
Reorganization of Parvalbumin Immunopositive Perisomatic Innervation of Principal Cells in Focal Cortical Dysplasia Type IIB in Human Epileptic Patients

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Dependence of Immunostaining on Fixation and Post-Mortem Delay
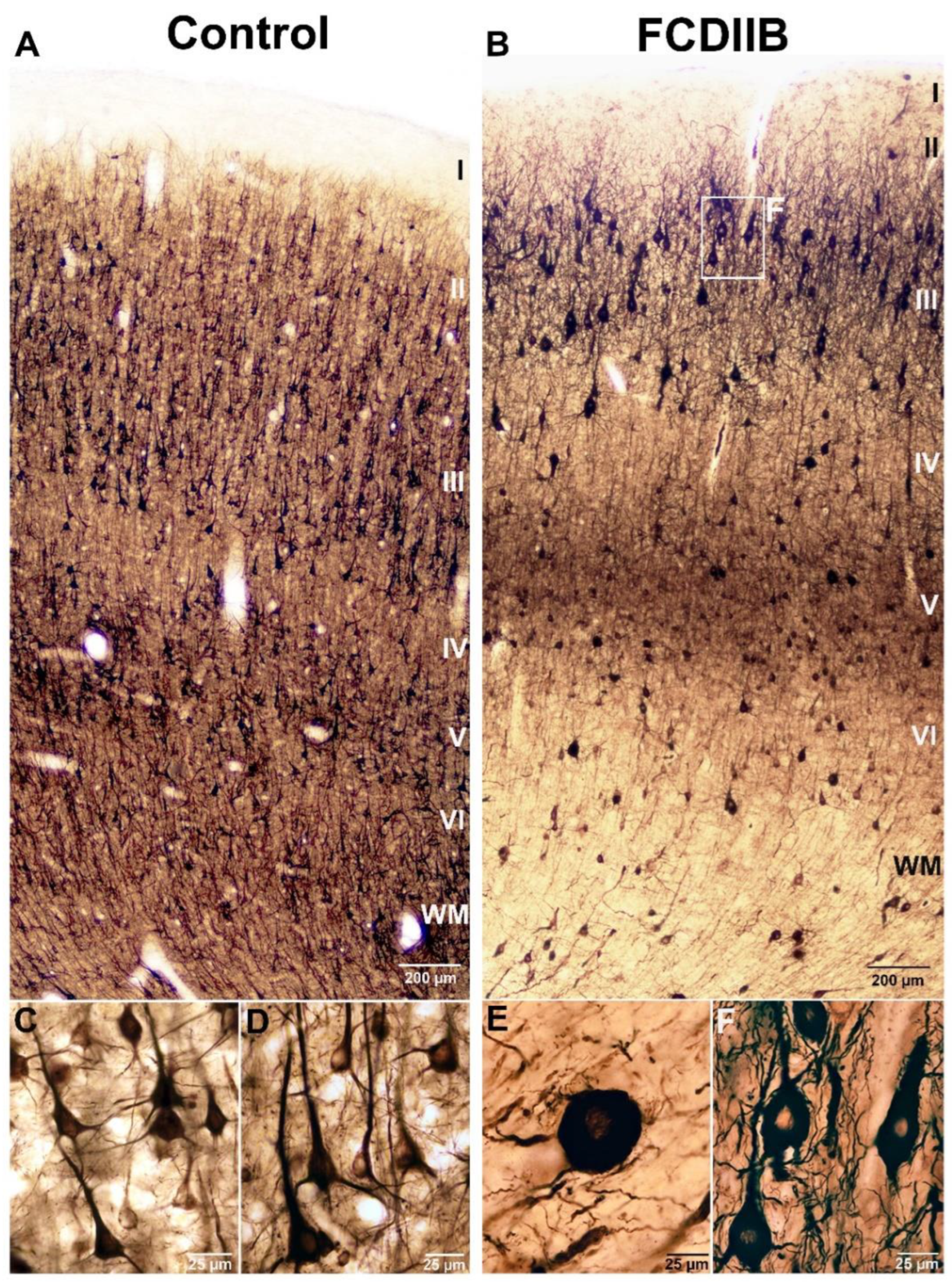
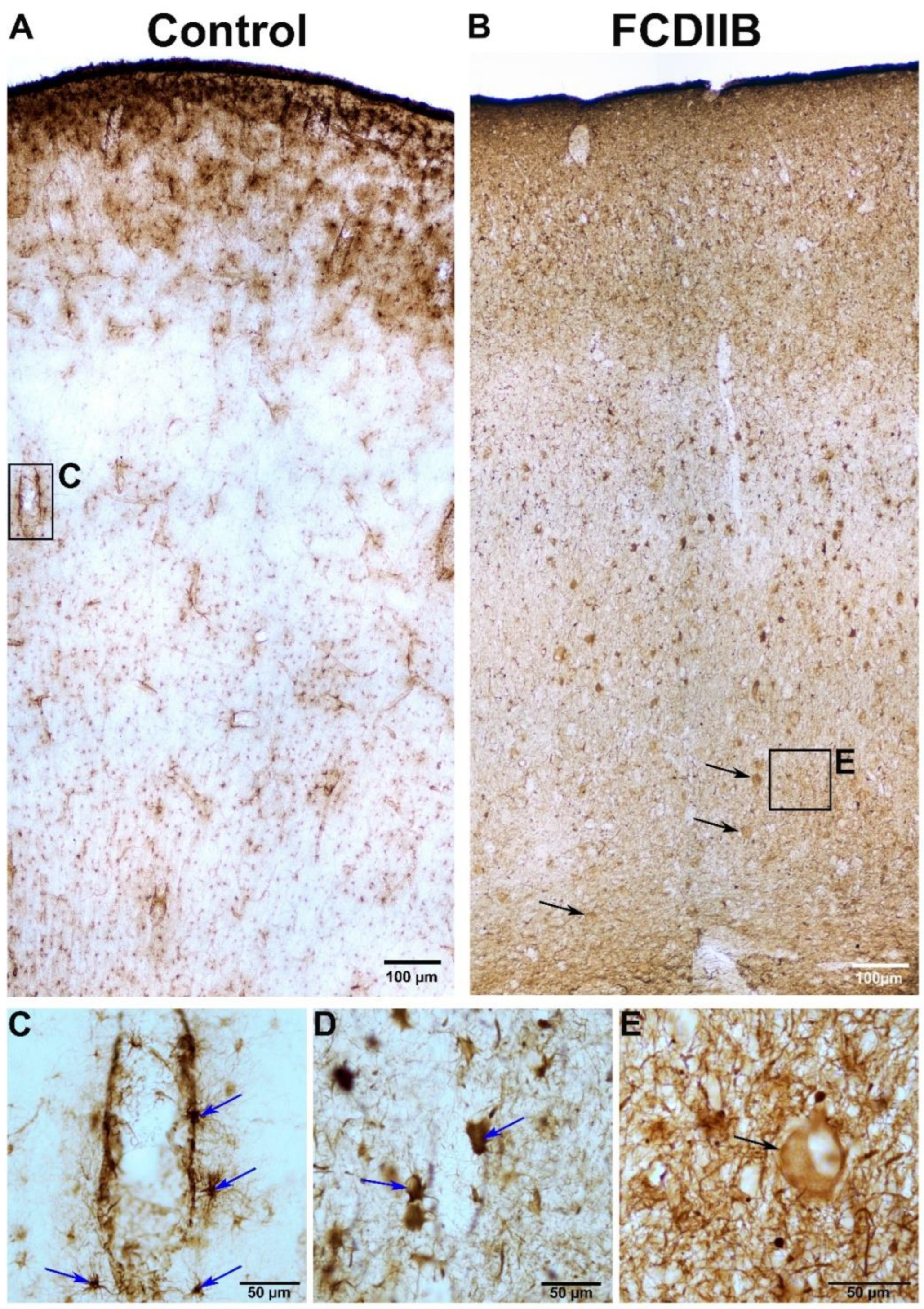
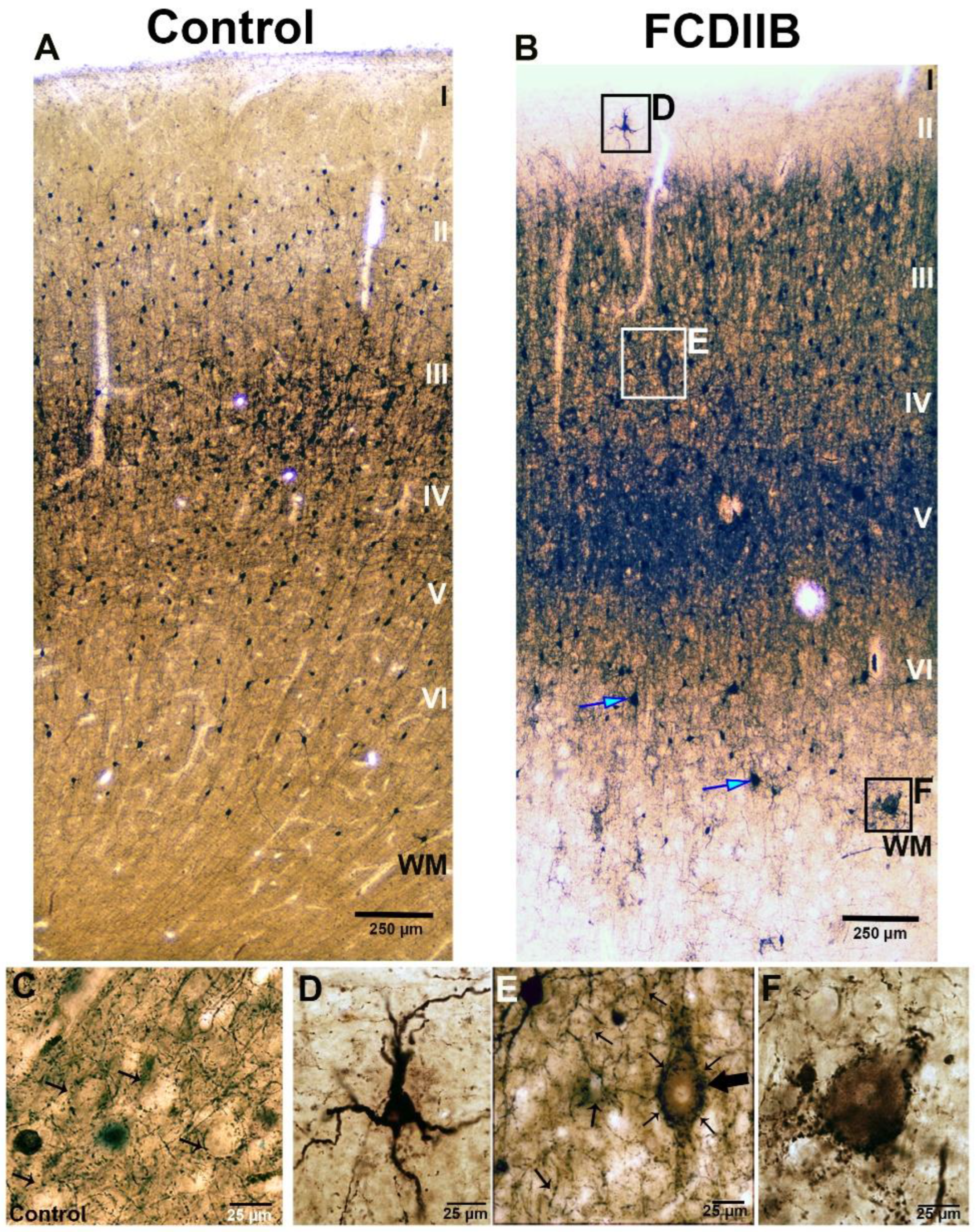
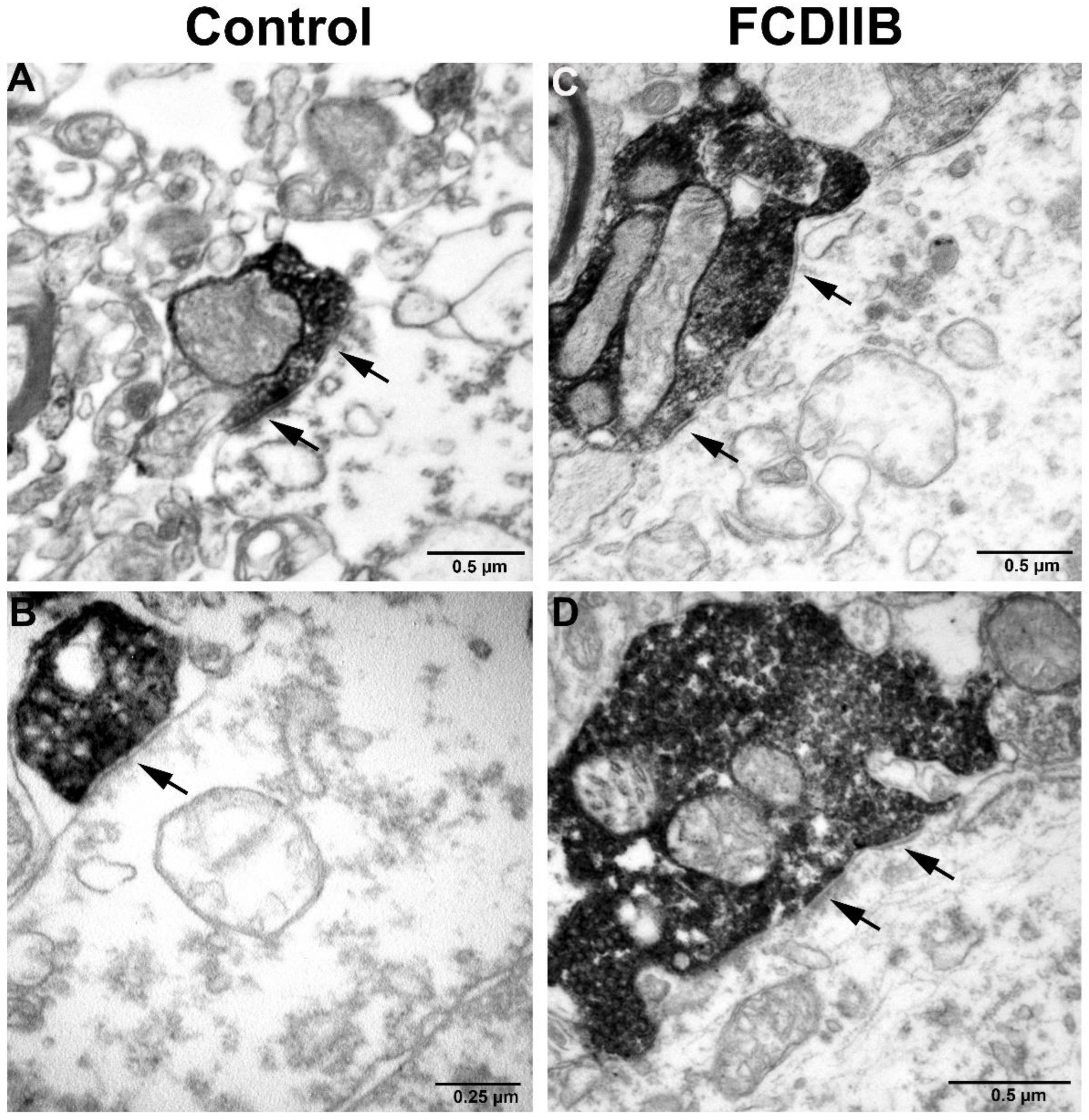
2.2. Cortical Structure and Examined Cell Types in Control and FCD Samples
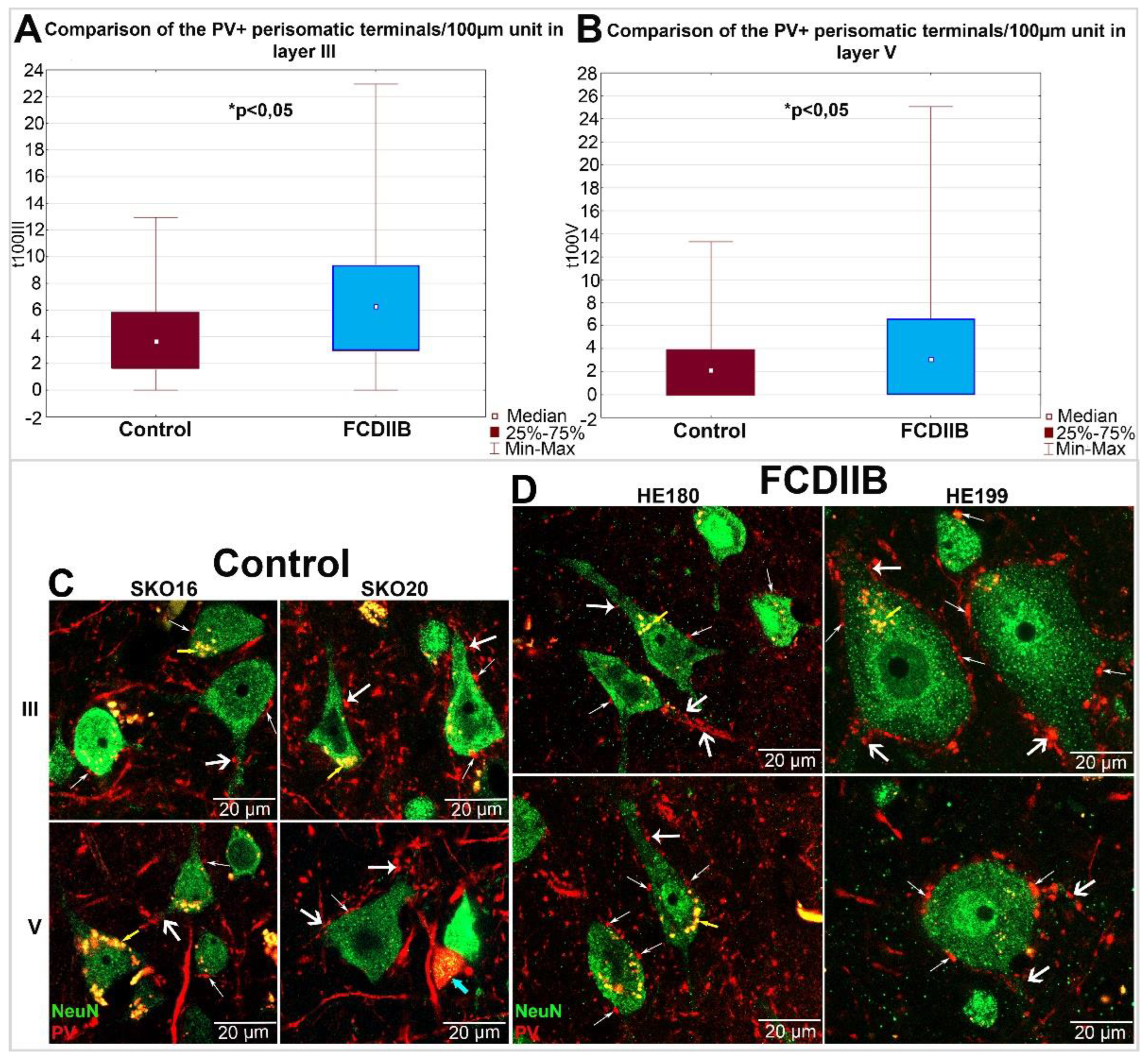
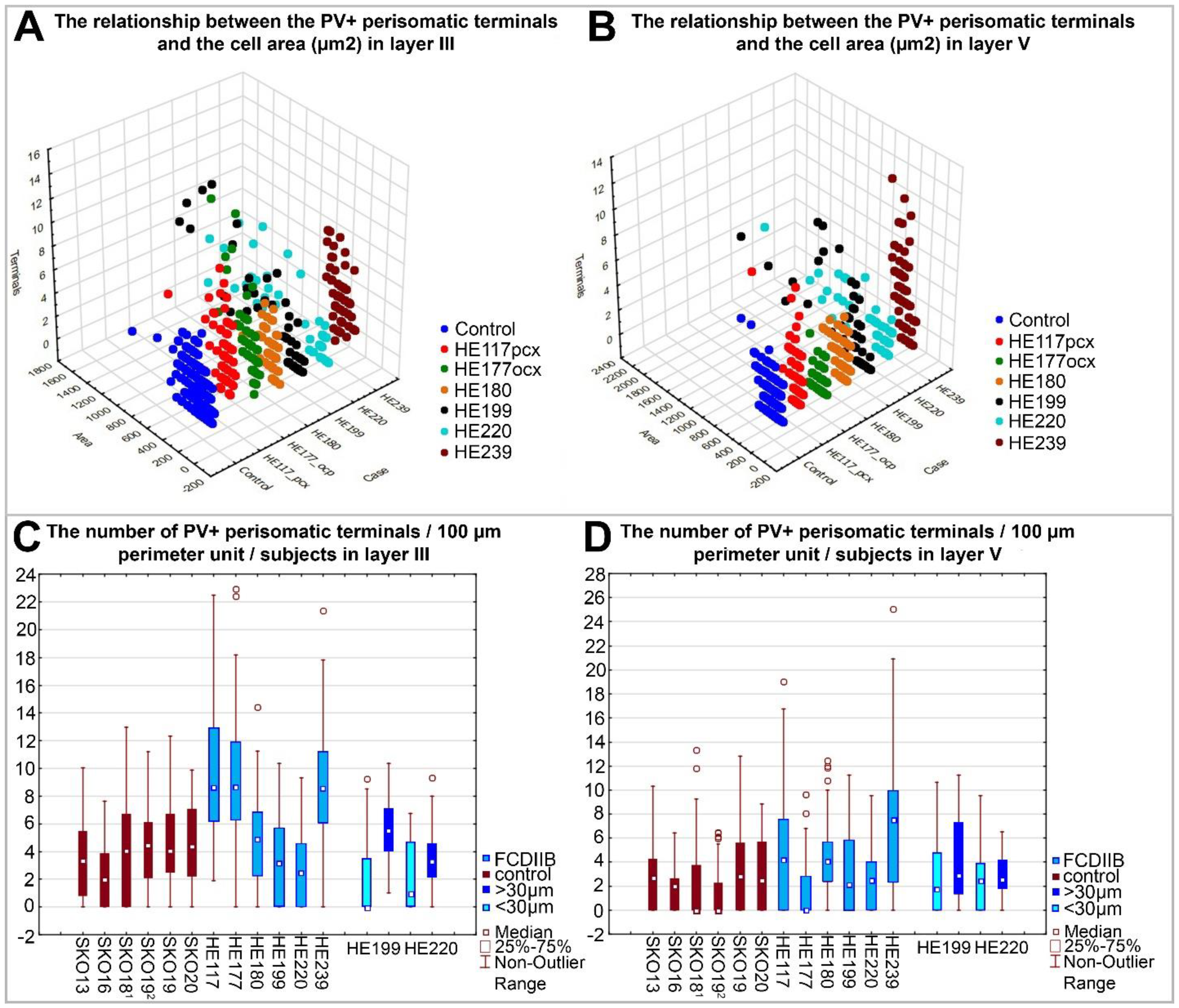
2.3. Quantitative Analyses of PV-Immunopositive Perisomatic Terminals in the Cortices of Control and FCDIIB Cases
3. Discussion
Limitations
4. Materials and Methods
4.1. Obtaining the Human Tissue
4.2. Immunohistochemistry
4.3. Electron Microscopy
4.4. Confocal Microscopy, Quantitative Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdijadid, S.; Mathern, G.W.; Levine, M.S.; Cepeda, C. Basic mechanisms of epileptogenesis in pediatric cortical dysplasia. CNS Neurosci. Ther. 2015, 21, 92–103. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 2012, 135, 1348–1369. [Google Scholar] [CrossRef] [PubMed]
- Siedlecka, M.; Grajkowska, W.; Galus, R.; Dembowska-Baginska, B.; Jozwiak, J. Focal cortical dysplasia: Molecular disturbances and clinicopathological classification (Review). Int. J. Mol. Med. 2016, 38, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. The GABA excitatory/inhibitory developmental sequence: A personal journey. Neuroscience 2014, 279, 187–219. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.R. Gamma, fast, and ultrafast waves of the brain: Their relationships with epilepsy and behavior. Epilepsy Behav. 2008, 13, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Maglóczky, Z. Sprouting in human temporal lobe epilepsy: Excitatory pathways and axons of interneurons. Epilepsy Res. 2010, 89, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Calcagnotto, M.E.; Paredes, M.F.; Tihan, T.; Barbaro, N.M.; Baraban, S.C. Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J. Neurosci. 2005, 25, 9649–9657. [Google Scholar] [CrossRef] [Green Version]
- Tóth, K.; Erőss, L.; Vajda, J.; Halász, P.; Freund, T.F.; Maglóczky, Z. Loss and reorganization of calretinin-containing interneurons in the epileptic human hippocampus. Brain 2010, 133, 2763–2777. [Google Scholar] [CrossRef] [Green Version]
- Maglóczky, Z.; Freund, T.F. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005, 28, 334–340. [Google Scholar] [CrossRef]
- Wittner, L.; Maglóczky, Z. Synaptic Reorganization of the Perisomatic Inhibitory Network in Hippocampi of Temporal Lobe Epileptic Patients. BioMed Res. Int. 2017, 2017, 7154295. [Google Scholar] [CrossRef] [Green Version]
- Houser, C.R. Do structural changes in GABA neurons give rise to the epileptic state? Adv. Exp. Med. Biol. 2014, 813, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colciaghi, F.; Finardi, A.; Nobili, P.; Locatelli, D.; Spigolon, G.; Battaglia, G.S. Progressive brain damage, synaptic reorganization and NMDA activation in a model of epileptogenic cortical dysplasia. PLoS ONE 2014, 9, e89898. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Lupien-Meilleur, A.; Tazerart, S.; Lachance, M.; Samarova, E.; Araya, R.; Lacaille, J.-C.; Rossignol, E. Remodeled Cortical Inhibition PreventsMotor Seizures in Generalized Epilepsy. Ann. Neurol. 2018, 84, 436–451. [Google Scholar] [CrossRef]
- Rossini, L.; De Santis, D.; Mauceri, R.R.; Tesoriero, C.; Bentivoglio, M.; Maderna, E.; Maiorana, A.; Deleo, F.; de Curtis, M.; Tringali, G.; et al. Dendritic pathology, spine loss and synaptic reorganization in human cortex from epilepsy patients. Brain 2021, 144, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Gram, L. Monotherapy versus polytherapy in epilepsy—A reappraisal. CNS Drugs 1995, 3, 194–208. [Google Scholar] [CrossRef]
- Sander, J.W.A.S. Some Aspects of Prognosis in the Epilepsies: A Review. Epilepsia 1993, 34, 1007–1016. [Google Scholar] [CrossRef]
- Sheng, J.; Liu, S.; Qin, H.; Li, B.; Zhang, X. Drug-Resistant Epilepsy and Surgery. Curr. Neuropharmacol. 2018, 16, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Spreafico, R.; Haaker, G.; Coras, R.; Kobow, K.; Bien, C.G.; Pfafflin, M.; Elger, C.; Widman, G.; Schramm, J.; et al. Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N. Engl. J. Med. 2017, 377, 1648–1656. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Jin, L.; Dou, W.; Lu, Q.; Zhou, L.; Ren, H.; Zhao, Y.; Feng, F.; Guo, Y.; Gao, J. Type IIB focal cortical dysplasia with balloon cells in medial temporal lobe epilepsy: Clinical, neuroimaging, and histopathological findings. Epilepsy Res. 2019, 157, 106189. [Google Scholar] [CrossRef]
- Baud, M.O.; Perneger, T.; Racz, A.; Pensel, M.C.; Elger, C.; Rydenhag, B.; Malmgren, K.; Cross, J.H.; McKenna, G.; Tisdall, M.; et al. European trends in epilepsy surgery. Neurology 2018, 91, e96–e106. [Google Scholar] [CrossRef]
- Taylor, D.C.; Falconer, M.A.; Bruton, C.J.; Corsellis, J.A. Focal dysplasia of the cerebral cortex in epilepsy. J. Neurol. Neurosurg. Psychiatry 1971, 34, 369–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmini, A.; Najm, I.; Avanzini, G.; Babb, T.; Guerrini, R.; Foldvary-Schaefer, N.; Jackson, G.; Lüders, H.O.; Prayson, R.; Spreafico, R.; et al. Terminology and classification of the cortical dysplasias. Neurology 2004, 62, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Prayson, R.A. Classification and pathological characteristics of the cortical dysplasias. Childs Nerv. Syst. 2014, 30, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Iffland, P.H.; Crino, P.B. Focal Cortical Dysplasia: Gene Mutations, Cell Signaling, and Therapeutic Implications. Annu. Rev. Pathol. 2017, 12, 547–571. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, J.M.; Donkels, C.; Fauser, S.; Schulze-Bonhage, A.; Prinz, M.; Zentner, J.; Haas, C.A. Characterization of focal cortical dysplasia with balloon cells by layer-specific markers: Evidence for differential vulnerability of interneurons. Epilepsia 2017, 58, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Baulac, S. mTOR signaling pathway genes in focal epilepsies. Prog. Brain Res. 2016, 226, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.X.; Lin, K.; Kang, D.Z.; Liu, X.X.; Wang, X.F.; Zheng, S.F.; Yu, L.H.; Lin, Z.Y. Similar PDK1-AKT-mTOR pathway activation in balloon cells and dysmorphic neurons of type II focal cortical dysplasia with refractory epilepsy. Epilepsy Res. 2015, 112, 137–149. [Google Scholar] [CrossRef]
- Rossini, L.; Villani, F.; Granata, T.; Tassi, L.; Tringali, G.; Cardinale, F.; Aronica, E.; Spreafico, R.; Garbelli, R. FCD Type II and mTOR pathway: Evidence for different mechanisms involved in the pathogenesis of dysmorphic neurons. Epilepsy Res. 2017, 129, 146–156. [Google Scholar] [CrossRef]
- Majolo, F.; Marinowic, D.R.; Machado, D.C.; Da Costa, J.C. MTOR pathway in focal cortical dysplasia type 2: What do we know? Epilepsy Behav. 2018, 85, 157–163. [Google Scholar] [CrossRef]
- André, V.M.; Wu, N.; Yamazaki, I.; Nguyen, S.T.; Fisher, R.S.; Vinters, H.V.; Mathern, G.W.; Levine, M.S.; Cepeda, C. Cytomegalic interneurons: A new abnormal cell type in severe pediatric cortical dysplasia. J. Neuropathol. Exp. Neurol. 2007, 66, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Medici, V.; Rossini, L.; Deleo, F.; Tringali, G.; Tassi, L.; Cardinale, F.; Bramerio, M.; de Curtis, M.; Garbelli, R.; Spreafico, R. Different parvalbumin and GABA expression in human epileptogenic focal cortical dysplasia. Epilepsia 2016, 57, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Sousa, G.K.; Capitelli, C.S.; Dombroski, T.C.D.; Zanella, C.A.B.; Terra, V.C.; Velasco, T.R.; Machado, H.R.; Assirati, J.A.; Carlotti, C.G.; Alves, V.M.; et al. Identification and immunophenotype of abnormal cells present in focal cortical dysplasia type IIb. Surg. Exp. Pathol. 2018, 1, 9. [Google Scholar] [CrossRef]
- Spreafico, R.; Pasquier, B.; Minotti, L.; Garbelli, R.; Kahane, P.; Grand, S.; Benabid, A.L.; Tassi, L.; Avanzini, G.; Battaglia, G.; et al. Immunocytochemical investigation on dysplastic human tissue from epileptic patients. Epilepsy Res. 1998, 32, 34–48. [Google Scholar] [CrossRef]
- Cepeda, C.; Hurst, R.S.; Flores-Hernández, J.; Hernández-Echeagaray, E.; Klapstein, G.J.; Boylan, M.K.; Calvert, C.R.; Jocoy, E.L.; Nguyen, O.K.; André, V.M.; et al. Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J. Neurosci. Res. 2003, 72, 472–486. [Google Scholar] [CrossRef]
- Cepeda, C.A.V.; Vinters, H.V.; Levine, M.S.; Mathern, G.W. Are cytomegalic neurons and balloon cells generators of epileptic activity in pediatric cortical dysplasia? Epilepsia 2005, 46 (Suppl. 5), 82–88. [Google Scholar] [CrossRef]
- Kuchukhidze, G.; Wieselthaler-Holzl, A.; Drexel, M.; Unterberger, I.; Luef, G.; Ortler, M.; Becker, A.J.; Trinka, E.; Sperk, G. Calcium-binding proteins in focal cortical dysplasia. Epilepsia 2015, 56, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, T.; Sukigara, S.; Saito, T.; Otsuki, T.; Takahashi, A.; Kaneko, Y.; Kaido, T.; Saito, Y.; Sato, N.; Kimura, Y.; et al. Delayed Maturation and Differentiation of Neurons in Focal Cortical Dysplasia With the Transmantle Sign: Analysis of Layer-Specific Marker Expression. J. Neuropathol. Exp. Neurol. 2012, 71, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Fauser, S.; Häussler, U.; Donkels, C.; Huber, S.; Nakagawa, J.; Prinz, M.; Schulze-Bonhage, A.; Zentner, J.; Haas, C.A. Disorganization of neocortical lamination in focal cortical dysplasia is brain-region dependent: Evidence from layer-specific marker expression. Acta Neuropathol. Commun. 2013, 1, 47. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Pineda, M.; Tallada, M.; Oliver, B.; Russi, A.; Oller, L.; Noboa, R.; Zújar, M.J.; Alcántara, S. Abnormal local-circuit neurons in epilepsia partialis continua associated with focal cortical dysplasia. Acta Neuropathol. 1992, 83, 647–652. [Google Scholar] [CrossRef]
- Spreafico, R.; Battaglia, G.; Arcelli, P.; Andermann, F.; Dubeau, F.; Palmini, A.; Olivier, A.; Villemure, J.G.; Tampieri, D.; Avanzini, G.; et al. Cortical dysplasia: An immunocytochemical study of three patients. Neurology 1998, 50, 27–36. [Google Scholar] [CrossRef]
- Garbelli, R.; Munari, C.; De Biasi, S.; Vitellaro-Zuccarello, L.; Galli, C.; Bramerio, M.; Mai, R.; Battaglia, G.; Spreafico, R. Taylor’s cortical dysplasia: A confocal and ultrastructural immunohistochemical study. Brain Pathol. 1999, 9, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Ozbas-Gerçeker, F.; Redeker, S.; Ramkema, M.; Spliet, W.G.; van Rijen, P.C.; Leenstra, S.; Gorter, J.A.; Troost, D. Expression and cellular distribution of high- and low-affinity neurotrophin receptors in malformations of cortical development. Acta Neuropathol. 2004, 108, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Seress, L.; Abrahám, H.; Dóczi, T.; Pokorny, J.; Klemm, J.; Bakay, R.A. Axosomatic synapses on granule cells are preserved in human non-infiltrating tumour or lesion-related and mesial temporal sclerotic epilepsy, but markedly reduced in tumour-infiltrated dentate gyrus with or without epilepsy. Prague Med. Rep. 2004, 105, 357–368. [Google Scholar]
- Tóth, K.; Wittner, L.; Urbán, Z.; Doyle, W.K.; Buzsáki, G.; Shigemoto, R.; Freund, T.F.; Maglóczky, Z. Morphology and synaptic input of substance P receptor-immunoreactive interneurons in control and epileptic human hippocampus. Neuroscience 2007, 144, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, K.; Maglóczky, Z. The vulnerability of calretinin-containing hippocampal interneurons to temporal lobe epilepsy. Front. Neuroanat. 2014, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, B.P.; Chaunsali, L.; Campbell, S.L.; Patel, D.C.; Goode, A.E.; Sontheimer, H. Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat. Commun. 2018, 9, 4724. [Google Scholar] [CrossRef] [Green Version]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef]
- Yuen, T.; Morokoff, A.; Bjorksten, A.; D’Abaco, G.; Paradiso, L.; Finch, S.; Wong, D.; Reid, C.; Powell, K.; Drummond, K.; et al. Glutamate is associated with a higher risk of seizures in patients with gliomas. Neurology 2012, 79, 883–889. [Google Scholar] [CrossRef]
- Liubinas, S.V.; O’Brien, T.J.; Moffat, B.M.; Drummond, K.J.; Morokoff, A.P.; Kaye, A.H. Tumour associated epilepsy and glutamate excitotoxicity in patients with gliomas. J. Clin. Neurosci. 2014, 21, 899–908. [Google Scholar] [CrossRef]
- Wittner, L.; Erőss, L.; Czirják, S.; Halász, P.; Freund, T.F.; Maglóczky, Z. Surviving CA1 pyramidal cells receive intact perisomatic inhibitory input in the human epileptic hippocampus. Brain 2005, 128, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Wittner, L.; Maglóczky, Z.; Borhegyi, Z.; Halász, P.; Tóth, S.; Erőss, L.; Szabó, Z.; Freund, T.F. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 2001, 108, 587–600. [Google Scholar] [CrossRef]
- Gonzalez-Riano, C.; Tapia-González, S.; García, A.; Muñoz, A.; DeFelipe, J.; Barbas, C. Metabolomics and neuroanatomical evaluation of post-mortem changes in the hippocampus. Brain Struct. Funct. 2017, 222, 2831–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittner, L.; Erőss, L.; Szabó, Z.; Tóth, S.; Czirják, S.; Halász, P.; Freund, T.F.; Maglóczky, Z. Synaptic reorganization of calbindin-positive neurons in the human hippocampal CA1 region in temporal lobe epilepsy. Neuroscience 2002, 115, 961–978. [Google Scholar] [CrossRef]
- Ludányi, A.; Eross, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condé, F.; Lund, J.S.; Jacobowitz, D.M.; Baimbridge, K.G.; Lewis, D.A. Local circuit neurons immunoreactive for calretinin, calbindin D-28k or parvalbumin in monkey prefrontal cortex: Distribution and morphology. J. Comp. Neurol. 1994, 341, 95–116. [Google Scholar] [CrossRef]
- Hof, P.R.; Cox, K.; Young, W.G.; Celio, M.R.; Rogers, J.; Morrison, J.H. Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1991, 50, 451–462. [Google Scholar] [CrossRef]
- Maglóczky, Z.; Halász, P.; Vajda, J.; Czirják, S.; Freund, T.F. Loss of Calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience 1997, 76, 377–385. [Google Scholar] [CrossRef]
- Ferrer, I.; Soriano, E.; Tuñón, T.; Fonseca, M.; Guionnet, N. Parvalbumin immunoreactive neurons in normal human temporal neocortex and in patients with Alzheimer’s disease. J. Neurol. Sci. 1991, 106, 135–141. [Google Scholar] [CrossRef]
- Kim, S.H.; Choi, J. Pathological Classification of Focal Cortical Dysplasia (FCD): Personal Comments for Well Understanding FCD Classification. J. Korean Neurosurg. Soc. 2019, 62, 288–295. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, T.F.; Katona, I. Perisomatic inhibition. Neuron 2007, 56, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, T.F. Interneuron Diversity series: Rhythm and mood in perisomatic inhibition. Trends Neurosci. 2003, 26, 489–495. [Google Scholar] [CrossRef]
- Buzsáki, G. Rhythms of the Brain; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Volman, V.; Behrens, M.M.; Sejnowski, T.J. Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. J. Neurosci. 2011, 31, 18137–18148. [Google Scholar] [CrossRef]
- Neubrandt, M.; Oláh, V.J.; Brunner, J.; Marosi, E.L.; Soltesz, I.; Szabadics, J. Single Bursts of Individual Granule Cells Functionally Rearrange Feedforward Inhibition. J. Neurosci. 2018, 38, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Prince, D.A.; Jacobs, K. Inhibitory function in two models of chronic epileptogenesis. Epilepsy Res. 1998, 32, 83–92. [Google Scholar] [CrossRef]
- Motalli, R.; Louvel, J.; Tancredi, V.; Kurcewicz, I.; Wan-Chow-Wah, D.; Pumain, R.; Avoli, M. GABA(B) receptor activation promotes seizure activity in the juvenile rat hippocampus. J. Neurophysiol. 1999, 82, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Yekhlef, L.; Breschi, G.L.; Lagostena, L.; Russo, G.; Taverna, S. Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J. Neurophysiol. 2015, 113, 1616–1630. [Google Scholar] [CrossRef] [Green Version]
- Colciaghi, F.; Finardi, A.; Frasca, A.; Balosso, S.; Nobili, P.; Carriero, G.; Locatelli, D.; Vezzani, A.; Battaglia, G. Status epilepticus-induced pathologic plasticity in a rat model of focal cortical dysplasia. Brain 2011, 134, 2828–2843. [Google Scholar] [CrossRef] [Green Version]
- Finardi, A.; Colciaghi, F.; Castana, L.; Locatelli, D.; Marras, C.E.; Nobili, P.; Fratelli, M.; Bramerio, M.A.; Lorusso, G.; Battaglia, G.S. Long-duration epilepsy affects cell morphology and glutamatergic synapses in type IIB focal cortical dysplasia. Acta Neuropathol. 2013, 126, 219–235. [Google Scholar] [CrossRef]
- Baho, E.; Chattopadhyaya, B.; Lavertu-Jolin, M.; Mazziotti, R.; Awad, P.N.; Chehrazi, P.N.; Groleau, M.; Jahannault-Talignani, C.; Sanon, N.T.; Vaucher, E.T.; et al. p75 Neurotrophin Receptor Activation Regulates the Timing of the Maturation of Cortical Parvalbumin Interneuron Connectivity and Promotes Juvenile-like Plasticity in Adult Visual Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 4489–4510. [Google Scholar] [CrossRef] [Green Version]
- Paz, J.T.; Huguenard, J.R. Microcircuits and their interactions in epilepsy: Is the focus out of focus? Nat. Neurosci. 2015, 18, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, V.; Nicoll, R.A. GABA generates excitement. Neuron 2003, 37, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Miles, R.; Tóth, K.; Gulyás, A.I.; Hájos, N.; Freund, T.F. Differences between somatic and dendritic inhibition in the hippocampus. Neuron 1996, 16, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Brodmann, K. Vergleichende Lokalisationslehre der Grosshirnrinde in Ihren Prinzipien Dargestellt auf Grund des Zellenbaues; Leipzig: Barth, Germany, 1909. [Google Scholar]
- Schnell, S.A.; Staines, W.A.; Wessendorf, M.W. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J. Histochem. Cytochem. 1999, 47, 719–730. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Case | Age (Years) | Gender | Post-Mortem Interval (Hours and Minutes) | Sampling Area | |
|---|---|---|---|---|---|
| SKO13 | 60 | female | 3:25 | R BA46 | |
| SKO16 | 72 | male | 2:22 | L BA46 | |
| SKO18 | 85 | male | 2:52 | R BA7 | |
| SKO19 | 61 | female | 2:53 | R BA18, L BA46 | |
| SKO20 | 27 | male | 3:45 | L BA46 | |
| Mean of control subjects | 61 | - | 3:05 | - | |
| FCD Case | Age (Years)-At the Time of Surgery | Duration of Epilepsy (Years) | Gender | Pathological Classification | Removed Brain Area |
| HE117 | 26 | 16 | male | FCDIIB | R pcx/BA7 |
| HE177 | 34.5 | 33 | female | FCDIIB | R ocx/BA18 |
| HE180 | 17 | 15 | female | FCDIIB | R fcx/BA46 |
| HE199 | 35 | 33.5 | female | FCDIIB | R fcx/BA46 |
| HE220 | 48 | 39 | male | FCDIIB | L fcx/BA46 |
| HE239 | 42 | 39.5 | male | FCDIIB | L fcx/BA46 |
| Mean of epileptic subjects | 33.75 | 29.33 | - | - | - |
| Control Case | Control (Number of Perisomatic Terminals/Number of Cells) | FCD Case | FCD (Number of Perisomatic Terminals/Number of Cells) | |
|---|---|---|---|---|
| Layer III | SKO13R BA46 | 104/60 | HE117 pcx/BA7 | 255/58 |
| SKO16L BA46 | 66/58 | HE177 ocx/BA18 | 265/61 | |
| SKO18R BA7 | 105/51 | HE180 fcx/BA46 | 121/50 | |
| SKO19R BA18 | 90/54 | HE199 fcx/BA46 | 132/48 | |
| SKO19L BA46 | 118/60 | HE220 fcx/BA46 | 98/49 | |
| SKO20L BA46 | 108/53 | HE239 fcx/BA46 | 251/61 | |
| Layer III mean number of terminals/cells/subject | - | 98.5/56 | - | 187/54.5 |
| Layer III mean number of terminals/1 cell | - | 1.76 | 3.43 | |
| Layer III total number of terminals/cells/all subjects | - | 591/336 | - | 1122/327 |
| Layer V | SKO13R BA46 | 81/63 | HE117 pcx/BA7 | 138/61 |
| SKO16L BA46 | 36/52 | HE177 ocx/BA18 | 39/52 | |
| SKO18R BA7 | 41/49 | HE180 fcx/BA46 | 113/61 | |
| SKO19R BA18 | 61/57 | HE199 fcx/BA46 | 110/48 | |
| SKO19L BA46 | 36/60 | HE220 fcx/BA46 | 77/48 | |
| SKO20L BA46 | 65/51 | HE239 fcx/BA46 | 225/61 | |
| Layer V mean number of terminals/cells/subject | - | 53.33/55.33 | - | 117/55.17 |
| Layer V mean number of terminals/1 cell | - | 0.96 | - | 2.12 |
| Layer V total number of terminals/cells/all subjects | - | 320/332 | - | 702/331 |
| Total number of terminals and cells in layers III and V | - | 911/668 | - | 1824/658 |
| Samples | The Mean Number of PV-Immunopositive Terminals/100 µm of Soma Perimeter | |
|---|---|---|
| Layer III | Layer V | |
| HE199 and HE220 fcx/BA46 normal-sized cells 1 | 2.01 ± 2.50 | 2.67 ± 3.03 |
| HE199 and HE220 fcx/BA46 giant cells 2 | 4.68 ± 2.68 | 3.53 ± 2.96 |
| SKO13R, SKO16L, SKO19L, SKO20L fcx/BA46 3 | 3.63 ± 2.94 | 2.19 ± 2.48 |
| Samples | Comparisons of the Number of PV-Immunopositive Terminals/100 µm of the Perimeter with Mann-Whitney U Test 1 | |
|---|---|---|
| Layer III | Layer V | |
| HE117/SKO18 pcx/BA7 | U = 595.000, N1 = 58, N2 = 51, p = 2.22 × 10−8 | U = 922.000, N1 = 61, N2 = 49, p = 4.92 × 10−4 |
| HE177/SKO19 ocx/BA18 | U = 550.500, N1 = 61, N2 = 54, p = 8.23 × 10−11 | U = 1097.000, N1 = 52, N2 = 57, p = 1.92 × 10−2 |
| HE239/SKO13 fcx/BA46 | U = 577.500, N1 = 61, N2 = 60, p = 5.39 × 10−12 | U = 903.500, N1 = 61, N2 = 63, p = 1.53 × 10−7 |
| HE180/SKO20 fcx/BA46 | U = 1268,0000, N1 = 50, N2 = 53, p = 7.10 × 10−1 | U = 1257,000, N1 = 61, N2 = 51, p = 8.17 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szekeres-Paraczky, C.; Szocsics, P.; Erőss, L.; Fabó, D.; Mód, L.; Maglóczky, Z. Reorganization of Parvalbumin Immunopositive Perisomatic Innervation of Principal Cells in Focal Cortical Dysplasia Type IIB in Human Epileptic Patients. Int. J. Mol. Sci. 2022, 23, 4746. https://doi.org/10.3390/ijms23094746
Szekeres-Paraczky C, Szocsics P, Erőss L, Fabó D, Mód L, Maglóczky Z. Reorganization of Parvalbumin Immunopositive Perisomatic Innervation of Principal Cells in Focal Cortical Dysplasia Type IIB in Human Epileptic Patients. International Journal of Molecular Sciences. 2022; 23(9):4746. https://doi.org/10.3390/ijms23094746
Chicago/Turabian StyleSzekeres-Paraczky, Cecília, Péter Szocsics, Loránd Erőss, Dániel Fabó, László Mód, and Zsófia Maglóczky. 2022. "Reorganization of Parvalbumin Immunopositive Perisomatic Innervation of Principal Cells in Focal Cortical Dysplasia Type IIB in Human Epileptic Patients" International Journal of Molecular Sciences 23, no. 9: 4746. https://doi.org/10.3390/ijms23094746