, C.C.; Liao, K.F. Herpes zoster correlates with increased risk of Parkinson’s disease in older people: A population-based cohort study in Taiwan. Medicine 2017, 96, e6075. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.L.; Shahida, S.; Madan, N.; Rao, S.; Khardori, N. Transient parkinsonism in West Nile virus encephalitis. Am. J. Med. 2003, 115, 252–253. [Google Scholar] [CrossRef]
- Murgod, U.A.; Muthane, U.B.; Ravi, V.; Radhesh, S.; Desai, A. Persistent movement disorders following Japanese encephalitis. Neurology 2001, 57, 2313–2315. [Google Scholar] [CrossRef]
- Das, K.; Ghosh, M.; Nag, C.; Nandy, S.P.; Banerjee, M.; Datta, M.; Devi, G.; Chaterjee, G. Role of familial, environmental and occupational factors in the development of Parkinson’s disease. Neuro-Degener. Dis. 2011, 8, 345–351. [Google Scholar] [CrossRef]
- Espay, A.J.; Henderson, K.K. Postencephalitic parkinsonism and basal ganglia necrosis due to Epstein-Barr virus infection. Neurology 2011, 76, 1529–1530. [Google Scholar] [CrossRef]
- Toovey, S.; Jick, S.S.; Meier, C.R. Parkinson’s disease or Parkinson symptoms following seasonal influenza. Influenza Other Respir. Viruses 2011, 5, 328–333. [Google Scholar] [CrossRef]
- Cocoros, N.M.; Svensson, E.; Szépligeti, S.K.; Vestergaard, S.V.; Szentkúti, P.; Thomsen, R.W.; Borghammer, P.; Sørensen, H.T.; Henderson, V.W. Long-term Risk of Parkinson Disease Following Influenza and Other Infections. JAMA Neurol. 2021, 78, 1461–1470. [Google Scholar] [CrossRef]
- Rohn, T.T.; Catlin, L.W. Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson’s disease brain. PLoS ONE 2011, 6, e20495. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed]
- Sadasivan, S.; Zanin, M.; O’Brien, K.; Schultz-Cherry, S.; Smeyne, R.J. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS ONE 2015, 10, e0124047. [Google Scholar] [CrossRef] [PubMed]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef] [PubMed]
- Kasen, A.; Houck, C.; Burmeister, A.R.; Sha, Q.; Brundin, L.; Brundin, P. Upregulation of α-synuclein following immune activation: Possible trigger of Parkinson’s disease. Neurobiol. Dis. 2022, 166, 105654. [Google Scholar] [CrossRef]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J. Virol. 2015, 90, 2767–2782. [Google Scholar] [CrossRef]
- Caggiu, E.; Paulus, K.; Arru, G.; Piredda, R.; Sechi, G.P.; Sechi, L.A. Humoral cross reactivity between α-synuclein and herpes simplex-1 epitope in Parkinson’s disease, a triggering role in the disease? J. Neuroimmunol. 2016, 291, 110–114. [Google Scholar] [CrossRef]
- Caggiu, E.; Paulus, K.; Galleri, G.; Arru, G.; Manetti, R.; Sechi, G.P.; Sechi, L.A. Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in Parkinson’s disease. J. Neuroimmunol. 2017, 310, 26–31. [Google Scholar] [CrossRef]
- Fazzini, E.; Fleming, J.; Fahn, S. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 1992, 7, 153–158. [Google Scholar] [CrossRef]
- Almaghaslah, D.; Kandasamy, G.; Almanasef, M.; Vasudevan, R.; Chandramohan, S. Review on the coronavirus disease (COVID-19) pandemic: Its outbreak and current status. Int. J. Clin. Pract. 2020, 74, e13637. [Google Scholar] [CrossRef]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health-The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.A.; Quandelacy, T.M.; Kada, S.; Prasad, P.V.; Steele, M.; Brooks, J.T.; Slayton, R.B.; Biggerstaff, M.; Butler, J.C. SARS-CoV-2 Transmission From People Without COVID-19 Symptoms. JAMA Netw. Open 2021, 4, e2035057. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ayeh, S.K.; Chidambaram, V.; Karakousis, P.C. Modes of transmission of SARS-CoV-2 and evidence for preventive behavioral interventions. BMC Infect. Dis. 2021, 21, 496. [Google Scholar] [CrossRef] [PubMed]
- Harapan, B.N.; Yoo, H.J. Neurological symptoms, manifestations, and complications associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 19 (COVID-19). J. Neurol. 2021, 268, 3059–3071. [Google Scholar] [CrossRef]
- Paterson, R.W.; Brown, R.L.; Benjamin, L.; Nortley, R.; Wiethoff, S.; Bharucha, T.; Jayaseelan, D.L.; Kumar, G.; Raftopoulos, R.E.; Zambreanu, L.; et al. The emerging spectrum of COVID-19 neurology: Clinical, radiological and laboratory findings. Brain J. Neurol. 2020, 143, 3104–3120. [Google Scholar] [CrossRef]
- Beghi, E.; Giussani, G.; Westenberg, E.; Allegri, R.; Garcia-Azorin, D.; Guekht, A.; Frontera, J.; Kivipelto, M.; Mangialasche, F.; Mukaetova-Ladinska, E.B.; et al. Acute and post-acute neurological manifestations of COVID-19: Present findings, critical appraisal, and future directions. J. Neurol. 2022, 269, 2265–2274. [Google Scholar] [CrossRef]
- Taquet, M.; Dercon, Q.; Luciano, S.; Geddes, J.R.; Husain, M.; Harrison, P.J. Incidence, co-occurrence, and evolution of long-COVID features: A 6-month retrospective cohort study of 273,618 survivors of COVID-19. PLoS Med. 2021, 18, e1003773. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence (NICE). National Institute for Health and Care Excellence: Clinical Guidelines. In COVID-19 Rapid Guideline: Managing the Long-Term Effects of COVID-19; National Institute for Health and Care Excellence (NICE): London, UK, 2020. [Google Scholar]
- Stephenson, J. New Federal Guidance Says COVID-19’s Long-term Effects Can Qualify as a Disability. JAMA Health Forum 2021, 2, e212820. [Google Scholar] [CrossRef]
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202. [Google Scholar] [CrossRef]
- Camargo-Martínez, W.; Lozada-Martínez, I.; Escobar-Collazos, A.; Navarro-Coronado, A.; Moscote-Salazar, L.; Pacheco-Hernández, A.; Janjua, T.; Bosque-Varela, P. Post-COVID 19 neurological syndrome: Implications for sequelae’s treatment. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2021, 88, 219–225. [Google Scholar] [CrossRef]
- Shah, W.; Hillman, T.; Playford, E.D.; Hishmeh, L. Managing the long term effects of covid-19: Summary of NICE, SIGN, and RCGP rapid guideline. BMJ 2021, 372, n136. [Google Scholar] [CrossRef] [PubMed]
- Khorramdelazad, H.; Kazemi, M.H.; Najafi, A.; Keykhaee, M.; Zolfaghari Emameh, R.; Falak, R. Immunopathological similarities between COVID-19 and influenza: Investigating the consequences of Co-infection. Microb. Pathog. 2021, 152, 104554. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Rao, A.R.; Hidayathullah, S.M.; Hegde, K.; Adhikari, P. Parkinsonism: An emerging post COVID sequelae. IDCases 2022, 27, e01388. [Google Scholar] [CrossRef] [PubMed]
- Boura, I.; Chaudhuri, K.R. Coronavirus Disease 2019 and Related Parkinsonism: The Clinical Evidence Thus Far. Mov. Disord. Clin. Pract. 2022, 9, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Cilia, R.; Bonvegna, S.; Straccia, G.; Andreasi, N.G.; Elia, A.E.; Romito, L.M.; Devigili, G.; Cereda, E.; Eleopra, R. Effects of COVID-19 on Parkinson’s Disease Clinical Features: A Community-Based Case-Control Study. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 1287–1292. [Google Scholar] [CrossRef]
- Brown, E.G.; Chahine, L.M.; Goldman, S.M.; Korell, M.; Mann, E.; Kinel, D.R.; Arnedo, V.; Marek, K.L.; Tanner, C.M. The Effect of the COVID-19 Pandemic on People with Parkinson’s Disease. J. Parkinson’s Dis. 2020, 10, 1365–1377. [Google Scholar] [CrossRef]
- Montenegro, P.; Moral, I.; Puy, A.; Cordero, E.; Chantada, N.; Cuixart, L.; Brotons, C. Prevalence of Post COVID-19 Condition in Primary Care: A Cross Sectional Study. Int. J. Environ. Res. Public Health 2022, 19, 1836. [Google Scholar] [CrossRef]
- Nasserie, T.; Hittle, M.; Goodman, S.N. Assessment of the Frequency and Variety of Persistent Symptoms Among Patients With COVID-19: A Systematic Review. JAMA Netw. Open 2021, 4, e2111417. [Google Scholar] [CrossRef]
- Leta, V.; Rodríguez-Violante, M.; Abundes, A.; Rukavina, K.; Teo, J.T.; Falup-Pecurariu, C.; Irincu, L.; Rota, S.; Bhidayasiri, R.; Storch, A.; et al. Parkinson’s Disease and Post-COVID-19 Syndrome: The Parkinson’s Long-COVID Spectrum. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1287–1289. [Google Scholar] [CrossRef]
- Doty, R.L. Olfactory dysfunction in Parkinson disease. Nat. Rev. Neurol. 2012, 8, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Tarakad, A.; Jankovic, J. Anosmia and Ageusia in Parkinson’s Disease. Int. Rev. Neurobiol. 2017, 133, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Augustin, M.; Schommers, P.; Stecher, M.; Dewald, F.; Gieselmann, L.; Gruell, H.; Horn, C.; Vanshylla, K.; Cristanziano, V.D.; Osebold, L.; et al. Post-COVID syndrome in non-hospitalised patients with COVID-19: A longitudinal prospective cohort study. Lancet Reg. Health. Eur. 2021, 6, 100122. [Google Scholar] [CrossRef]
- Vavougios, G.D. Potentially irreversible olfactory and gustatory impairments in COVID-19: Indolent vs. fulminant SARS-CoV-2 neuroinfection. Brain Behav. Immun. 2020, 87, 107–108. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.J.; Fletcher, R.B.; et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef]
- Hubbard, P.S.; Esiri, M.M.; Reading, M.; McShane, R.; Nagy, Z. Alpha-synuclein pathology in the olfactory pathways of dementia patients. J. Anat. 2007, 211, 117–124. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Parkinson’s Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef]
- Beach, T.G.; White, C.L., 3rd; Hladik, C.L.; Sabbagh, M.N.; Connor, D.J.; Shill, H.A.; Sue, L.I.; Sasse, J.; Bachalakuri, J.; Henry-Watson, J.; et al. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009, 117, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Cersosimo, M.G.; Raina, G.B.; Pecci, C.; Pellene, A.; Calandra, C.R.; Gutiérrez, C.; Micheli, F.E.; Benarroch, E.E. Gastrointestinal manifestations in Parkinson’s disease: Prevalence and occurrence before motor symptoms. J. Neurol. 2013, 260, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liao, J.; Liu, X.; Zhong, Y.; Cai, X.; Long, L. Review: The Role of Intestinal Dysbiosis in Parkinson’s Disease. Front. Cell. Infect. Microbiol. 2021, 11, 615075. [Google Scholar] [CrossRef] [PubMed]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef]
- Shannon, K.M.; Keshavarzian, A.; Dodiya, H.B.; Jakate, S.; Kordower, J.H. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 716–719. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Anselmi, L.; Toti, L.; Bove, C.; Hampton, J.; Travagli, R.A. A Nigro-Vagal Pathway Controls Gastric Motility and Is Affected in a Rat Model of Parkinsonism. Gastroenterology 2017, 153, 1581–1593. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Martí, M.J.; Hernández, I.; Valldeoriola, F.; Reñé, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1010–1018. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Chaves Andrade, M.; Souza de Faria, R.; Avelino Mota Nobre, S. COVID-19: Can the symptomatic SARS-CoV-2 infection affect the homeostasis of the gut-brain-microbiota axis? Med. Hypotheses 2020, 144, 110206. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, Z.; Zhang, M.; Liu, S.; Zhou, L.; Yang, C.; Liu, C. The Role of the Gastrointestinal System in Neuroinvasion by SARS-CoV-2. Front. Neurosci. 2021, 15, 694446. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Wang, X.; Zhang, G.; Guo, M.; Ma, H.; Zhao, D.; Sun, Y.; He, J.; Liu, L.; Zhang, K.; et al. Re-detectable positive SARS-CoV-2 RNA tests in patients who recovered from COVID-19 with intestinal infection. Protein Cell 2021, 12, 230–235. [Google Scholar] [CrossRef]
- Kaźmierczak-Siedlecka, K.; Vitale, E.; Makarewicz, W. COVID-19-gastrointestinal and gut microbiota-related aspects. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10853–10859. [Google Scholar] [CrossRef]
- Viana, S.D.; Nunes, S.; Reis, F. ACE2 imbalance as a key player for the poor outcomes in COVID-19 patients with age-related comorbidities-Role of gut microbiota dysbiosis. Ageing Res. Rev. 2020, 62, 101123. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; La Fata, G.; Steinert, R.E.; Weber, P. Relationship between the gut microbiome and brain function. Nutr. Rev. 2018, 76, 481–496. [Google Scholar] [CrossRef]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef]
- Majde, J.A. Neuroinflammation resulting from covert brain invasion by common viruses-a potential role in local and global neurodegeneration. Med. Hypotheses 2010, 75, 204–213. [Google Scholar] [CrossRef]
- Almutairi, M.M.; Sivandzade, F.; Albekairi, T.H.; Alqahtani, F.; Cucullo, L. Neuroinflammation and Its Impact on the Pathogenesis of COVID-19. Front. Med. 2021, 8, 745789. [Google Scholar] [CrossRef] [PubMed]
- Bauer, L.; Laksono, B.M.; de Vrij, F.M.S.; Kushner, S.A.; Harschnitz, O.; van Riel, D. The neuroinvasiveness, neurotropism, and neurovirulence of SARS-CoV-2. Trends Neurosci. 2022, 45, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Yachou, Y.; El Idrissi, A.; Belapasov, V.; Ait Benali, S. Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: Understanding the neurological manifestations in COVID-19 patients. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2020, 41, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, G.; Fragoso, G.; Sciutto, E. Neuroinflammation in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) infection: Pathogenesis and clinical manifestations. Curr. Opin. Pharmacol. 2022, 63, 102181. [Google Scholar] [CrossRef]
- Rowaiye, A.B.; Okpalefe, O.A.; Onuh Adejoke, O.; Ogidigo, J.O.; Hannah Oladipo, O.; Ogu, A.C.; Oli, A.N.; Olofinase, S.; Onyekwere, O.; Rabiu Abubakar, A.; et al. Attenuating the Effects of Novel COVID-19 (SARS-CoV-2) Infection-Induced Cytokine Storm and the Implications. J. Inflamm. Res. 2021, 14, 1487–1510. [Google Scholar] [CrossRef]
- Olbei, M.; Hautefort, I.; Modos, D.; Treveil, A.; Poletti, M.; Gul, L.; Shannon-Lowe, C.D.; Korcsmaros, T. SARS-CoV-2 Causes a Different Cytokine Response Compared to Other Cytokine Storm-Causing Respiratory Viruses in Severely Ill Patients. Front. Immunol. 2021, 12, 629193. [Google Scholar] [CrossRef]
- Islam, H.; Chamberlain, T.C.; Mui, A.L.; Little, J.P. Elevated Interleukin-10 Levels in COVID-19: Potentiation of Pro-Inflammatory Responses or Impaired Anti-Inflammatory Action? Front. Immunol. 2021, 12, 677008. [Google Scholar] [CrossRef]
- Santa Cruz, A.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Castro, A.G.; Silvestre, R. Interleukin-6 Is a Biomarker for the Development of Fatal Severe Acute Respiratory Syndrome Coronavirus 2 Pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef]
- Lu, Q.; Zhu, Z.; Tan, C.; Zhou, H.; Hu, Y.; Shen, G.; Zhu, P.; Yang, G.; Xie, X. Changes of serum IL-10, IL-1β, IL-6, MCP-1, TNF-α, IP-10 and IL-4 in COVID-19 patients. Int. J. Clin. Pract. 2021, 75, e14462. [Google Scholar] [CrossRef]
- Mehta, P.; Fajgenbaum, D.C. Is severe COVID-19 a cytokine storm syndrome: A hyperinflammatory debate. Curr. Opin. Rheumatol. 2021, 33, 419–430. [Google Scholar] [CrossRef]
- Chen, H.; O’Reilly, E.J.; Schwarzschild, M.A.; Ascherio, A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am. J. Epidemiol. 2008, 167, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Hsu, R.J.; Yu, W.C.; Peng, G.R.; Ye, C.H.; Hu, S.; Chong, P.C.T.; Yap, K.Y.; Lee, J.Y.C.; Lin, W.C.; Yu, S.H. The Role of Cytokines and Chemokines in Severe Acute Respiratory Syndrome Coronavirus 2 Infections. Front. Immunol. 2022, 13, 832394. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef]
- Klein, R.S.; Garber, C.; Funk, K.E.; Salimi, H.; Soung, A.; Kanmogne, M.; Manivasagam, S.; Agner, S.; Cain, M. Neuroinflammation During RNA Viral Infections. Annu. Rev. Immunol. 2019, 37, 73–95. [Google Scholar] [CrossRef]
- Yang, R.C.; Huang, K.; Zhang, H.P.; Li, L.; Zhang, Y.F.; Tan, C.; Chen, H.C.; Jin, M.L.; Wang, X.R. SARS-CoV-2 productively infects human brain microvascular endothelial cells. J. Neuroinflamm. 2022, 19, 149. [Google Scholar] [CrossRef]
- Raghavan, S.; Kenchappa, D.B.; Leo, M.D. SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Front. Cardiovasc. Med. 2021, 8, 687783. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Mahajan, S.D. SARS-COV2 Alters Blood Brain Barrier Integrity Contributing to Neuro-Inflammation. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2021, 16, 4–6. [Google Scholar] [CrossRef]
- John, G.R.; Lee, S.C.; Brosnan, C.F. Cytokines: Powerful regulators of glial cell activation. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2003, 9, 10–22. [Google Scholar] [CrossRef]
- da Fonseca, A.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Kielian, T. Neuron-astrocyte interactions in neurodegenerative diseases: Role of neuroinflammation. Clin. Exp. Neuroimmunol. 2015, 6, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Nguyen, K.H.; Ball, J.B.; Hopkins, S.; Kelley, T.; Baratta, M.V.; Fleshner, M.; Maier, S.F. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav. Immun. 2022, 100, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, Z.L.; Klenja, D.; Janjua, N.; Cami-Kobeci, G.; Ahmed, B.Y. COVID-19 and Parkinson’s Disease: Shared Inflammatory Pathways Under Oxidative Stress. Brain Sci. 2020, 10, 807. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Jahan, S.; Khan, A.; Siddiqui, A.J.; Redhu, N.S.; Wahajuddin; Khan, J.; Banwas, S.; Alshehri, B.; Alaidarous, M. Neurological Manifestation of SARS-CoV-2 Induced Inflammation and Possible Therapeutic Strategies Against COVID-19. Mol. Neurobiol. 2021, 58, 3417–3434. [Google Scholar] [CrossRef]
- Sahu, M.R.; Rani, L.; Subba, R.; Mondal, A.C. Cellular senescence in the aging brain: A promising target for neurodegenerative diseases. Mech. Ageing Dev. 2022, 204, 111675. [Google Scholar] [CrossRef]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Tripathi, U.; Nchioua, R.; Prata, L.; Zhu, Y.; Gerdes, E.O.W.; Giorgadze, N.; Pirtskhalava, T.; Parker, E.; Xue, A.; Espindola-Netto, J.M.; et al. SARS-CoV-2 causes senescence in human cells and exacerbates the senescence-associated secretory phenotype through TLR-3. Aging 2021, 13, 21838–21854. [Google Scholar] [CrossRef]
- Kandhaya-Pillai, R.; Yang, X.; Tchkonia, T.; Martin, G.M.; Kirkland, J.L.; Oshima, J. TNF-α/IFN-γ synergy amplifies senescence-associated inflammation and SARS-CoV-2 receptor expression via hyper-activated JAK/STAT1. Aging Cell 2022, 21, e13646. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Di Benedetto, S. How Immunosenescence and Inflammaging May Contribute to Hyperinflammatory Syndrome in COVID-19. Int. J. Mol. Sci. 2021, 22, 12539. [Google Scholar] [CrossRef]
- Matschke, J.; Lütgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schröder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; de Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Vavougios, G.D.; Breza, M.; Mavridis, T.; Krogfelt, K.A. FYN, SARS-CoV-2, and IFITM3 in the neurobiology of Alzheimer’s disease. Brain Disord. 2021, 3, 100022. [Google Scholar] [CrossRef]
- Clairembault, T.; Kamphuis, W.; Leclair-Visonneau, L.; Rolli-Derkinderen, M.; Coron, E.; Neunlist, M.; Hol, E.M.; Derkinderen, P. Enteric GFAP expression and phosphorylation in Parkinson’s disease. J. Neurochem. 2014, 130, 805–815. [Google Scholar] [CrossRef]
- Rodriguez-Perez, A.I.; Garrido-Gil, P.; Pedrosa, M.A.; Garcia-Garrote, M.; Valenzuela, R.; Navarro, G.; Franco, R.; Labandeira-Garcia, J.L. Angiotensin type 2 receptors: Role in aging and neuroinflammation in the substantia nigra. Brain Behav. Immun. 2020, 87, 256–271. [Google Scholar] [CrossRef]
- Paul, D.; Mohankumar, S.K.; Thomas, R.S.; Kheng, C.B.; Basavan, D. Potential Implications of Angiotensin-converting Enzyme 2 Blockades on Neuroinflammation in SARS-CoV-2 Infection. Curr. Drug Targets 2022, 23, 364–372. [Google Scholar] [CrossRef]
- Williams, A.; Branscome, H.; Khatkar, P.; Mensah, G.A.; Al Sharif, S.; Pinto, D.O.; DeMarino, C.; Kashanchi, F. A comprehensive review of COVID-19 biology, diagnostics, therapeutics, and disease impacting the central nervous system. J. Neurovirol. 2021, 27, 667–690. [Google Scholar] [CrossRef]
- Pavel, A.; Murray, D.K.; Stoessl, A.J. COVID-19 and selective vulnerability to Parkinson’s disease. Lancet Neurol. 2020, 19, 719. [Google Scholar] [CrossRef]
- Klingenstein, M.; Klingenstein, S.; Neckel, P.H.; Mack, A.F.; Wagner, A.P.; Kleger, A.; Liebau, S.; Milazzo, A. Evidence of SARS-CoV2 Entry Protein ACE2 in the Human Nose and Olfactory Bulb. Cells Tissues Organs 2020, 209, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The Spatial and Cell-Type Distribution of SARS-CoV-2 Receptor ACE2 in the Human and Mouse Brains. Front. Neurol. 2020, 11, 573095. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Du, T.; Hong, W.; Chen, L.; Que, H.; Lu, S.; Peng, X. Neurological complications and infection mechanism of SARS-COV-2. Signal Transduct. Target. Ther. 2021, 6, 406. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffré, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136.e7. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. [Google Scholar] [CrossRef]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Paolo, T.D.; Tremblay, M. Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions? Front. Cell. Neurosci. 2018, 12, 282. [Google Scholar] [CrossRef]
- Estrada, E. Cascading from SARS-CoV-2 to Parkinson’s Disease through Protein-Protein Interactions. Viruses 2021, 13, 897. [Google Scholar] [CrossRef]
- Wen, H.; Zhan, L.; Chen, S.; Long, L.; Xu, E. Rab7 may be a novel therapeutic target for neurologic diseases as a key regulator in autophagy. J. Neurosci. Res. 2017, 95, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Park, J.H.; Chung, K.C. The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep. 2020, 53, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Irfan, M.; Ahsan, H.; Ahmed, A.; Kaushik, A.C.; Khan, A.S.; Chinnasamy, S.; Ali, A.; Wei, D.Q. Structures of SARS-CoV-2 RNA-Binding Proteins and Therapeutic Targets. Intervirology 2021, 64, 55–68. [Google Scholar] [CrossRef]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M. Interactions between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem. Neurosci. 2022, 13, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Malkus, K.A.; Tsika, E.; Ischiropoulos, H. Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: How neurons are lost in the Bermuda triangle. Mol. Neurodegener. 2009, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.A.; Ostaszewski, M.; Matsuoka, Y.; Ghosh, S.; Glaab, E.; Trefois, C.; Crespo, I.; Perumal, T.M.; Jurkowski, W.; Antony, P.M.; et al. Integrating pathways of Parkinson’s disease in a molecular interaction map. Mol. Neurobiol. 2014, 49, 88–102. [Google Scholar] [CrossRef]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Muqit, M.M.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’ disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.F. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 2014, 70, 204–213. [Google Scholar] [CrossRef]
- Hattori, N.; Tanaka, M.; Ozawa, T.; Mizuno, Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann. Neurol. 1991, 30, 563–571. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef]
- Gatti, P.; Ilamathi, H.S.; Todkar, K.; Germain, M. Mitochondria Targeted Viral Replication and Survival Strategies-Prospective on SARS-CoV-2. Front. Pharmacol. 2020, 11, 578599. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef]
- Burtscher, J.; Cappellano, G.; Omori, A.; Koshiba, T.; Millet, G.P. Mitochondria: In the Cross Fire of SARS-CoV-2 and Immunity. iScience 2020, 23, 101631. [Google Scholar] [CrossRef]
- Morowitz, J.M.; Pogson, K.B.; Roque, D.A.; Church, F.C. Role of SARS-CoV-2 in Modifying Neurodegenerative Processes in Parkinson’s Disease: A Narrative Review. Brain Sci. 2022, 12, 536. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and Oxidative Stress. Biochem. Biokhimiia 2020, 85, 1543–1553. [Google Scholar] [CrossRef]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front. Microbiol. 2021, 12, 780768. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef]
- Shi, T.T.; Yang, F.Y.; Liu, C.; Cao, X.; Lu, J.; Zhang, X.L.; Yuan, M.X.; Chen, C.; Yang, J.K. Angiotensin-converting enzyme 2 regulates mitochondrial function in pancreatic β-cells. Biochem. Biophys. Res. Commun. 2018, 495, 860–866. [Google Scholar] [CrossRef]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef]
- Clough, E.; Inigo, J.; Chandra, D.; Chaves, L.; Reynolds, J.L.; Aalinkeel, R.; Schwartz, S.A.; Khmaladze, A.; Mahajan, S.D. Mitochondrial Dynamics in SARS-COV2 Spike Protein Treated Human Microglia: Implications for Neuro-COVID. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2021, 16, 770–784. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Matsubara, K.; Nomura, Y. Endoplasmic reticulum stress and Parkinson’s disease: The role of HRD1 in averting apoptosis in neurodegenerative disease. Oxidative Med. Cell. Longev. 2013, 2013, 239854. [Google Scholar] [CrossRef]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, D.; Stabile, A.M.; Vacca, C.; Pistilli, A.; Rende, M.; Gioiello, A.; Cruciani, G.; Galli, F. Endoplasmic reticulum stress and NF-kB activation in SARS-CoV-2 infected cells and their response to antiviral therapy. IUBMB Life 2022, 74, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Dzakah, E.E.; Wang, H.; Tang, S. The ORF8 protein of SARS-CoV-2 induced endoplasmic reticulum stress and mediated immune evasion by antagonizing production of interferon beta. Virus Res. 2021, 296, 198350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gan, M.; Ebrahim, A.S.; Lin, W.L.; Melrose, H.L.; Yen, S.H. ER stress response plays an important role in aggregation of α-synuclein. Mol. Neurodegener. 2010, 5, 56. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.T.; Qian, X.; van der Velden, J.L.; Chia, S.B.; McMillan, D.H.; Flemer, S.; Hoffman, S.M.; Lahue, K.G.; Schneider, R.W.; Nolin, J.D.; et al. Glutathione S-transferase pi modulates NF-κB activation and pro-inflammatory responses in lung epithelial cells. Redox Biol. 2016, 8, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qiao, J.; You, Q.; Zong, S.; Peng, Q.; Liu, Y.; Hu, S.; Liu, W.; Li, S.; Shu, X.; et al. SARS-CoV-2 Nsp5 Activates NF-κB Pathway by Upregulating SUMOylation of MAVS. Front. Immunol. 2021, 12, 750969. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef]
- Dolatshahi, M.; Ranjbar Hameghavandi, M.H.; Sabahi, M.; Rostamkhani, S. Nuclear factor-kappa B (NF-κB) in pathophysiology of Parkinson disease: Diverse patterns and mechanisms contributing to neurodegeneration. Eur. J. Neurosci. 2021, 54, 4101–4123. [Google Scholar] [CrossRef]
- Bellucci, A.; Bubacco, L.; Longhena, F.; Parrella, E.; Faustini, G.; Porrini, V.; Bono, F.; Missale, C.; Pizzi, M. Nuclear Factor-κB Dysregulation and α-Synuclein Pathology: Critical Interplay in the Pathogenesis of Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-κB-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 725–732. [Google Scholar] [CrossRef]
- Lakshmana, M.K. SARS-CoV-2-induced autophagy dysregulation may cause neuronal dysfunction in COVID-19. Neural Regen. Res. 2022, 17, 1255–1256. [Google Scholar] [CrossRef]
- Singh, K.; Chen, Y.C.; Hassanzadeh, S.; Han, K.; Judy, J.T.; Seifuddin, F.; Tunc, I.; Sack, M.N.; Pirooznia, M. Network Analysis and Transcriptome Profiling Identify Autophagic and Mitochondrial Dysfunctions in SARS-CoV-2 Infection. Front. Genet. 2021, 12, 599261. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442.e5. [Google Scholar] [CrossRef]
- Hou, P.; Wang, X.; Wang, H.; Wang, T.; Yu, Z.; Xu, C.; Zhao, Y.; Wang, W.; Zhao, Y.; Chu, F.; et al. The ORF7a protein of SARS-CoV-2 initiates autophagy and limits autophagosome-lysosome fusion via degradation of SNAP29 to promote virus replication. Autophagy 2022, 144, 110206. [Google Scholar] [CrossRef]
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Jan, E.; Luo, H. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem. Biophys. Res. Commun. 2021, 540, 75–82. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- García-Pérez, B.E.; González-Rojas, J.A.; Salazar, M.I.; Torres-Torres, C.; Castrejón-Jiménez, N.S. Taming the Autophagy as a Strategy for Treating COVID-19. Cells 2020, 9, 2679. [Google Scholar] [CrossRef]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Zlotogorski-Hurvitz, A.; Dayan, D.; Chaushu, G.; Korvala, J.; Salo, T.; Sormunen, R.; Vered, M. Human saliva-derived exosomes: Comparing methods of isolation. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2015, 63, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; Lim, J.W.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert Rev. Proteom. 2009, 6, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Jung, M.K.; Mun, J.Y. Sample Preparation and Imaging of Exosomes by Transmission Electron Microscopy. J. Vis. Exp. JoVE 2018, 56482. [Google Scholar] [CrossRef]
- Borges, F.T.; Reis, L.A.; Schor, N. Extracellular vesicles: Structure, function, and potential clinical uses in renal diseases. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Med. E Biol. 2013, 46, 824–830. [Google Scholar] [CrossRef]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.K.; Fernandez, M.V.; Groves, N.S.; Freed, E.O.; van Engelenburg, S.B. Genomic tagging of endogenous human ESCRT-I complex preserves ESCRT-mediated membrane-remodeling functions. J. Biol. Chem. 2019, 294, 16266–16281. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. CCS 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Larssen, P.; Wik, L.; Czarnewski, P.; Eldh, M.; Löf, L.; Ronquist, K.G.; Dubois, L.; Freyhult, E.; Gallant, C.J.; Oelrich, J.; et al. Tracing Cellular Origin of Human Exosomes Using Multiplex Proximity Extension Assays. Mol. Cell. Proteom. MCP 2017, 16, 502–511. [Google Scholar] [CrossRef]
- Anastasi, F.; Masciandaro, S.M.; Carratore, R.D.; Dell’Anno, M.T.; Signore, G.; Falleni, A.; McDonnell, L.A.; Bongioanni, P. Proteomics Profiling of Neuron-Derived Small Extracellular Vesicles from Human Plasma: Enabling Single-Subject Analysis. Int. J. Mol. Sci. 2021, 22, 2951. [Google Scholar] [CrossRef]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- Hornung, S.; Dutta, S.; Bitan, G. CNS-Derived Blood Exosomes as a Promising Source of Biomarkers: Opportunities and Challenges. Front. Mol. Neurosci. 2020, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Bai, X.; Zhang, A.; Huang, J.; Xu, S.; Zhang, J. Role of Exosomes in Central Nervous System Diseases. Front. Mol. Neurosci. 2019, 12, 240. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Si, X.; Tian, J.; Chen, Y.; Yan, Y.; Pu, J.; Zhang, B. Central Nervous System-Derived Exosomal Alpha-Synuclein in Serum May Be a Biomarker in Parkinson’s Disease. Neuroscience 2019, 413, 308–316. [Google Scholar] [CrossRef]
- Olanow, C.W.; Brundin, P. Parkinson’s disease and alpha synuclein: Is Parkinson’s disease a prion-like disorder? Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 31–40. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Fan, R.Z.; Guo, M.; Luo, S.; Cui, M.; Tieu, K. Exosome release and neuropathology induced by α-synuclein: New insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol. Commun. 2019, 7, 184. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain J. Neurol. 2016, 139, 481–494. [Google Scholar] [CrossRef]
- Han, C.; Xiong, N.; Guo, X.; Huang, J.; Ma, K.; Liu, L.; Xia, Y.; Shen, Y.; Li, J.; Jiang, H.; et al. Exosomes from patients with Parkinson’s disease are pathological in mice. J. Mol. Med. 2019, 97, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Si, X.L.; Fang, Y.J.; Li, L.F.; Gu, L.Y.; Yin, X.Z.; Jun, T.; Yan, Y.P.; Pu, J.L.; Zhang, B.R. From inflammasome to Parkinson’s disease: Does the NLRP3 inflammasome facilitate exosome secretion and exosomal alpha-synuclein transmission in Parkinson’s disease? Exp. Neurol. 2021, 336, 113525. [Google Scholar] [CrossRef] [PubMed]
- Vandendriessche, C.; Bruggeman, A.; Van Cauwenberghe, C.; Vandenbroucke, R.E. Extracellular Vesicles in Alzheimer’s and Parkinson’s Disease: Small Entities with Large Consequences. Cells 2020, 9, 2485. [Google Scholar] [CrossRef]
- Li, K.L.; Huang, H.Y.; Ren, H.; Yang, X.L. Role of exosomes in the pathogenesis of inflammation in Parkinson’s disease. Neural Regen. Res. 2022, 17, 1898–1906. [Google Scholar] [CrossRef]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Brück, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, G.; Kou, L.; Yin, S.; Han, C.; Hu, J.; Wan, F.; Sun, Y.; Wu, J.; Li, Y.; et al. Reactive microglia enhance the transmission of exosomal α-synuclein via toll-like receptor 2. Brain J. Neurol. 2021, 144, 2024–2037. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain J. Neurol. 2020, 143, 1476–1497. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, D.; Cisternas-Olmedo, M.; Arcos, J.; Nassif, M.; Vidal, R.L. Contribution of Autophagy-Lysosomal Pathway in the Exosomal Secretion of Alpha-Synuclein and Its Impact in the Progression of Parkinson’s Disease. Front. Mol. Neurosci. 2022, 15, 805087. [Google Scholar] [CrossRef] [PubMed]
- Rattanavirotkul, N.; Kirschner, K.; Chandra, T. Induction and transmission of oncogene-induced senescence. Cell. Mol. Life Sci. CMLS 2021, 78, 843–852. [Google Scholar] [CrossRef]
- Borghesan, M.; Fafián-Labora, J.; Eleftheriadou, O.; Carpintero-Fernández, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximénez-Embún, P.; Lowe, R.; et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019, 27, 3956–3971.e6. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Fafián-Labora, J.A.; Rodríguez-Navarro, J.A.; O’Loghlen, A. Small Extracellular Vesicles Have GST Activity and Ameliorate Senescence-Related Tissue Damage. Cell Metab. 2020, 32, 71–86.e5. [Google Scholar] [CrossRef]
- Terlecki-Zaniewicz, L.; Lämmermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmüllner, R.; Dellago, H.; Skalicky, S.; Pum, D.; et al. Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging 2018, 10, 1103–1132. [Google Scholar] [CrossRef]
- Jankovic, J.; Rajput, A.H.; McDermott, M.P.; Perl, D.P. The evolution of diagnosis in early Parkinson disease. Parkinson Study Group. Arch. Neurol. 2000, 57, 369–372. [Google Scholar] [CrossRef]
- Adler, C.H.; Beach, T.G.; Hentz, J.G.; Shill, H.A.; Caviness, J.N.; Driver-Dunckley, E.; Sabbagh, M.N.; Sue, L.I.; Jacobson, S.A.; Belden, C.M.; et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: Clinicopathologic study. Neurology 2014, 83, 406–412. [Google Scholar] [CrossRef] [Green Version]
- Niu, M.; Li, Y.; Li, G.; Zhou, L.; Luo, N.; Yao, M.; Kang, W.; Liu, J. A longitudinal study on α-synuclein in plasma neuronal exosomes as a biomarker for Parkinson’s disease development and progression. Eur. J. Neurol. 2020, 27, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wu, Y.; Liu, G.; Jiang, Y.; Wang, X.; Wang, Z.; Feng, T. α-Synuclein in salivary extracellular vesicles as a potential biomarker of Parkinson’s disease. Neurosci. Lett. 2019, 696, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Kojima, M.; Kurosawa, T.; Sasaki, R.; Ichihara, S.; Hiraku, Y.; Tomimoto, H.; Murata, M.; Oikawa, S. Proteomic Profiling of Exosomal Proteins for Blood-based Biomarkers in Parkinson’s Disease. Neuroscience 2018, 392, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.H.; Chen, Z.T.; Zhou, R.L.; Zhang, X.; Ye, Q.Y.; Wang, Y.Z. Increased DJ-1 and α-Synuclein in Plasma Neural-Derived Exosomes as Potential Markers for Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 438. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Huang, Y.; Zhang, H.; Lu, H.; Zheng, J.C. Exosomal miRNAs in central nervous system diseases: Biomarkers, pathological mediators, protective factors and therapeutic agents. Prog. Neurobiol. 2019, 183, 101694. [Google Scholar] [CrossRef]
- Maciotta, S.; Meregalli, M.; Torrente, Y. The involvement of microRNAs in neurodegenerative diseases. Front. Cell. Neurosci. 2013, 7, 265. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.P.; Li, Y.X.; Zhu, X.H.; Du, X.G.; Zhou, M.; Li, W.B.; Deng, H.Y. Effect of Regulatory Network of Exosomes and microRNAs on Neurodegenerative Diseases. Chin. Med. J. 2018, 131, 2216–2225. [Google Scholar] [CrossRef]
- Paschon, V.; Takada, S.H.; Ikebara, J.M.; Sousa, E.; Raeisossadati, R.; Ulrich, H.; Kihara, A.H. Interplay Between Exosomes, microRNAs and Toll-Like Receptors in Brain Disorders. Mol. Neurobiol. 2016, 53, 2016–2028. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Gao, Q.; Ping, D.; Wang, Y.; Wu, W.; Lin, X.; Fang, Y.; Zhang, J.; Shao, A. The Role of Exosomal microRNAs and Oxidative Stress in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 3232869. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.F.; Qu, M.W.; Li, G.C.; Zhang, F.B.; Rui, H.C. Circulating exosomal miRNAs as diagnostic biomarkers in Parkinson’s disease. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5278–5283. [Google Scholar] [CrossRef]
- He, S.; Huang, L.; Shao, C.; Nie, T.; Xia, L.; Cui, B.; Lu, F.; Zhu, L.; Chen, B.; Yang, Q. Several miRNAs derived from serum extracellular vesicles are potential biomarkers for early diagnosis and progression of Parkinson’s disease. Transl. Neurodegener. 2021, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, P.; Ghate, V.; Nampoothiri, M.; Lewis, S. Multifunctional role of exosomes in viral diseases: From transmission to diagnosis and therapy. Cell. Signal. 2022, 94, 110325. [Google Scholar] [CrossRef]
- van Dongen, H.M.; Masoumi, N.; Witwer, K.W.; Pegtel, D.M. Extracellular Vesicles Exploit Viral Entry Routes for Cargo Delivery. Microbiol. Mol. Biol. Rev. MMBR 2016, 80, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Nolte-‘t Hoen, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef]
- Gunasekaran, M.; Bansal, S.; Ravichandran, R.; Sharma, M.; Perincheri, S.; Rodriguez, F.; Hachem, R.; Fisher, C.E.; Limaye, A.P.; Omar, A.; et al. Respiratory viral infection in lung transplantation induces exosomes that trigger chronic rejection. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2020, 39, 379–388. [Google Scholar] [CrossRef]
- Kuate, S.; Cinatl, J.; Doerr, H.W.; Uberla, K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology 2007, 362, 26–37. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deeks, S.G.; Mustapic, M.; Kapogiannis, D.; Henrich, T.J.; Lu, S.; Goldberg, S.A.; Hoh, R.; Chen, J.Y.; Martinez, E.O.; et al. SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19. Ann. Neurol. 2022, 91, 772–781. [Google Scholar] [CrossRef]
- Pesce, E.; Manfrini, N.; Cordiglieri, C.; Santi, S.; Bandera, A.; Gobbini, A.; Gruarin, P.; Favalli, A.; Bombaci, M.; Cuomo, A.; et al. Exosomes Recovered From the Plasma of COVID-19 Patients Expose SARS-CoV-2 Spike-Derived Fragments and Contribute to the Adaptive Immune Response. Front. Immunol. 2021, 12, 785941. [Google Scholar] [CrossRef] [PubMed]
- Barberis, E.; Vanella, V.V.; Falasca, M.; Caneapero, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; De Giorgis, V.; Puricelli, C.; Vaschetto, R.; et al. Circulating Exosomes Are Strongly Involved in SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8, 632290. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Booth, A.M.; Hildreth, J.E. The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 10592–10597. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Bihl, J. Exosome-Mediated Transfer of ACE2 (Angiotensin-Converting Enzyme 2) from Endothelial Progenitor Cells Promotes Survival and Function of Endothelial Cell. Oxidative Med. Cell. Longev. 2020, 2020, 4213541. [Google Scholar] [CrossRef]
- Inal, J.M. Decoy ACE2-expressing extracellular vesicles that competitively bind SARS-CoV-2 as a possible COVID-19 therapy. Clin. Sci. 2020, 134, 1301–1304. [Google Scholar] [CrossRef]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef]
- Böker, K.O.; Lemus-Diaz, N.; Rinaldi Ferreira, R.; Schiller, L.; Schneider, S.; Gruber, J. The Impact of the CD9 Tetraspanin on Lentivirus Infectivity and Exosome Secretion. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 634–647. [Google Scholar] [CrossRef]
- Earnest, J.T.; Hantak, M.P.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. The tetraspanin CD9 facilitates MERS-coronavirus entry by scaffolding host cell receptors and proteases. PLoS Pathog. 2017, 13, e1006546. [Google Scholar] [CrossRef]
- Lam, S.M.; Zhang, C.; Wang, Z.; Ni, Z.; Zhang, S.; Yang, S.; Huang, X.; Mo, L.; Li, J.; Lee, B.; et al. A multi-omics investigation of the composition and function of extracellular vesicles along the temporal trajectory of COVID-19. Nat. Metab. 2021, 3, 909–922. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, T.; Zhang, B. Exosomes in Parkinson’s Disease. Neurosci. Bull. 2017, 33, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.D.; MacLean, A.G. Extracellular Vesicles as a Means of Viral Immune Evasion, CNS Invasion, and Glia-Induced Neurodegeneration. Front. Cell. Neurosci. 2021, 15, 695899. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Spencer, S.J. Obesity and neuroinflammation: A pathway to cognitive impairment. Brain Behav. Immun. 2014, 42, 10–21. [Google Scholar] [CrossRef]
- Fuggle, N.R.; Howe, F.A.; Allen, R.L.; Sofat, N. New insights into the impact of neuro-inflammation in rheumatoid arthritis. Front. Neurosci. 2014, 8, 357. [Google Scholar] [CrossRef]
- Henry, C.J.; Huang, Y.; Wynne, A.M.; Godbout, J.P. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 2009, 23, 309–317. [Google Scholar] [CrossRef]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef]
- Li, J.J.; Wang, B.; Kodali, M.C.; Chen, C.; Kim, E.; Patters, B.J.; Lan, L.; Kumar, S.; Wang, X.; Yue, J.; et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflamm. 2018, 15, 8. [Google Scholar] [CrossRef]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflamm. 2014, 11, 68. [Google Scholar] [CrossRef]
- Bianco, F.; Pravettoni, E.; Colombo, A.; Schenk, U.; Möller, T.; Matteoli, M.; Verderio, C. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J. Immunol. 2005, 174, 7268–7277. [Google Scholar] [CrossRef]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef]
- Sur, S.; Khatun, M.; Steele, R.; Isbell, T.S.; Ray, R.; Ray, R.B. Exosomes from COVID-19 Patients Carry Tenascin-C and Fibrinogen-β in Triggering Inflammatory Signals in Cells of Distant Organ. Int. J. Mol. Sci. 2021, 22, 3184. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.J.; Li, W.; Lim, S.L.; Campbell, I.L. The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Ottum, P.A.; Arellano, G.; Reyes, L.I.; Iruretagoyena, M.; Naves, R. Opposing Roles of Interferon-Gamma on Cells of the Central Nervous System in Autoimmune Neuroinflammation. Front. Immunol. 2015, 6, 539. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Banerjea, A.C. SARS-CoV-2 Spike Targets USP33-IRF9 Axis via Exosomal miR-148a to Activate Human Microglia. Front. Immunol. 2021, 12, 656700. [Google Scholar] [CrossRef]
- Williams, G.P.; Schonhoff, A.M.; Jurkuvenaite, A.; Gallups, N.J.; Standaert, D.G.; Harms, A.S. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain J. Neurol. 2021, 144, 2047–2059. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Fang, P.; Schachner, M.; Shen, Y.Q. HMGB1 in development and diseases of the central nervous system. Mol. Neurobiol. 2012, 45, 499–506. [Google Scholar] [CrossRef]
- Santoro, M.; Maetzler, W.; Stathakos, P.; Martin, H.L.; Hobert, M.A.; Rattay, T.W.; Gasser, T.; Forrester, J.V.; Berg, D.; Tracey, K.J.; et al. In-vivo evidence that high mobility group box 1 exerts deleterious effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model and Parkinson’s disease which can be attenuated by glycyrrhizin. Neurobiol. Dis. 2016, 91, 59–68. [Google Scholar] [CrossRef]
- Sasaki, T.; Liu, K.; Agari, T.; Yasuhara, T.; Morimoto, J.; Okazaki, M.; Takeuchi, H.; Toyoshima, A.; Sasada, S.; Shinko, A.; et al. Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp. Neurol. 2016, 275 Pt 1, 220–231. [Google Scholar] [CrossRef]
- Canaslan, S.; Schmitz, M.; Villar-Piqué, A.; Maass, F.; Gmitterová, K.; Varges, D.; Lingor, P.; Llorens, F.; Hermann, P.; Zerr, I. Detection of Cerebrospinal Fluid Neurofilament Light Chain as a Marker for Alpha-Synucleinopathies. Front. Aging Neurosci. 2021, 13, 717930. [Google Scholar] [CrossRef]
- Bäckström, D.; Linder, J.; Jakobson Mo, S.; Riklund, K.; Zetterberg, H.; Blennow, K.; Forsgren, L.; Lenfeldt, N. NfL as a biomarker for neurodegeneration and survival in Parkinson disease. Neurology 2020, 95, e827–e838. [Google Scholar] [CrossRef] [PubMed]
- Srpova, B.; Uher, T.; Hrnciarova, T.; Barro, C.; Andelova, M.; Michalak, Z.; Vaneckova, M.; Krasensky, J.; Noskova, L.; Havrdova, E.K.; et al. Serum neurofilament light chain reflects inflammation-driven neurodegeneration and predicts delayed brain volume loss in early stage of multiple sclerosis. Mult. Scler. 2021, 27, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Tang, N.; Peluso, M.J.; Iyer, N.S.; Torres, L.; Donatelli, J.L.; Munter, S.E.; Nixon, C.C.; Rutishauser, R.L.; Rodriguez-Barraquer, I.; et al. Characterization and Biomarker Analyses of Post-COVID-19 Complications and Neurological Manifestations. Cells 2021, 10, 386. [Google Scholar] [CrossRef]
- Butowt, R.; von Bartheld, C.S. The route of SARS-CoV-2 to brain infection: Have we been barking up the wrong tree? Mol. Neurodegener. 2022, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Bulfamante, G.; Bocci, T.; Falleni, M.; Campiglio, L.; Coppola, S.; Tosi, D.; Chiumello, D.; Priori, A. Brainstem neuropathology in two cases of COVID-19: SARS-CoV-2 trafficking between brain and lung. J. Neurol. 2021, 268, 4486–4491. [Google Scholar] [CrossRef] [PubMed]
- Rangon, C.M.; Krantic, S.; Moyse, E.; Fougère, B. The Vagal Autonomic Pathway of COVID-19 at the Crossroad of Alzheimer’s Disease and Aging: A Review of Knowledge. J. Alzheimer’s Dis. Rep. 2020, 4, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Fenrich, M.; Mrdenovic, S.; Balog, M.; Tomic, S.; Zjalic, M.; Roncevic, A.; Mandic, D.; Debeljak, Z.; Heffer, M. SARS-CoV-2 Dissemination Through Peripheral Nerves Explains Multiple Organ Injury. Front. Cell. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef]
- Sarubbo, F.; El Haji, K.; Vidal-Balle, A.; Bargay Lleonart, J. Neurological consequences of COVID-19 and brain related pathogenic mechanisms: A new challenge for neuroscience. Brain Behav. Immun.-Health 2022, 19, 100399. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Alvarez-Fischer, D.; Arias-Carrión, O.; Djufri, M.; Windolph, A.; Keber, U.; Borta, A.; Ries, V.; Schwarting, R.K.; Scheller, D.; et al. A new dopaminergic nigro-olfactory projection. Acta Neuropathol. 2015, 130, 333–348. [Google Scholar] [CrossRef]
- Otake, K.; Oiso, Y.; Mitsuma, T.; Hirooka, Y.; Adachi, K. Hypothalamic dysfunction in Parkinson’s disease patients. Acta Med. Hung. 1994, 50, 3–13. [Google Scholar]
- Greene, J.G. Causes and consequences of degeneration of the dorsal motor nucleus of the vagus nerve in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D.; Al-Dalahmah, O.; Banu, M.A.; Boehme, A.K.; Boubour, A.L.; Bruce, S.S.; Chong, A.M.; Claassen, J.; et al. COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain J. Neurol. 2021, 144, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Käufer, C.; Schreiber, C.S.; Hartke, A.S.; Denden, I.; Stanelle-Bertram, S.; Beck, S.; Kouassi, N.M.; Beythien, G.; Becker, K.; Schreiner, T.; et al. Microgliosis and neuronal proteinopathy in brain persist beyond viral clearance in SARS-CoV-2 hamster model. EBioMedicine 2022, 79, 103999. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Paramasivam, P.; Kamath, M.; Sharma, A.; Rome, S.; Murugesan, R. Genetic Exchange of Lung-Derived Exosome to Brain Causing Neuronal Changes on COVID-19 Infection. Mol. Neurobiol. 2021, 58, 5356–5368. [Google Scholar] [CrossRef]
- Rahman, A.; Tabassum, T.; Araf, Y.; Al Nahid, A.; Ullah, M.A.; Hosen, M.J. Silent hypoxia in COVID-19: Pathomechanism and possible management strategy. Mol. Biol. Rep. 2021, 48, 3863–3869. [Google Scholar] [CrossRef]
- Butturini, E.; Boriero, D.; Carcereri de Prati, A.; Mariotto, S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 2019, 669, 22–30. [Google Scholar] [CrossRef]
- Lashgari, N.A.; Roudsari, N.M.; Momtaz, S.; Sathyapalan, T.; Abdolghaffari, A.H.; Sahebkar, A. The involvement of JAK/STAT signaling pathway in the treatment of Parkinson’s disease. J. Neuroimmunol. 2021, 361, 577758. [Google Scholar] [CrossRef]
- Musgrove, R.E.; Helwig, M.; Bae, E.J.; Aboutalebi, H.; Lee, S.J.; Ulusoy, A.; Di Monte, D.A. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. J. Clin. Investig. 2019, 129, 3738–3753. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int. J. Mol. Sci. 2022, 23, 3394. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mysiris, D.S.; Vavougios, G.D.; Karamichali, E.; Papoutsopoulou, S.; Stavrou, V.T.; Papayianni, E.; Boutlas, S.; Mavridis, T.; Foka, P.; Zarogiannis, S.G.; et al. Post-COVID-19 Parkinsonism and Parkinson’s Disease Pathogenesis: The Exosomal Cargo Hypothesis. Int. J. Mol. Sci. 2022, 23, 9739. https://doi.org/10.3390/ijms23179739
Mysiris DS, Vavougios GD, Karamichali E, Papoutsopoulou S, Stavrou VT, Papayianni E, Boutlas S, Mavridis T, Foka P, Zarogiannis SG, et al. Post-COVID-19 Parkinsonism and Parkinson’s Disease Pathogenesis: The Exosomal Cargo Hypothesis. International Journal of Molecular Sciences. 2022; 23(17):9739. https://doi.org/10.3390/ijms23179739
Chicago/Turabian StyleMysiris, Dimitrios S., George D. Vavougios, Eirini Karamichali, Stamatia Papoutsopoulou, Vasileios T. Stavrou, Eirini Papayianni, Stylianos Boutlas, Theodoros Mavridis, Pelagia Foka, Sotirios G. Zarogiannis, and et al. 2022. "Post-COVID-19 Parkinsonism and Parkinson’s Disease Pathogenesis: The Exosomal Cargo Hypothesis" International Journal of Molecular Sciences 23, no. 17: 9739. https://doi.org/10.3390/ijms23179739