
NSCLC as the Paradigm of Precision Medicine at Its Finest: The Rise of New Druggable Molecular Targets for Advanced Disease

 ,
,  ,
,
Abstract
:1. Introduction: Driver Alterations beyond EGFR, ALK and ROS1: What Do We Know So Far?
2. An Overview of the Main Rising Driver Alterations and Their Clinical Implications
2.1. Deregulation of MET Signalling Pathway
2.2. RET Rearrangements
2.3. NTRK1, NTRK2, NTRK3 Fusions
2.4. KRAS Mutations
2.5. HER2 Mutations and Amplifications
2.6. Current Evidence and Ongoing Trials Testing Combination Strategies
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37,513,025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Inamura, K. Lung Cancer: Understanding Its Molecular Pathology and the 2015 WHO Classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.-W.; Kato, T.; et al. Osimertinib in Resected EGFR -Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Cona, M.S.; Indini, A.; Testi, A.; Cresta, S.; Signorelli, D.; Garassino, M.C.; Sinno, V.; Sesana, S.; Pelosi, G.; de Braud, F.G.; et al. Druggable aberrations in solid tumors: An overview on ALK and ROS-1 status. Ann. Oncol. 2015, 26, vi143. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.I.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Kinno, T.; Tsuta, K.; Shiraishi, K.; Mizukami, T.; Suzuki, M.; Yoshida, A.; Suzuki, K.; Asamura, H.; Furuta, K.; Kohno, T.; et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 138–142. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Camidge, D.R.; Gadgeel, S.M.; Rosell, R.; Dziadziuszko, R.; Kim, D.W.; Pérol, M.; Ou, S.H.I.; Ahn, J.S.; Shaw, A.T.; et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann. Oncol. 2020, 31, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, H.R.; Ahn, M.J.; Yang, J.C.H.; Han, J.Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; García Campelo, M.R.; Kim, D.W.; et al. Brigatinib Versus Crizotinib in Advanced ALK Inhibitor-Naive ALK-Positive Non-Small Cell Lung Cancer: Second Interim Analysis of the Phase III ALTA-1L Trial. J. Clin. Oncol. 2020, 38, 3592–3603. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Hashemi, S.M.S.; Mazieres, J.; Kim, T.M.; Quoix, E.; Souquet, P.J.; Barlesi, F.; Baik, C.; et al. Phase 2 Study of Dabrafenib Plus Trametinib in Patients With BRAF V600E-Mutant Metastatic NSCLC: Updated 5-Year Survival Rates and Genomic Analysis. J. Thorac. Oncol. 2022, 17, 103–115. [Google Scholar] [CrossRef]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef]
- Farago, A.F.; Taylor, M.S.; Doebele, R.C.; Zhu, V.W.; Kummar, S.; Spira, A.I.; Boyle, T.A.; Haura, E.B.; Arcila, M.E.; Benayed, R.; et al. Clinicopathologic Features of Non–Small-Cell Lung Cancer Harboring an NTRK Gene Fusion. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Rolfo, C. NTRK gene fusions: A rough diamond ready to sparkle. Lancet Oncol. 2020, 21, 472–474. [Google Scholar] [CrossRef]
- Wang, R.; Hu, H.; Pan, Y.; Li, Y.; Ye, T.; Li, C.; Luo, X.; Wang, L.; Li, H.; Zhang, Y.; et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 4352–4359. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 patients with lung cancer harboring MET Exon 14 skipping alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non–Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Tsongalis, P.D.; Peterson, J.D.; de Abreu, F.B.; Black, C.C.; Gutmann, E.J.; Liu, X.; Tafe, L.J.; Amos, C.I.; Tsongalis, G.J. The potential utility of re-mining results of somatic mutation testing: KRAS status in lung adenocarcinoma. Cancer Genet. 2016, 209, 195–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Library of Medicine. MET MET Proto-Oncogene, Receptor Tyrosine Kinase [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=4233 (accessed on 7 June 2022).
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-Dependent Solid Tumors: Molecular Diagnosis and Targeted Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569. [Google Scholar] [CrossRef] [PubMed]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET Gene Copy Number Leads to Shorter Survival in Patients with Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Bubendorf, L.; Dafni, U.; Schöbel, M.; Finn, S.P.; Tischler, V.; Sejda, A.; Marchetti, A.; Thunnissen, E.; Verbeken, E.K.; Warth, A.; et al. Prevalence and clinical association of MET gene overexpression and amplification in patients with NSCLC: Results from the European Thoracic Oncology Platform (ETOP) Lungscape project. Lung Cancer 2017, 111, 143–149. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [Green Version]
- Casadevall, D.; Gimeno, J.; Clavé, S.; Taus, Á.; Pijuan, L.; Arumí, M.; Lorenzo, M.; Menéndez, S.; Cañadas, I.; Albanell, J.; et al. MET expression and copy number heterogeneity in nonsquamous non-small cell lung cancer (nsNSCLC). Oncotarget 2015, 6, 16215. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029. [Google Scholar] [CrossRef]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Barón, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number–Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltschukat, S.; Engstler, B.S.; Huang, A.; Hao, H.X.; Tam, A.; Wang, H.Q.; Liang, J.; DiMare, M.T.; Bhang, H.E.C.; Wang, Y.; et al. Capmatinib (INC280) is active against models of non–small cell lung cancer and other cancer types with defined mechanisms of MET activation. Clin. Cancer Res. 2019, 25, 3164–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14–Mutated or MET -Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ou, S.-H.I.; Weiss, J.; Ades, S.; Conte, U.; Tang, Y.; Wang, S.C.-E.; Murphy, D.; et al. Crizotinib in patients (pts) with MET-amplified non-small cell lung cancer (NSCLC): Updated safety and efficacy findings from a phase 1 trial. J. Clin. Oncol. 2018, 36, 9062. [Google Scholar] [CrossRef]
- Landi, L.; Chiari, R.; Tiseo, M.; D’Inca, F.; Dazzi, C.; Chella, A.; Delmonte, A.; Bonanno, L.; Giannarelli, D.; Cortinovis, D.L.; et al. Crizotinib in MET-deregulated or ROS1-rearranged pretreated non–small cell lung cancer (METROS): A phase II, prospective, multicenter, two-arms trial. Clin. Cancer Res. 2019, 25, 7312–7319. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.C. MET receptor juxtamembrane exon 14 alternative spliced variant: Novel cancer genomic predictive biomarker. Cancer Discov. 2015, 5, 802. [Google Scholar] [CrossRef] [Green Version]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB Domain Binding Site on the Met Receptor Tyrosine Kinase Converts It into a Transforming Protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET exon 14 mutations in Non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Mathieu, L.N.; Larkins, E.; Akinboro, O.; Roy, P.; Amatya, A.K.; Fiero, M.H.; Mishra-Kalyani, P.S.; Helms, W.S.; Myers, C.E.; Skinner, A.M.; et al. FDA Approval Summary: Capmatinib and Tepotinib for the Treatment of Metastatic NSCLC Harboring MET Exon 14 Skipping Mutations or Alterations. Clin. Cancer Res. 2022, 28, 249–254. [Google Scholar] [CrossRef]
- European Medicines Agency. Tepmetko. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/tepmetko#authorisation-details-section (accessed on 4 June 2022).
- European Medicines Agency. Tabrecta: Pending EC Decision. Available online: https://www.ema.europa.eu/en/medicines/human/summaries-opinion/tabrecta#key-facts-section (accessed on 4 June 2022).
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, N.; et al. Once-daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non-small-cell lung cancers harbouring MET exon 14 skipping alterations: A multicentre, single-arm, open-label, phase 2 study. Lancet Respir. Med. 2021, 9, 1154–1164. [Google Scholar] [CrossRef]
- Camidge, D.R.; Janku, F.; Martinez-Bueno, A.; Catenacci, D.V.T.; Lee, J.; Lee, S.-H.; Dowlati, A.; Rohrberg, K.S.; Navarro, A.; Moon, Y.W.; et al. Safety and preliminary clinical activity of the MET antibody mixture, Sym015 in advanced non-small cell lung cancer (NSCLC) patients with MET amplification/exon 14 deletion (METAmp/Ex14∆). J. Clin. Oncol. 2020, 38, 9510. [Google Scholar] [CrossRef]
- Camidge, D.R.; Moiseenko, F.; Cicin, I.; Horinouchi, H.; Filippova, E.; Bar, J.; Lu, S.; Tomasini, P.; Ocampo, C.; Sullivan, D.; et al. Abstract CT179: Telisotuzumab vedotin (teliso-v) monotherapy in patients with previously treated c-Met+ advanced non-small cell lung cancer. Cancer Res. 2021, 81, CT179. [Google Scholar] [CrossRef]
- National Library of Medicine. RET Ret Proto-Oncogene [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=5979 (accessed on 7 June 2022).
- Tan, A.C.; Lai, G.G.Y.; Tan, G.S.; Poon, S.Y.; Doble, B.; Lim, T.H.; Aung, Z.W.; Takano, A.; Tan, W.L.; Ang, M.K.; et al. Utility of incorporating next-generation sequencing (NGS) in an Asian non-small cell lung cancer (NSCLC) population: Incremental yield of actionable alterations and cost-effectiveness analysis. Lung Cancer 2020, 139, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Gautschi, O.; Milia, J.; Filleron, T.; Wolf, J.; Carbone, D.P.; Owen, D.; Camidge, R.; Narayanan, V.; Doebele, R.C.; Besse, B.; et al. Targeting RET in patients with RET-rearranged lung cancers: Results from the global, multicenter RET registry. J. Clin. Oncol. 2017, 35, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Gainor, J.F.; Curigliano, G.; Kim, D.W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.W.; et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): A multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021, 22, 959–969. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration (FDA). FDA Approves Selpercatinib for Lung and Thyroid Cancers with RET Gene Mutations or Fusions. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selpercatinib-lung-and-thyroid-cancers-ret-gene-mutations-or-fusions (accessed on 8 May 2022).
- U.S. Food & Drug Administration (FDA). FDA Approves Pralsetinib for Lung Cancer with RET Gene Fusions. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pralsetinib-lung-cancer-ret-gene-fusions (accessed on 8 May 2022).
- European Medicines Agency. Gavreto. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/gavreto (accessed on 4 June 2022).
- European Medicines Agency. Retsevmo. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/retsevmo (accessed on 4 June 2022).
- Drilon, A.E.; Zhai, D.; Rogers, E.; Deng, W.; Zhang, X.; Ung, J.; Lee, D.; Rodon, L.; Graber, A.; Zimmerman, Z.F.; et al. The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. J. Clin. Oncol. 2020, 38, 3616. [Google Scholar] [CrossRef]
- Schoffski, P.; Cho, B.C.; Italiano, A.; Loong, H.H.F.; Massard, C.; Rodriguez, L.M.; Shih, J.-Y.; Subbiah, V.; Verlingue, L.; Andreas, K.; et al. BOS172738, a highly potent and selective RET inhibitor, for the treatment of RET-altered tumors including RET-fusion+ NSCLC and RET-mutant MTC: Phase 1 study results. J. Clin. Oncol. 2021, 39, 3008. [Google Scholar] [CrossRef]
- Lin, J.J.; Liu, S.V.; McCoach, C.E.; Zhu, V.W.; Tan, A.C.; Yoda, S.; Peterson, J.; Do, A.; Prutisto-Chang, K.; Dagogo-Jack, I.; et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann. Oncol. 2020, 31, 1725–1733. [Google Scholar] [CrossRef]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, A.; Le, A.T.; Doebele, R.C. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [Green Version]
- Haratake, N.; Seto, T. NTRK Fusion-positive Non–small-cell Lung Cancer: The Diagnosis and Targeted Therapy. Clin. Lung Cancer 2021, 22, 1–5. [Google Scholar] [CrossRef]
- Boulle, F.; Kenis, G.; Cazorla, M.; Hamon, M.; Steinbusch, H.W.M.; Lanfumey, L.; van den Hove, D.L.A. TrkB inhibition as a therapeutic target for CNS-related disorders. Prog. Neurobiol. 2012, 98, 197–206. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Passiglia, F.; Caparica, R.; Giovannetti, E.; Giallombardo, M.; Listi, A.; Diana, P.; Cirrincione, G.; Caglevic, C.; Raez, L.E.; Russo, A.; et al. The potential of neurotrophic tyrosine kinase (NTRK) inhibitors for treating lung cancer. Expert Opin. Investig. Drugs 2016, 25, 385–392. [Google Scholar] [CrossRef]
- Russo, A.; Lopes, A.R.; McCusker, M.G.; Garrigues, S.G.; Ricciardi, G.R.; Arensmeyer, K.E.; Scilla, K.A.; Mehra, R.; Rolfo, C. New Targets in Lung Cancer (Excluding EGFR, ALK, ROS1). Curr. Oncol. Rep. 2020, 22, 48. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.Y.; et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731. [Google Scholar] [CrossRef]
- European Medicines Agency. Vitrakvi. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vitrakvi (accessed on 4 June 2022).
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- European Medicines Agency. Rozlytrek. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/rozlytrek (accessed on 4 June 2022).
- Dziadziuszko, R.; Krebs, M.G.; de Braud, F.; Siena, S.; Drilon, A.; Doebele, R.C.; Patel, M.R.; Chul Cho, B.; Liu, S.V.; Ahn, M.J.; et al. Updated Integrated Analysis of the Efficacy and Safety of Entrectinib in Locally Advanced or Metastatic ROS1 Fusion–Positive Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 1253–1263. [Google Scholar] [CrossRef]
- Ou, S.H.I.; Fujiwara, Y.; Shaw, A.T.; Yamamoto, N.; Nakagawa, K.; Fan, F.; Hao, Y.; Gao, Y.; Jänne, P.A.; Seto, T. Efficacy of Taletrectinib (AB-106/DS-6051b) in ROS1+ NSCLC: An Updated Pooled Analysis of U.S. and Japan Phase 1 Studies. JTO Clin. Res. Rep. 2021, 2, 100108. [Google Scholar] [CrossRef]
- Zhou, C.; Fan, H.; Wang, Y.; Wu, H.; Yang, N.; Li, K.; Wang, X.; Qin, X.; Yu, Q.; Fang, Y.; et al. Taletrectinib (AB-106; DS-6051b) in metastatic non-small cell lung cancer (NSCLC) patients with ROS1 fusion: Preliminary results of TRUST. J. Clin. Oncol. 2021, 39, 9066. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior trk kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Ou, S.H.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (Tpx-0005) is a next-generation ros1/trk/alk inhibitor that potently inhibits ros1/trk/alk solvent-front mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Anonymous. Combating Acquired TRK Inhibitor Resistance. Cancer Discov. 2019, 9, 684–685. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2669–2974. [Google Scholar] [CrossRef] [Green Version]
- Boch, C.; Kollmeier, J.; Roth, A.; Stephan-Falkenau, S.; Misch, D.; Grüning, W.; Bauer, T.T.; Mairinger, T. The frequency of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC): Routine screening data for central Europe from a cohort study. BMJ Open 2013, 3, e002560. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Leighl, N.B.; Tsao, M.S.; Shepherd, F.A. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J. Thorac. Oncol. 2013, 8, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- European Medicines Agency. Lumykras. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lumykras (accessed on 4 June 2022).
- Hallin, J.; Engstrom, L.D.; Hargi, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS G12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRASG12C Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, JCO-21. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022. [Google Scholar] [CrossRef]
- Cision PR Newswire. Mirati Therapeutics Submits Marketing Authorization Application to the European Medicines Agency for Investigational Adagrasib as a Treatment for Previously-Treated KRASG12C-mutated Non-Small Cell Lung Cancer. Available online: https://www.prnewswire.com/news-releases/mirati-therapeutics-submits-marketing-authorization-application-to-the-european-medicines-agency-for-investigational-adagrasib-as-a-treatment-for-previously-treated-krasg12c-mutated-non-small-cell-lung-cancer-301551654.html (accessed on 4 June 2022).
- National Library of Medicine. ERBB2 erb-b2 Receptor Tyrosine Kinase 2 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/2064#gene-expression (accessed on 7 May 2022).
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, V.; Fleitas, T.; Tarazona, N.; Cejalvo, J.M.; Gimeno-Valiente, F.; Martinez-Ciarpaglini, C.; Huerta, M.; Roselló, S.; Castillo, J.; Roda, D.; et al. Towards precision oncology for HER2 blockade in gastroesophageal adenocarcinoma. Ann. Oncol. 2019, 30, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Lara, P.N.; Laptalo, L.; Longmate, J.; Lau, D.H.M.; Gandour-Edwards, R.; Gumerlock, P.H.; Doroshow, J.H.; Gandara, D.R. Trastuzumab plus Docetaxel in HER2/neu–Positive Non–Small-Cell Lung Cancer: A California Cancer Consortium Screening and Phase II Trial. Clin. Lung Cancer 2004, 5, 231–236. [Google Scholar] [CrossRef]
- Zinner, R.G.; Glisson, B.S.; Fossella, F.V.; Pisters, K.M.W.; Kies, M.S.; Lee, P.M.; Massarelli, E.; Sabloff, B.; Fritsche, H.A.; Ro, J.Y.; et al. Trastuzumab in combination with cisplatin and gemcitabine in patients with Her2-overexpressing, untreated, advanced non-small cell lung cancer: Report of a phase II trial and findings regarding optimal identification of patients with Her2-overexpressing disease. Lung Cancer 2004, 44, 99–110. [Google Scholar] [CrossRef]
- Krug, L.M.; Miller, V.A.; Patel, J.; Crapanzano, J.; Azzoli, C.G.; Gomez, J.; Kris, M.G.; Heelan, R.T.; Pizzo, B.; Tyson, L.; et al. Randomized phase II study of weekly docetaxel plus trastuzumab versus weekly paclitaxel plus trastuzumab in patients with previously untreated advanced nonsmall cell lung carcinoma. Cancer 2005, 104, 2149–2155. [Google Scholar] [CrossRef]
- Hotta, K.; Aoe, K.; Kozuki, T.; Ohashi, K.; Ninomiya, K.; Ichihara, E.; Kubo, T.; Ninomiya, T.; Chikamori, K.; Harada, D.; et al. A Phase II Study of Trastuzumab Emtansine in HER2-Positive Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 273–279. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Stahel, R.; Bubendorf, L.; Bonomi, P.; Villegas, A.; Kowalski, D.M.; Baik, C.S.; Isla, D.; De Castro Carpeno, J.; Garrido, P.; et al. Trastuzumab Emtansine (T-DM1) in Patients with Previously Treated HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Efficacy, Safety, and Biomarkers. Clin. Cancer Res. 2019, 25, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O’Meara, S.; Smith, R.; Parker, A.; et al. Intragenic ERBB2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Shen, R.; Buonocore, D.; Olah, Z.T.; Ni, A.; Ginsberg, M.S.; Ulaner, G.A.; Offin, M.; Feldman, D.; Hembrough, T.; et al. Ado-trastuzumab emtansine for patients with HER2-mutant lung cancers: Results from a phase II basket trial. J. Clin. Oncol. 2018, 36, 2532–2537. [Google Scholar] [CrossRef]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2 -Mutant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251. [Google Scholar] [CrossRef]
- Nakagawa, K.; Nagasaka, M.; Felip, E.; Pacheco, J.; Baik, C.; Goto, Y.; Saltos, A.; Li, B.; Udagawa, H.; Gadgeel, S.; et al. OA04.05 Trastuzumab Deruxtecan in HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Interim Results of DESTINY-Lung01. J. Thorac. Oncol. 2021, 16, S109–S110. [Google Scholar] [CrossRef]
- Lai, W.V.; Lebas, L.; Barnes, T.A.; Milia, J.; Ni, A.; Gautschi, O.; Peters, S.; Ferrara, R.; Plodkowski, A.J.; Kavanagh, J.; et al. Afatinib in patients with metastatic or recurrent HER2-mutant lung cancers: A retrospective international multicentre study. Eur. J. Cancer 2019, 109, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Curioni-Fontecedro, A.; Nechushtan, H.; Shih, J.Y.; Liao, W.Y.; Gautschi, O.; Spataro, V.; Unk, M.; Chih-Hsin Yang, J.; Lorence, R.M.; et al. Activity of Afatinib in Heavily Pretreated Patients With ERBB2 Mutation-Positive Advanced NSCLC: Findings From a Global Named Patient Use Program. J. Thorac. Oncol. 2018, 13, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Grève, J.; Moran, T.; Graas, M.P.; Galdermans, D.; Vuylsteke, P.; Canon, J.L.; Schallier, D.; Decoster, L.; Teugels, E.; Massey, D.; et al. Phase II study of afatinib, an irreversible ErbB family blocker, in demographically and genotypically defined lung adenocarcinoma. Lung Cancer 2015, 88, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Smit, E.F.; Dafni, U.; Wolf, J.; Wasąg, B.; Biernat, W.; Finn, S.P.; Kammler, R.; Tsourti, Z.; Rabaglio, M.; et al. Afatinib in NSCLC With HER2 Mutations: Results of the Prospective, Open-Label Phase II NICHE Trial of European Thoracic Oncology Platform (ETOP). J. Thorac. Oncol. 2019, 14, 1086–1094. [Google Scholar] [CrossRef]
- Kris, M.G.; Camidge, D.R.; Giaccone, G.; Hida, T.; Li, B.T.; O’Connell, J.; Taylor, I.; Zhang, H.; Arcila, M.E.; Goldberg, Z.; et al. Targeting HER2 aberrations as actionable drivers in lung cancers: Phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann. Oncol. 2015, 26, 1421–1427. [Google Scholar] [CrossRef]
- Mazéres, J.; Barlesi, F.; Filleron, T.; Besse, B.; Monnet, I.; Beau-Faller, M.; Peters, S.; Dansin, E.; Früh, M.; Pless, M.; et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EUHER2 cohort. Ann. Oncol. 2016, 27, 281–286. [Google Scholar] [CrossRef]
- Elamin, Y.Y.; Robichaux, J.P.; Carter, B.W.; Altan, M.; Gibbons, D.L.; Fossella, F.V.; Lam, V.K.; Patel, A.B.; Negrao, M.V.; Le, X.; et al. Poziotinib for Patients With HER2 Exon 20 Mutant Non–Small-Cell Lung Cancer: Results From a Phase II Trial. J. Clin. Oncol. 2022, 40, 702–709. [Google Scholar] [CrossRef]
- Le, X.; Cornelissen, R.; Garassino, M.; Clarke, J.M.; Tchekmedyian, N.; Goldman, J.W.; Leu, S.Y.; Bhat, G.; Lebel, F.; Heymach, J.V.; et al. Poziotinib in Non-Small-Cell Lung Cancer Harboring HER2 Exon 20 Insertion Mutations After Prior Therapies: ZENITH20-2 Trial. J. Clin. Oncol. 2022, 40, 710–718. [Google Scholar] [CrossRef]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients With EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, e214761, Erratum in JAMA Oncol. 2022. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, T.; Qin, Z.; Jiang, J.; Wang, Q.; Yang, S.; Rivard, C.; Gao, G.; Ng, T.L.; Tu, M.M.; et al. HER2 exon 20 insertions in non-small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Li, X.; Wang, Q.; Gao, G.; Zhang, Y.; Chen, J.; Shu, Y.; Hu, Y.; Fan, Y.; Fang, J.; et al. Pyrotinib in HER2-Mutant Advanced Lung Adenocarcinoma After Platinum-Based Chemotherapy: A Multicenter, Open-Label, Single-Arm, Phase II Study. J. Clin. Oncol. 2020, 38, 2753–2761. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Offin, M.; Hembrough, T.; Cecchi, F.; Shen, R.; Olah, Z.; Panora, E.; Myers, M.; Brzostowski, E.; Buonocore, D.; et al. P1.13-44 Safety, PK, and Preliminary Antitumor Activity of the Oral EGFR/HER2 Exon 20 Inhibitor TAK-788 in NSCLC. J. Thorac. Oncol. 2018, 13, S599. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Joon, O.P.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.H.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.W.; Soo, R.A.; Kim, S.W.; Pan, H.; et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): An open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar] [CrossRef]
- McCoach, C.E.; Yu, A.; Gandara, D.R.; Riess, J.W.; Vang, D.P.; Li, T.; Lara, P.N.; Gubens, M.; Lara, F.; Mack, P.C.; et al. Phase I/II Study of Capmatinib Plus Erlotinib in Patients With MET-Positive Non–Small-Cell Lung Cancer. JCO Precis. Oncol. 2021, 1, 177–190. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.H.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Cortinovis, D.L.; Majem, M.; Johnson, M.L.; Mardjuadi, F.I.; Zhao, X.; Siripurapu, S.V.; Jiang, Z.; Wolf, J. Efficacy and safety of capmatinib plus pembrolizumab in treatment (tx)-naïve patients with advanced non–small cell lung cancer (NSCLC) with high tumor PD-L1 expression: Results of a randomized, open-label, multicenter, phase 2 study. J. Clin. Oncol. 2022, 40, 9118. [Google Scholar] [CrossRef]
- Briere, D.M.; Li, S.; Calinisan, A.; Sudhakar, N.; Aranda, R.; Hargis, L.; Peng, D.H.; Deng, J.; Engstrom, L.D.; Hallin, J.; et al. The KRAS G12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol. Cancer Ther. 2021, 20, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Wallrabenstein, T.; Del Rio, J.; Templeton, A.J.; Buess, M. Much has changed in the last decade except overall survival: A Swiss single center analysis of treatment and survival in patients with stage IV non-small cell lung cancer. PLoS ONE 2020, 15, e0233768. [Google Scholar] [CrossRef] [PubMed]
- Spini, A.; Gini, R.; Rosellini, P.; Singier, A.; Bellan, C.; Pascucci, A.; Leoncini, L.; Mathieu, C.; Martellucci, I.; Furiesi, F.; et al. First-Line Pharmacotherapies and Survival among Patients Diagnosed with Non-Resectable NSCLC: A Real-Life Setting Study with Gender Prospective. Cancers 2021, 13, 6129. [Google Scholar] [CrossRef] [PubMed]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Kerr, K.M.; Bubendorf, L.; Edelman, M.J.; Marchetti, A.; Mok, T.; Novello, S.; O’Byrne, K.; Stahel, R.; Peters, S.; Felip, E.; et al. Second ESMO consensus conference on lung cancer: Pathology and molecular biomarkers for non-small-cell lung cancer. Ann. Oncol. 2014, 25, 1681–1690. [Google Scholar] [CrossRef]
- Joshi, A.; Zanwar, S.; Noronha, V.; Patil, V.M.; Chougule, A.; Kumar, R.; Janu, A.; Mahajan, A.; Kapoor, A.; Prabhash, K. EGFR mutation in squamous cell carcinoma of the lung: Does it carry the same connotation as in adenocarcinomas? Onco. Targets. Ther. 2017, 10, 1859. [Google Scholar] [CrossRef] [Green Version]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Rekhtman, N.; Paik, P.K.; Arcila, M.E.; Tafe, L.J.; Oxnard, G.R.; Moreira, A.L.; Travis, W.D.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: Lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clin. Cancer Res. 2012, 18, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- Kenmotsu, H.; Serizawa, M.; Koh, Y.; Isaka, M.; Takahashi, T.; Taira, T.; Ono, A.; Maniwa, T.; Takahashi, S.; Mori, K.; et al. Prospective genetic profiling of squamous cell lung cancer and adenosquamous carcinoma in Japanese patients by multitarget assays. BMC Cancer 2014, 14, 786. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Choi, Y.L.; Song, J.Y.; Zhu, Y.; Xu, Q.; Zhang, F.; Jiang, L.; Cheng, J.; Zheng, G.; Mao, M. ALK, ROS1 and RET rearrangements in lung squamous cell carcinoma are very rare. Lung Cancer 2016, 94, 22–27. [Google Scholar] [CrossRef]
- Sheikine, Y.; Pavlick, D.; Klempner, S.J.; Trabucco, S.E.; Chung, J.H.; Rosenzweig, M.; Wang, K.; Velcheti, V.; Frampton, G.M.; Peled, N.; et al. BRAF in Lung Cancers: Analysis of Patient Cases Reveals Recurrent BRAF Mutations, Fusions, Kinase Duplications, and Concurrent Alterations. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.K.; Tran, H.T.; Banks, K.C.; Lanman, R.B.; Rinsurongkawong, W.; Peled, N.; Lewis, J.; Lee, J.J.; Roth, J.; Roarty, E.B.; et al. Targeted Tissue and Cell-Free Tumor DNA Sequencing of Advanced Lung Squamous-Cell Carcinoma Reveals Clinically Significant Prevalence of Actionable Alterations. Clin. Lung Cancer 2019, 20, 30–36.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, H.G.; Ho, A.T.N.; Altibi, A.M.A.; Nakazawa, T.; Katoh, R.; Kondo, T. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer—A systematic review and meta-analysis. Lung Cancer 2018, 123, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.D.; Lee, S.E.; Oh, D.Y.; Yu, D.B.; Jeong, H.M.; Kim, J.; Hong, S.; Jung, H.S.; Oh, E.; Song, J.Y.; et al. MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: Clinicopathologic Implications and Prognostic Values. J. Thorac. Oncol. 2017, 12, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Jänne, P.; Skokan, M.; Finocchiaro, G.; Rossi, E.; Ligorio, C.; Zucali, P.; Terracciano, L.; Toschi, L.; Roncalli, M.; et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann. Oncol. 2008, 20, 298–304. [Google Scholar] [CrossRef]
- Pillai, R.N.; Behera, M.; Berry, L.D.; Rossi, M.R.; Kris, M.G.; Johnson, B.E.; Bunn, P.A.; Ramalingam, S.S.; Khuri, F.R. HER2 Mutations in Lung Adenocarcinomas: A Report from the Lung Cancer Mutation Consortium. Cancer 2017, 123, 4099. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, G.R.R.; Russo, A.; Franchina, T.; Ferraro, G.; Zanghì, M.; Picone, A.; Scimone, A.; Adamo, V. NSCLC and HER2: Between Lights and Shadows. J. Thorac. Oncol. 2014, 9, 1750–1762. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Ji, X.; Su, C. HER2-Altered Non-Small Cell Lung Cancer: Biology, Clinicopathologic Features, and Emerging Therapies. Front. Oncol. 2022, 12, 860313. [Google Scholar] [CrossRef]
- Ninomiya, K.; Hata, T.; Yoshioka, H.; Ohashi, K.; Bessho, A.; Hosokawa, S.; Ishikawa, N.; Yamasaki, M.; Shibayama, T.; Aoe, K.; et al. A Prospective Cohort Study to Define the Clinical Features and Outcome of Lung Cancers Harboring HER2 Aberration in Japan (HER2-CS STUDY). Chest 2019, 156, 357–366. [Google Scholar] [CrossRef]
- Lin, C.; Wang, S.; Xie, W.; Chang, J.; Gan, Y. The RET fusion gene and its correlation with demographic and clinicopathological features of non-small cell lung cancer: A meta-analysis. Cancer Biol. Ther. 2015, 16, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, S.; Scheel, A.H.; Scheffler, M.; Schultheis, A.M.; Gautschi, O.; Aebersold, F.; Diebold, J.; Pall, G.; Rothschild, S.; Bubendorf, L.; et al. Clinicopathological Characteristics of RET Rearranged Lung Cancer in European Patients. J. Thorac. Oncol. 2016, 11, 122–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Reis, H.; Metzenmacher, M.; Goetz, M.; Savvidou, N.; Darwiche, K.; Aigner, C.; Herold, T.; Eberhardt, W.E.; Skiba, C.; Hense, J.; et al. MET Expression in Advanced Non-Small-Cell Lung Cancer: Effect on Clinical Outcomes of Chemotherapy, Targeted Therapy, and Immunotherapy. Clin. Lung Cancer 2018, 19, e441–e463. [Google Scholar] [CrossRef]
- Hegde, A.; Andreev-Drakhlin, A.Y.; Roszik, J.; Huang, L.; Liu, S.; Hess, K.; Cabanillas, M.; Hu, M.I.; Busaidy, N.L.; Sherman, S.I.; et al. Responsiveness to immune checkpoint inhibitors versus other systemic therapies in RET-aberrant malignancies. ESMO Open 2020, 5, e000799. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, Z.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of Acquired Resistance to First-Line Osimertinib: Preliminary Data from the Phase III FLAURA Study. Available online: https://oncologypro.esmo.org/meeting-resources/esmo-2018-congress/Mechanisms-of-acquired-resistance-to-first-line-osimertinib-preliminary-data-from-the-phase-III-FLAURA-study (accessed on 8 May 2022).


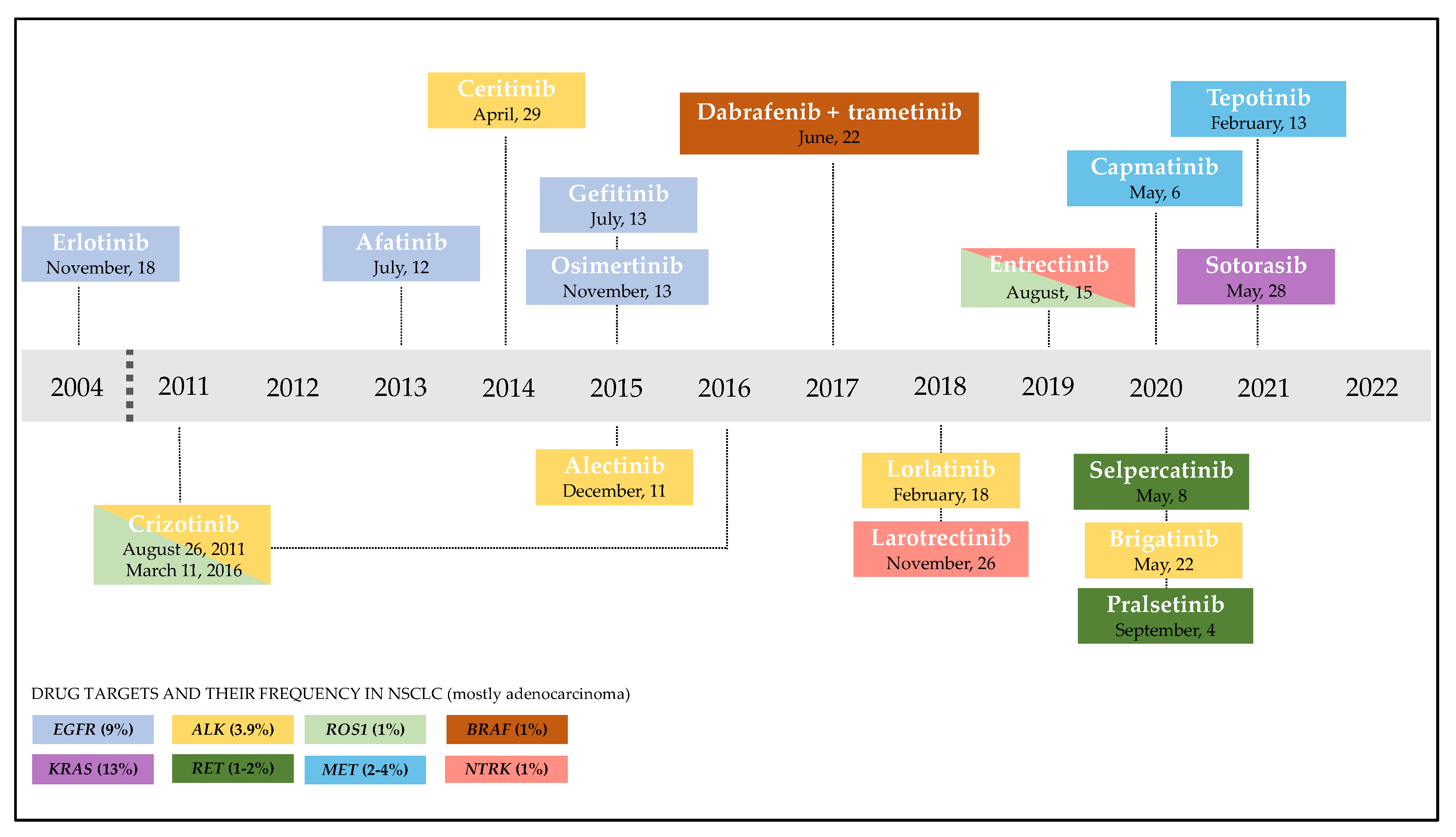{kind=link}
| Clinical Trial | Trial Type | Driver Mutation | Treatment Arms | Clinical Outcomes | Most Frequent AEs (All Grades) |
|---|---|---|---|---|---|
| GEOMETRY mono-1 | Phase 2, multicenter, multi-cohort, single-arm, non-randomized, open-label study | METex14 skipping mutation | Capmatinib | Pretreated pts: ORR 41%, DoR 9.7, mPFS 5.4 m. Treatment naïve: ORR 68%, DoR 12.6, mPFS 12.4 m. | Peripheral edema, nausea, vomiting, creatinine increase. |
| VISION | Phase 2, multicenter, multi-cohort, single-arm, non-randomized, open-label study | METex14 skipping mutation | Tepotinib | Combined biopsy: ORR by ICR 46%, DoR 11.1 m, mPFS 8.5 m. Liquid biopsy: ORR by ICR 48%, DoR 9.9 m, mPFS 8.5 m. Tissue biopsy: ORR by ICR 50%, DoR 15.7 m, mPFS 11 m. | Peripheral edema, nausea, diarrhea, creatinine increase. |
| LIBRETTO-001 | Phase 1/2 trial, international, open-label study | RET fusion | Selpercatinib | ORR by ICR 64%, DoR 17.5 m, mPFS 18.4 m. | Diarrhea, AST increase, dry mouth, hypertension, fatigue. |
| ARROW | Phase 1/2, multi-cohort, international, open-label study | RET fusion | Pralsetinib | Pretreated: ORR 61%, DoR NR, mPFS 17.1 m. Treatment naïve: ORR 70%, DoR 9.0 m, 9.1 m. | AST/ALT increase, anemia, leucopenia, fatigue, constipation. |
| LOXO-TRK-14001 | Phase 1/2, multi-cohort, international, open-label study | NTRK gene fusion | Larotrectinib * | Overall population: ORR 75%, DoR NR, mPFS NR. (7 lung cancer pts enrolled) | AST/ALT increase, anemia, neutropenia, weight increase. |
| SCOUT/ NAVIGATE | Phase 1/2, multicenter, multi-cohort, single-arm, non-randomized, open-label study | NTRK gene fusion | Larotrectinib * | ORR 30%, DCR 70%, mPFS 18.3 m, mOS NR. | AST/ALT increase, leucopenia, neutropenia, vomiting. |
| STARTRK-1, STARTRK-2, ALKA-372-001 (Pooled analysis) | Phase 1/2, multicenter, single-arm, non-randomized, open-label study | NTRK gene fusion, ROS1 rearrangement | Entrectinib | Overall population: ORR 59.3%, DoR 12.9, mPFS 12.9 m, mOS 23.9 m. | Fatigue, dysgeusia, paresthesia, nausea, myalgia. |
| CodeBreak 100 | Phase 2, multicenter, international, single arm, open-label study | KRASG12C mutation | Sotorasib | ORR by ICR 37.4%, DCR 80.5%, mPFS 6.7 m. | AST/ALT increase, leucopenia, anemia, diarrhea, myalgia, nausea, fatigue, hepatotoxicity, cough. |
| NCT Number | Gene Alteration | Experimental Drug | Phase | Study Design | Current Status |
|---|---|---|---|---|---|
| NCT05015608 | MET amplification | Savolitinib | 3 | Randomized: savolitinib + osimertinib vs. pemetrexed + cisplatin/carboplatin | Recruiting |
| NCT04338243 | MET amplification | Glumetinib | 1/2 | Single group assignment | Unknown |
| NCT02435121 | MET amplification | SAR125844 | 2 | Single group assignment | Completed |
| NCT02544633 | MET mutation, amplification | MGCD265 | 2 | Single group assignment | Completed |
| NCT03175224 | METex14, amplification, fusion | APL-101 | 1/2 | Single group assignment | Recruiting |
| NCT04270591 | MET mutation, amplification | Glumetinib | 1/2 | Single group assignment | Active, not recruiting |
| NCT02648724 | MET amplification | Sym015 | 1/2 | Single group assignment | Completed |
| NCT03539536 | MET amplification | Telisotuzumab vedotin | 2 | Single group assignment | Recruiting |
| NCT05163249 | MET amplification | Savolitinib | 2 | Randomized: osimertinib ± savolitinib | Not yet recruiting |
| NCT03993873 | MET mutation, amplification | TPX-0022 | 1/2 | Single group assignment | Recruiting |
| NCT Number | Gene Alteration | Experimental Drug | Phase | Study Design | Current Status |
|---|---|---|---|---|---|
| NCT04683250 | RET alterations | TAS0953/HM06 | 1/2 | Sequential assignment | Recruiting |
| NCT03784378 | RET mutation | RXDX-105 | 1 | Single group assignment | Completed |
| NCT03780517 | RET alterations | BOS172738 | 1 | Sequential assignment | Active, not recruiting |
| NCT04161391 | RET fusion, mutation | TPX-0046 | 1/2 | Single group assignment | Recruiting |
| NCT01877811 | RET rearrangement, fusion | RXDX-105 | 1 | Single group assignment | Completed |
| NCT Number | Gene Alteration | Experimental Drug | Phase | Study Design | Current Status |
|---|---|---|---|---|---|
| NCT03170206 | KRAS mutation | Binimetinib plus palbociclib | 1/2 | Single group assignment | Recruiting |
| NCT05374538 | KRASG12C mutation | VIC-1911 plus sotorasib | 1 | Single group assignment | Not yet recruiting |
| NCT04685135 | KRASG12C mutation | Adagrasib | 3 | Randomized: adagrasib vs. docetaxel | Recruiting |
| NCT04735068 | KRAS mutation | Binimetinib | 2 | Single group assignment | Recruiting |
| NCT05132075 | KRASG12C mutation | JDQ443 | 3 | Randomized: JDQ443 vs. docetaxel | Recruiting |
| NCT04967079 | KRAS mutation | Trametinib plus anlotinib | 1 | Single group assignment | Recruiting |
| NCT04620330 | KRAS mutation | VS-6766 | 2 | Randomized: VS-6766 ± defactinib | Recruiting |
| NCT01933932 | KRAS mutation | Selumetinib | 3 | Randomized: docetaxel + selumetinib/placebo | Active, not recruiting |
| NCT04613596 | KRASG12C mutation | Adagrasib | 2 | Randomized: adagrasib ± pembrolizumab | Recruiting |
| NCT03299088 | KRAS mutation | Trametinib plus pembrolizumab | 1 | Single group assignment | Active, not recruiting |
| NCT03520842 | KRAS mutation | Regorafenib plus methotrexate | 1 | Single group assignment | Active, not recruiting |
| NCT05276726 | KRASG12C mutation | JAB 21822 | 1 | Sequential assignment | Not yet recruiting |
| NCT03681483 | KRAS mutation | RO5126766 | 1 | Single group assignment | Active, not recruiting |
| NCT02642042 | KRAS mutation | Trametinib plus docetaxel | 2 | Single group assignment | Active, not recruiting |
| NCT03808558 | KRAS mutation | TVB-2640 | 2 | Single group assignment | Recruiting |
| NCT05375994 | KRASG12C mutation | Defactinib plus adagrasib | 1/2 | Sequential assignment | Not yet recruiting |
| NCT04449874 | KRASG12C mutation | GDC-6036 | 2 | Single group assignment | Recruiting |
| NCT05379985 | KRASG12 mutation | RMC-6236 | 1 | Single group assignment | Not yet recruiting |
| NCT04965818 | KRAS mutation | Futibatinib and binimetinib | 1/2 | Single group assignment | Recruiting |
| NCT04263090 | KRAS mutation | Rigosetib | 1/2 | Sequential assignment | Recruiting |
| NCT04699188 | KRASG12C mutation | JDQ443, TNO155, tislelizumab | 1/2 | Sequential assignment | Recruiting |
| NCT05288205 | KRASG12C mutation | JAB-21822, JAB-3312 | 1/2 | Sequential assignment | Not yet recruiting |
| NCT05358249 | KRASG12C mutation | JDQ443 | 1/2 | Sequential assignment | Not yet recruiting |
| NCT05054725 | KRASG12C mutation | RMC-4630 plus sotorasib | 2 | Sequential assignment | Recruiting |
| NCT04956640 | KRASG12C mutation | LY3537982 | 1 | Single group assignment | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelotti, A.; de Scordilli, M.; Bertoli, E.; De Carlo, E.; Del Conte, A.; Bearz, A. NSCLC as the Paradigm of Precision Medicine at Its Finest: The Rise of New Druggable Molecular Targets for Advanced Disease. Int. J. Mol. Sci. 2022, 23, 6748. https://doi.org/10.3390/ijms23126748
Michelotti A, de Scordilli M, Bertoli E, De Carlo E, Del Conte A, Bearz A. NSCLC as the Paradigm of Precision Medicine at Its Finest: The Rise of New Druggable Molecular Targets for Advanced Disease. International Journal of Molecular Sciences. 2022; 23(12):6748. https://doi.org/10.3390/ijms23126748
Chicago/Turabian StyleMichelotti, Anna, Marco de Scordilli, Elisa Bertoli, Elisa De Carlo, Alessandro Del Conte, and Alessandra Bearz. 2022. "NSCLC as the Paradigm of Precision Medicine at Its Finest: The Rise of New Druggable Molecular Targets for Advanced Disease" International Journal of Molecular Sciences 23, no. 12: 6748. https://doi.org/10.3390/ijms23126748