
CRISPR/Cas9–Mediated Genome Editing for Pseudomonas fulva, a Novel Pseudomonas Species with Clinical, Animal, and Plant–Associated Isolates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
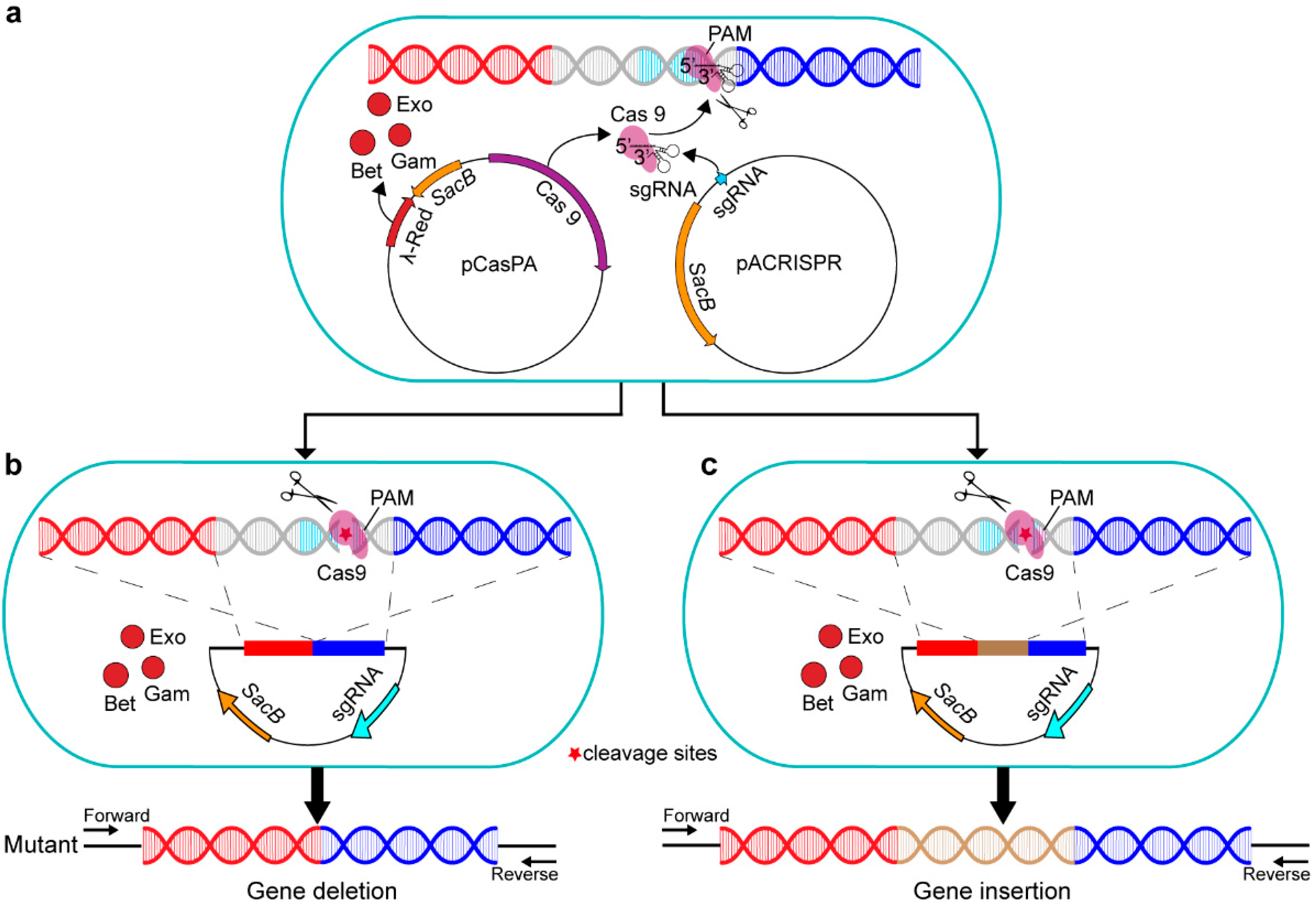
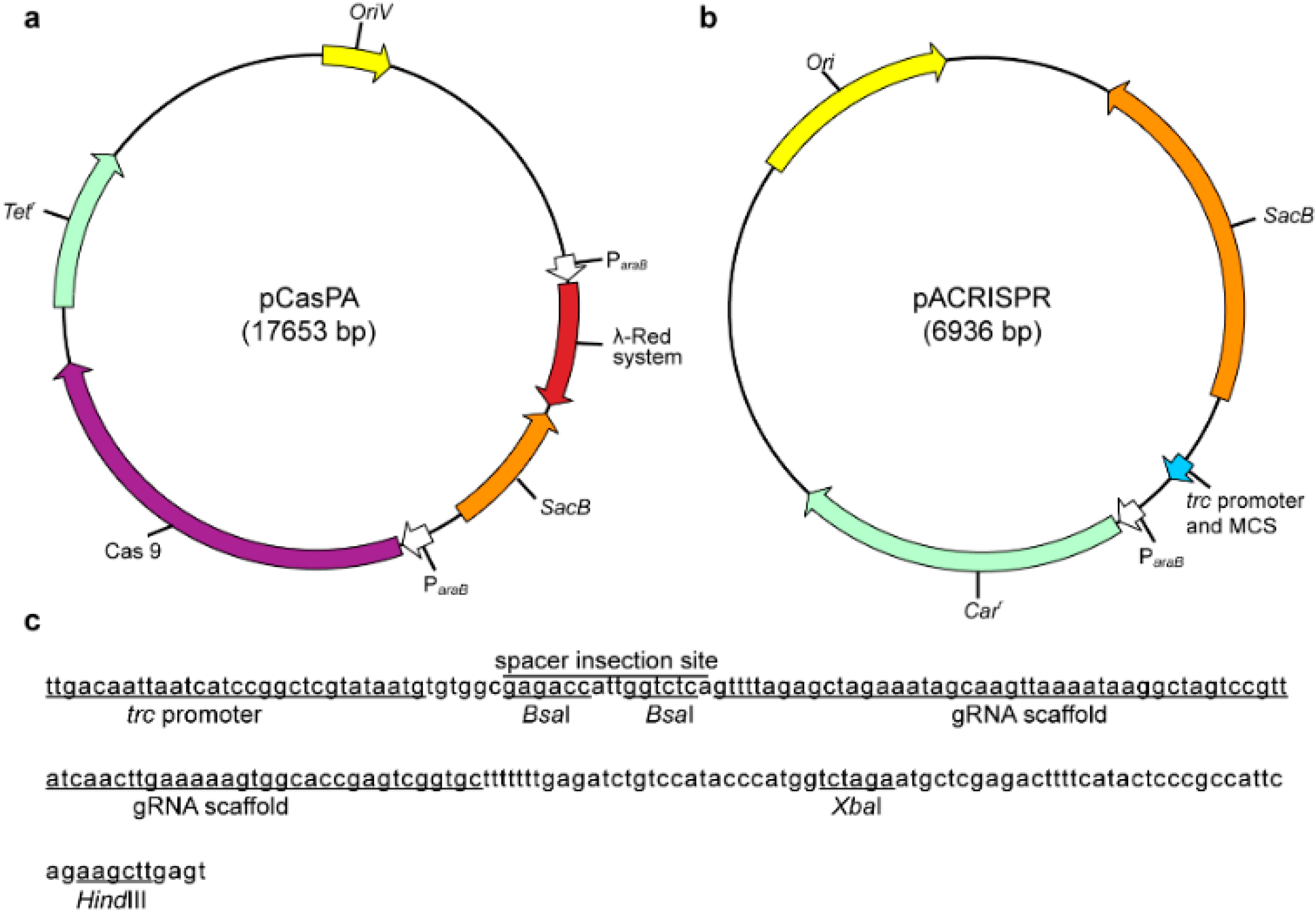
2.1. Construction of a Two–Plasmid CRISPR–Cas9 System in P. fulva
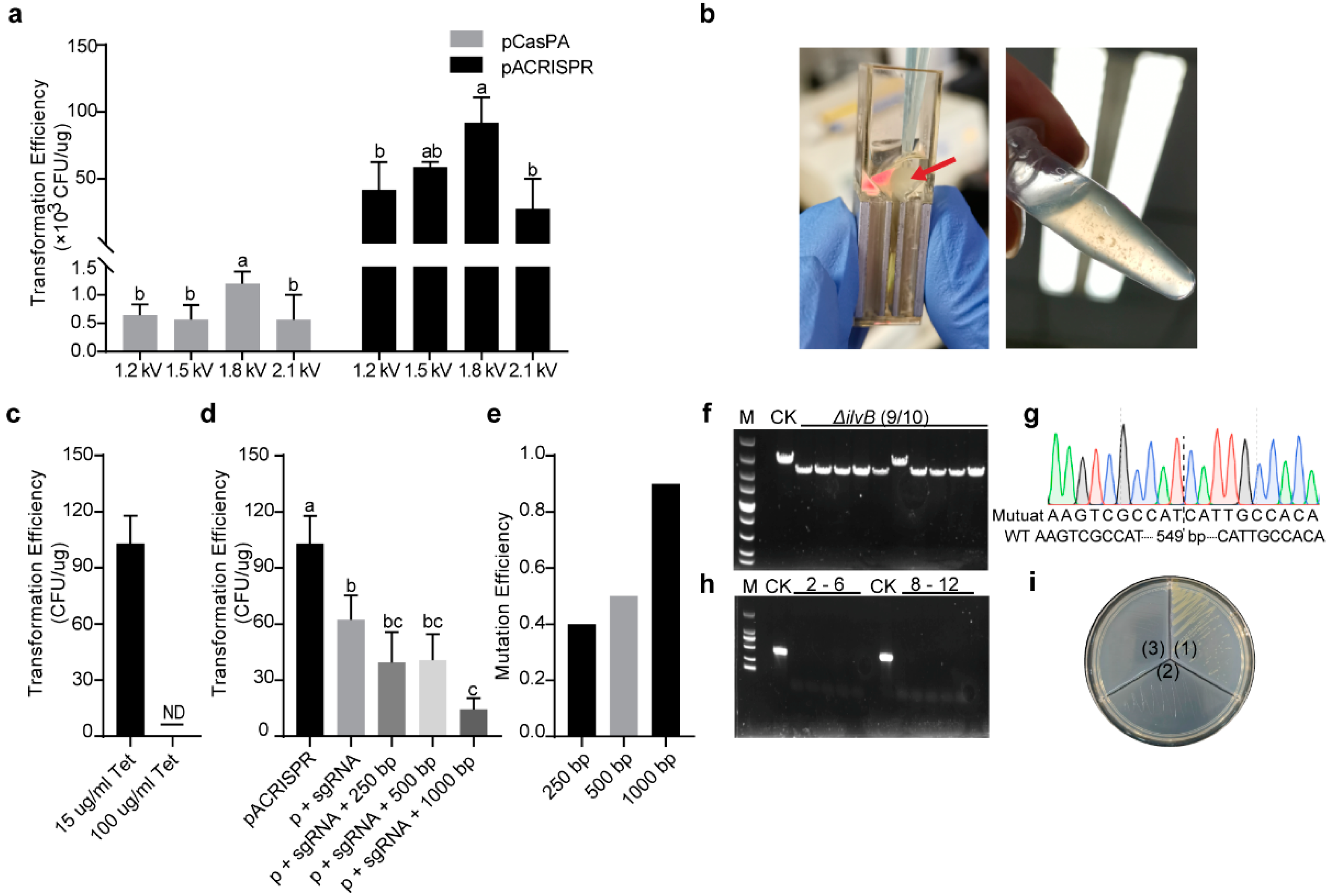
2.2. Estimation of Transformation Parameters for Higher Transformation Efficiency
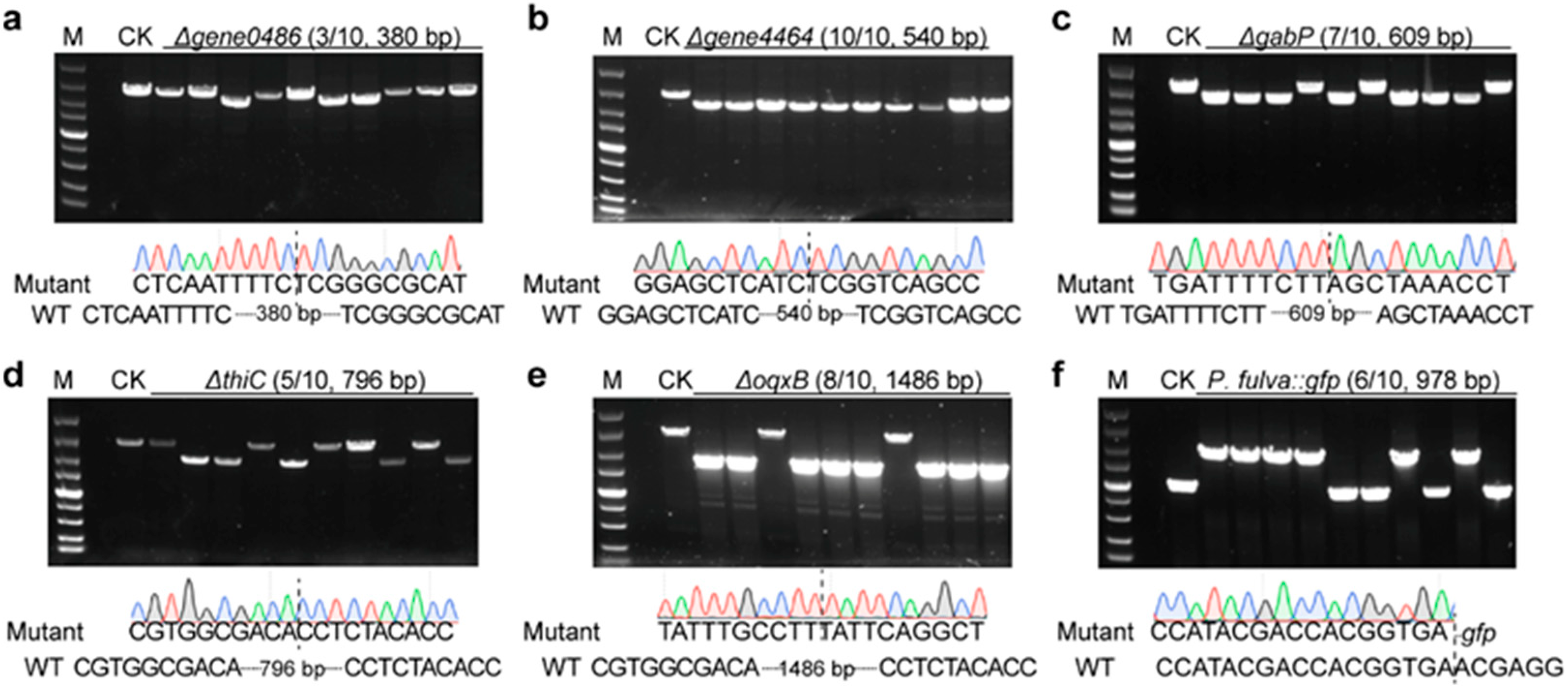
2.3. Analysis of Multigene Editing Efficiencies with the Established Two–Plasmid System in P. fulva
3. Materials and Methods
3.1. Bacteria, Plasmids, Primers, and Growth Conditions
3.2. Plasmid Construction
3.3. Preparation of Competent Cells
3.4. Selection of Electrotransformation Parameters
3.5. Gene Editing and Plasmid Curing
3.6. Measurement of the Growth Curve
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kahl, S.; Kleinsteuber, S.; Nivala, J.; van Afferden, M.; Reemtsma, T. Emerging Biodegradation of the Previously Persistent Artificial Sweetener Acesulfame in Biological Wastewater Treatment. Environ. Sci. Technol. 2018, 52, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Kuklinsky-Sobral, J.; Araujo, W.L.; Mendes, R.; Geraldi, I.O.; Pizzirani-Kleiner, A.A.; Azevedo, J.L. Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environ. Microbiol. 2004, 6, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Hong, Y.; Liu, J.; Gao, Y.Z.; Ma, Z.; Yang, B.; Ling, W.T.; Waigi, M.G. A PAH-degrading bacterial community enriched with contaminated agricultural soil and its utility for microbial bioremediation. Environ. Pollut. 2019, 251, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, D.M.; Dees, J.L.; Ibberson, C.B.; Huse, H.K.; Mathiesen, I.H.; Kirketerp-Møller, K.; Wolcott, R.D.; Rumbaugh, K.P.; Bjarnsholt, T.; Whiteley, M. Pseudomonas aeruginosa transcriptome during human infection. Proc. Natl. Acad. Sci. USA 2018, 115, E5125–E5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, H.; Loper, J.E. Genomics of secondary metabolite production by Pseudomonas spp. Nat. Prod. Rep. 2009, 26, 1408–1446. [Google Scholar] [CrossRef]
- Peix, A.; Ramírez-Bahena, M.-H.; Velázquez, E. Historical evolution and current status of the taxonomy of genus Pseudomonas. Infect. Genet. Evol. 2009, 9, 1132–1147. [Google Scholar] [CrossRef]
- Nikel, P.I.; Martínez-García, E.; de Lorenzo, V. Biotechnological domestication of pseudomonads using synthetic biology. Nat. Rev. Microbiol. 2014, 12, 368–379. [Google Scholar] [CrossRef]
- Xie, Y.; Shao, X.; Deng, X. Regulation of type III secretion system in Pseudomonas syringae. Environ. Microbiol. 2019, 21, 4465–4477. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.J.; Feng, Y.M.; Zhan, H.; Liu, J.; Yang, F.; Zhang, K.Y.; Zhang, L.H.; Chen, S.H. Characterization of a Pyrethroid-Degrading Pseudomonas fulva Strain P31 and Biochemical Degradation Pathway of D-Phenothrin. Front. Microbiol. 2018, 9, 1003. [Google Scholar] [CrossRef]
- Jariyal, M.; Jindal, V.; Mandal, K.; Gupta, V.K.; Singh, B. Bioremediation of organophosphorus pesticide phorate in soil by microbial consortia. Ecotoxicol. Environ. Saf. 2018, 159, 310–316. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Gu, G. Study on the Remediation of Phorate in Soil by Microbial Consortia. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Changchun, China, 21–23 August 2020; Volume 508, p. 012120. [Google Scholar] [CrossRef]
- Adeniji, A.A.; Babalola, O.O. Evaluation of Pseudomonas fulva PS9.1 and Bacillus velezensis NWUMFkBS10.5 as Candidate Plant Growth Promoters during Maize-Fusarium Interaction. Plants 2022, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Zheng, L.P.; Wang, J.W. Inducing perylenequinone production from a bambusicolous fungus Shiraia sp. S9 through co-culture with a fruiting body-associated bacterium Pseudomonas fulva SB1. Microb. Cell Fact. 2019, 18, 121. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.R.; Lien, C.Y.; Tsai, W.C.; Lai, W.A.; Hsu, C.W.; Tsai, N.W.; Chang, C.C.; Lu, C.H.; Chien, C.C.; Chang, W.N. The clinical characteristics of adult bacterial meningitis caused by non-Pseudomonas (Ps.) aeruginosa Pseudomonas species: A clinical comparison with Ps. aeruginosa meningitis. Kaohsiung J. Med. Sci. 2018, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Uddin, F.; Roulston, K.; McHugh, T.D.; Khan, T.A.; Sohail, M. Bacteremia in a human caused by an XDR strain of Pseudomonas fulva. J. Infect. Dev. Ctries. 2018, 12, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Uzuner, N.; Ozcan, N.; Kangul, H.; Kadandir, I.R. The First Case of Urosepsis Due to Multidrug Resistant Pseudomonas fulva. Flora Infeksiyon Hastalik. Ve Klin. Mikrobiyoloji Derg. 2020, 25, 269–274. [Google Scholar] [CrossRef]
- Hoang, T.T.; Karkhoff-Schweizer, R.R.; Kutchma, A.J.; Schweizer, H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 1998, 212, 77–86. [Google Scholar] [CrossRef]
- Xu, Z.; Li, M.; Li, Y.; Cao, H.; Miao, L.; Xu, Z.; Higuchi, Y.; Yamasaki, S.; Nishino, K.; Woo, P.C.Y.; et al. Native CRISPR-Cas-Mediated Genome Editing Enables Dissecting and Sensitizing Clinical Multidrug-Resistant P. aeruginosa. Cell Rep. 2019, 29, 1707–1717.e1703. [Google Scholar] [CrossRef]
- Cianfanelli, F.R.; Cunrath, O.; Bumann, D. Efficient dual-negative selection for bacterial genome editing. BMC Microbiol. 2020, 20, 129. [Google Scholar] [CrossRef]
- Xu, Z.; Li, Y.; Yan, A. Repurposing the Native Type I-F CRISPR-Cas System in Pseudomonas aeruginosa for Genome Editing. STAR Protoc. 2020, 1, 100039. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, Z.; Gao, X.; Li, J.; Shang, G. Combination of ssDNA recombineering and CRISPR-Cas9 for Pseudomonas putida KT2440 genome editing. Appl. Microbiol. Biotechnol. 2019, 103, 2783–2795. [Google Scholar] [CrossRef]
- Aparicio, T.; de Lorenzo, V.; Martínez-García, E. CRISPR/Cas9-Based Counterselection Boosts Recombineering Efficiency in Pseudomonas putida. Biotechnol. J. 2017, 13, 1700161. [Google Scholar] [CrossRef] [PubMed]
- Asin-Garcia, E.; Martin-Pascual, M.; Garcia-Morales, L.; van Kranenburg, R.; Martins Dos Santos, V.A.P. ReScribe: An Unrestrained Tool Combining Multiplex Recombineering and Minimal-PAM ScCas9 for Genome Recoding Pseudomonas putida. ACS Synth. Biol. 2021, 10, 2672–2688. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Zhao, M.; Wojcik, S.; Taiaroa, G.; Butler, M.; Poulter, R. The application of the CRISPR-Cas9 system in Pseudomonas syringae pv. actinidiae. J. Med. Microbiol. 2020, 69, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, W.J.; Chang, J.; Thireault, C.A.; Kimbrel, J. Recombineering and stable integration of the Pseudomonas syringae pv. syringae 61 hrp/hrc cluster into the genome of the soil bacterium Pseudomonas fluorescens Pf0-1. Plant J. 2010, 60, 919–928. [Google Scholar] [CrossRef]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Westra, E.R.; Dowling, A.J.; Broniewski, J.M.; van Houte, S. Evolution and ecology of CRISPR. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 307–331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zhang, Z.; Unver, T.; Zhang, B. CRISPR/Cas: A powerful tool for gene function study and crop improvement. J. Adv. Res. 2021, 29, 207–221. [Google Scholar] [CrossRef]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Li, Y.; Li, M.; Xiang, H.; Yan, A. Harnessing the type I CRISPR-Cas systems for genome editing in prokaryotes. Environ. Microbiol. 2021, 23, 542–558. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, D.J.; Ward, J.D.; Reiner, D.J.; Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 2013, 10, 1028–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Lessard, S.; Francioli, L.; Alfoldi, J.; Tardif, J.C.; Ellinor, P.T.; Macarthur, D.G.; Lettre, G.; Orkin, S.H.; Canver, M.C. Human genetic variation alters CRISPR-Cas9 on- and off-targeting specificity at therapeutically implicated loci. Proc. Natl. Acad. Sci. USA 2017, 114, E11257–E11266. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.H.; Lee, W.J.; An, J.H.; Lee, J.H.; Kim, Y.H.; Kim, H.; Oh, Y.; Park, Y.H.; Jin, Y.B.; Jun, B.H.; et al. Prediction-based highly sensitive CRISPR off-target validation using target-specific DNA enrichment. Nat. Commun. 2020, 11, 3596. [Google Scholar] [CrossRef]
- Xie, X.; Ma, X.; Zhu, Q.; Zeng, D.; Li, G.; Liu, Y.G. CRISPR-GE: A Convenient Software Toolkit for CRISPR-Based Genome Editing. Mol. Plant 2017, 10, 1246–1249. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zheng, Y.; Zhao, Z.; Liu, T.; Li, J. Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. Bioinformatics 2018, 34, i757–i765. [Google Scholar] [CrossRef] [Green Version]
- Minkenberg, B.; Zhang, J.; Xie, K.; Yang, Y. CRISPR-PLANT v2: An online resource for highly specific guide RNA spacers based on improved off-target analysis. Plant Biotechnol. J. 2019, 17, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Qian, F.; Yang, J.; Liu, Y.; Dong, F.; Xu, C.; Sun, B.; Chen, B.; Xu, X.; Li, Y.; et al. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat. Commun. 2017, 8, 15179. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.H.; van Pijkeren, J.P. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res. 2014, 42, e131. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef] [Green Version]
- Diallo, M.; Hocq, R.; Collas, F.; Chartier, G.; Wasels, F.; Wijaya, H.S.; Werten, M.W.T.; Wolbert, E.J.H.; Kengen, S.W.M.; van der Oost, J.; et al. Adaptation and application of a two-plasmid inducible CRISPR-Cas9 system in Clostridium beijerinckii. Methods 2020, 172, 51–60. [Google Scholar] [CrossRef]
- Narayanan, N.; Xu, Y.; Chou, C.P. High-level gene expression for recombinant penicillin acylase production using the araB promoter system in Escherichia coli. Biotechnol. Prog. 2006, 22, 1518–1523. [Google Scholar] [CrossRef]
- Brosius, J.; Erfle, M.; Storella, J. Spacing of the -10 and -35 regions in the tac promoter: Effect on its in vivo activity. J. Biol. Chem. 1985, 260, 3539–3541. [Google Scholar] [CrossRef]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden gate shuffling: A one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison 3rd, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, H.P. Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol. 1992, 6, 1195–1204. [Google Scholar] [CrossRef]
- Mruk, I.; Kaczorowski, T.; Witczak, A. Natural tuning of restriction endonuclease synthesis by cluster of rare arginine codons. Sci. Rep. 2019, 9, 5808. [Google Scholar] [CrossRef]
- Papagianni, M.; Papamichael, E.M. Plasmid transformation of Weissella paramesenteroides DX by electroporation. Anaerobe 2014, 30, 60–64. [Google Scholar] [CrossRef]
- Arai, T.; Aikawa, S.; Sudesh, K.; Kondo, T.; Kosugi, A. Electrotransformation of thermophilic bacterium Caldimonas manganoxidans. J. Microbiol. Methods 2022, 192, 106375. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Y.; Zhang, Y.; Pi, Y.; Gu, T.; Song, L.; Wang, Y.; Ji, Q. CRISPR/Cas9-based Genome Editing in Pseudomonas aeruginosa and Cytidine Deaminase-Mediated Base Editing in Pseudomonas Species. iScience 2018, 6, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Ausubel, F.M. Current Protocols in Molecular Biology; Greene Pub. Associates and Wiley-Interscience: New York, NY, USA, 1987; Volume 1. [Google Scholar]
- Nickoloff, J.A. Electroporation Protocols for Microorganisms; Springer Science & Business Media: Berlin, Germany, 1995; Volume 47. [Google Scholar]
- Teh, B.S.; Apel, J.; Shao, Y.; Boland, W. Colonization of the Intestinal Tract of the Polyphagous Pest Spodoptera littoralis with the GFP-Tagged Indigenous Gut Bacterium Enterococcus mundtii. Front. Microbiol. 2016, 7, 928. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; He, J.; Muhammad, A.; Shao, Y. CRISPR/Cas9–Mediated Genome Editing for Pseudomonas fulva, a Novel Pseudomonas Species with Clinical, Animal, and Plant–Associated Isolates. Int. J. Mol. Sci. 2022, 23, 5443. https://doi.org/10.3390/ijms23105443
Zhang N, He J, Muhammad A, Shao Y. CRISPR/Cas9–Mediated Genome Editing for Pseudomonas fulva, a Novel Pseudomonas Species with Clinical, Animal, and Plant–Associated Isolates. International Journal of Molecular Sciences. 2022; 23(10):5443. https://doi.org/10.3390/ijms23105443
Chicago/Turabian StyleZhang, Nan, Jintao He, Abrar Muhammad, and Yongqi Shao. 2022. "CRISPR/Cas9–Mediated Genome Editing for Pseudomonas fulva, a Novel Pseudomonas Species with Clinical, Animal, and Plant–Associated Isolates" International Journal of Molecular Sciences 23, no. 10: 5443. https://doi.org/10.3390/ijms23105443