
Novel Hybrid 1,2,4- and 1,2,3-Triazoles Targeting Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase (InhA): Design, Synthesis, and Molecular Docking

 ,
,  , , , ,
, , , ,
Abstract
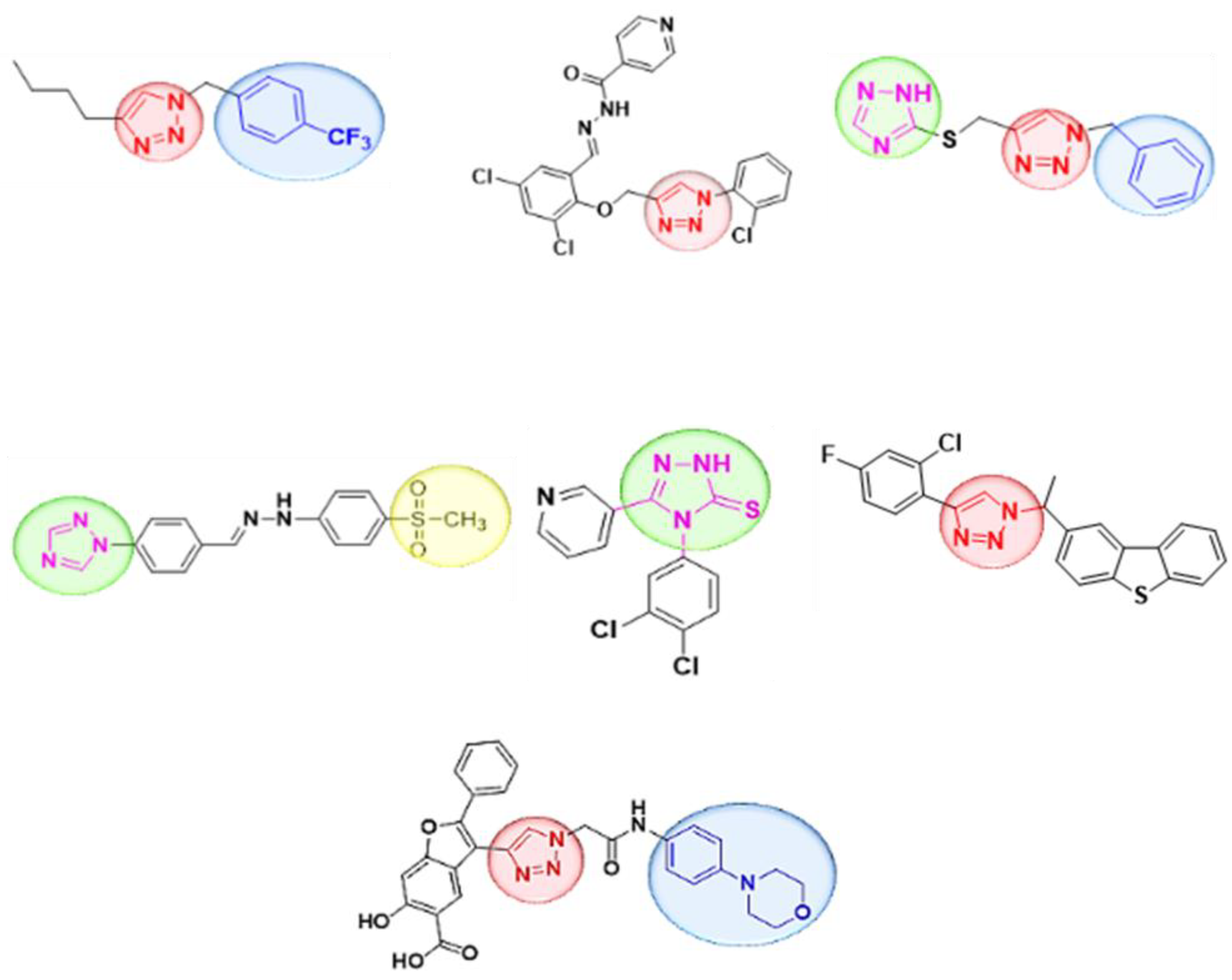
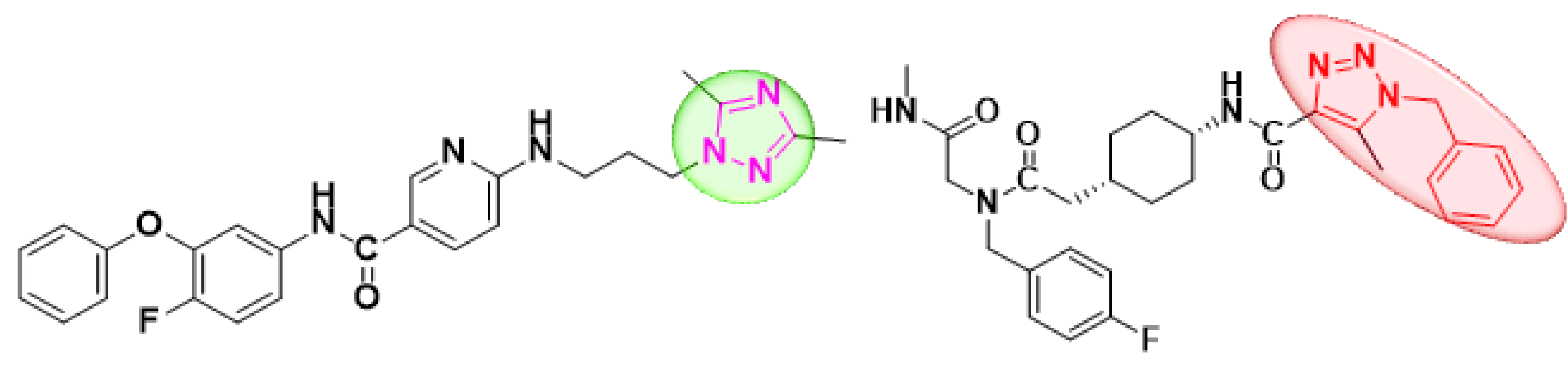
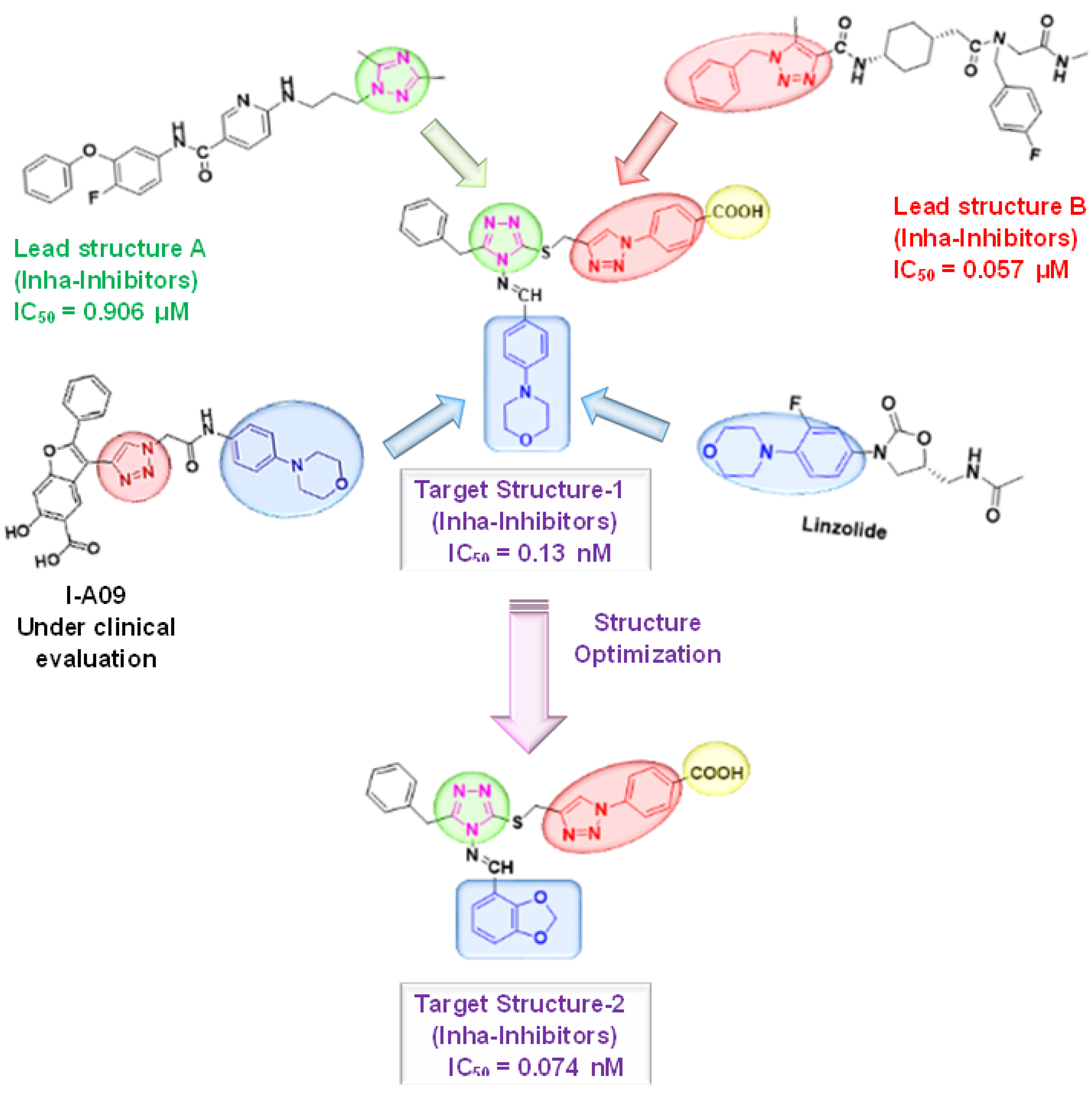
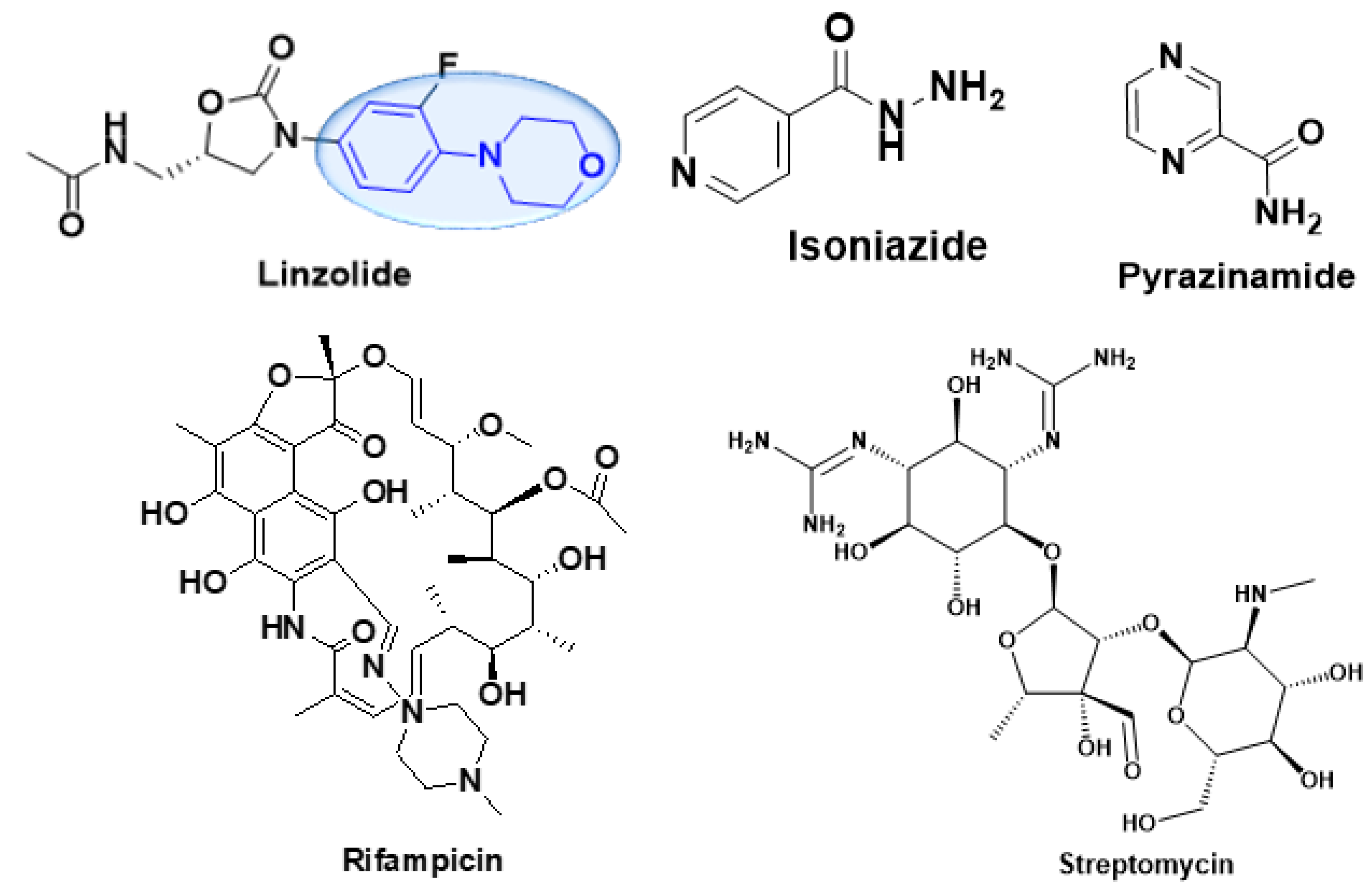
:1. Introduction
2. Results and Discussion
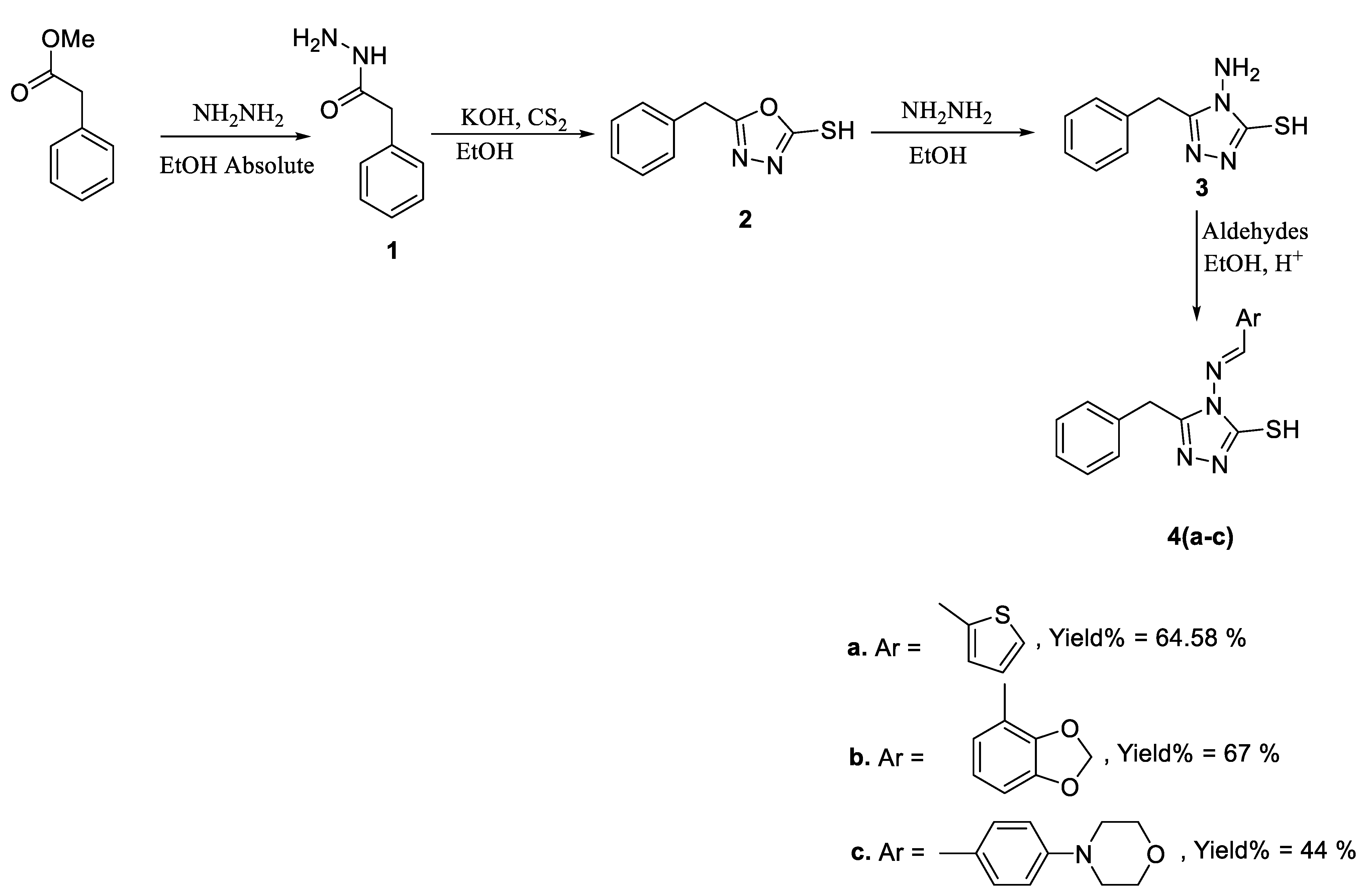
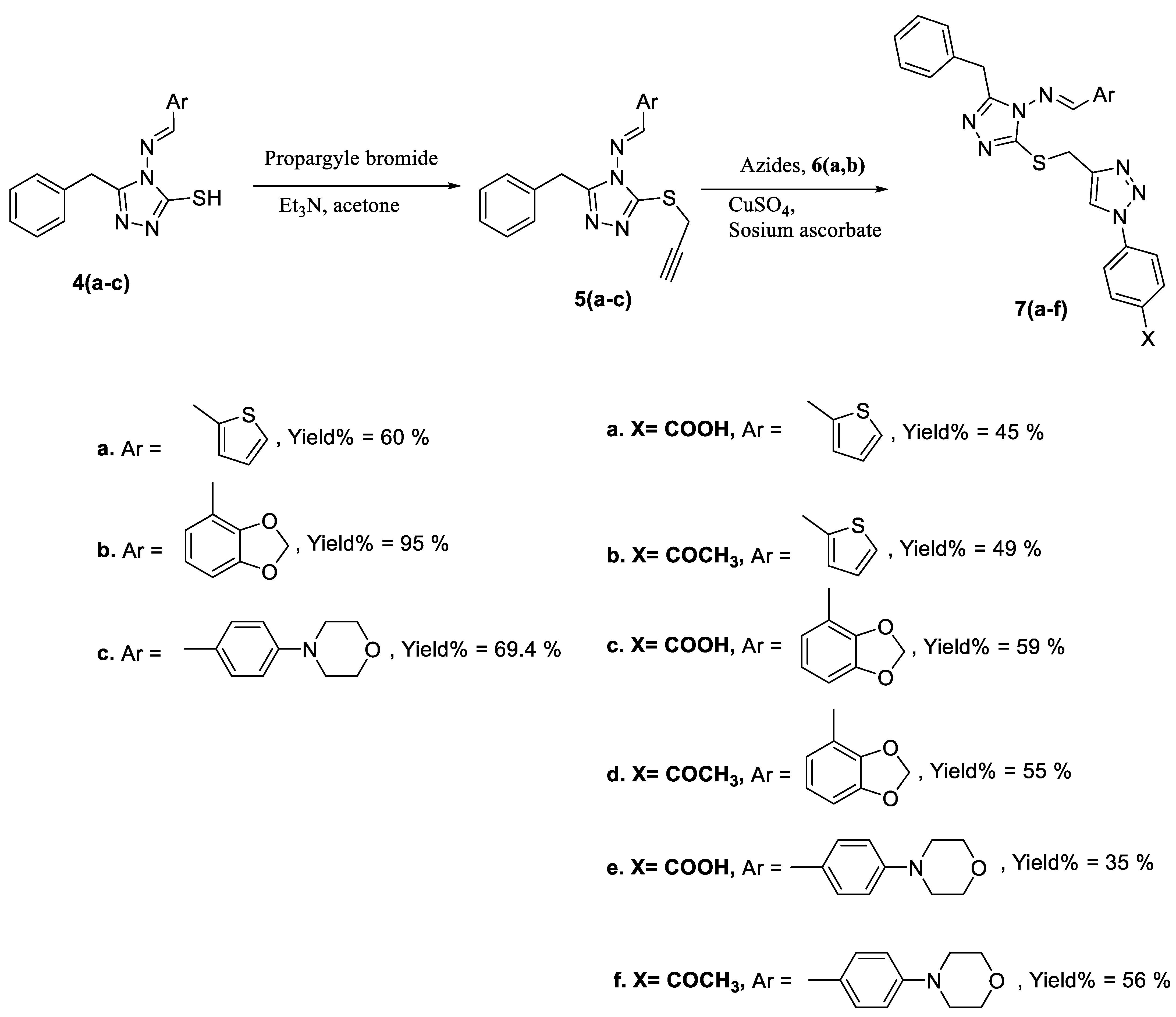
2.1. Chemistry
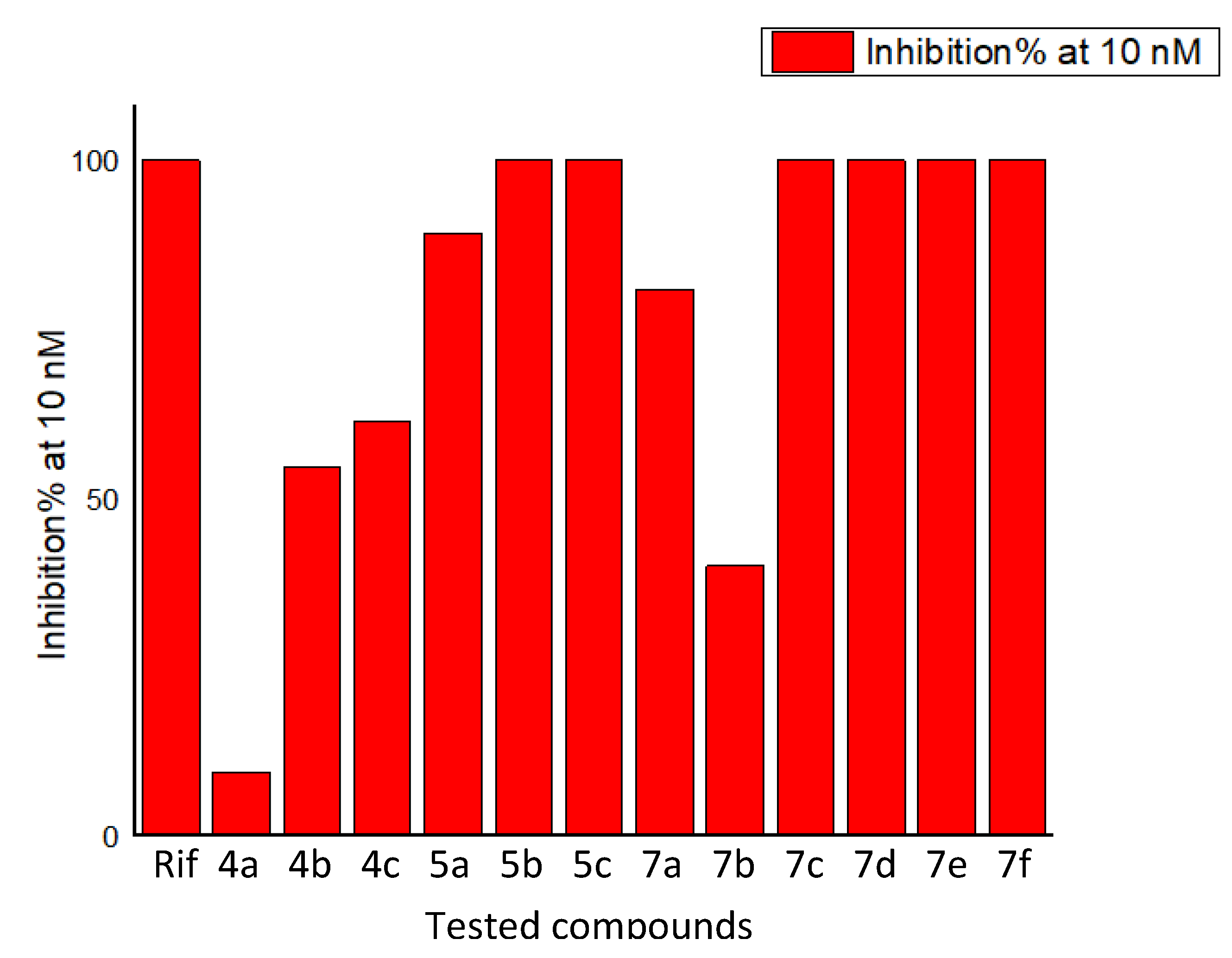
2.2. Biological Evaluation
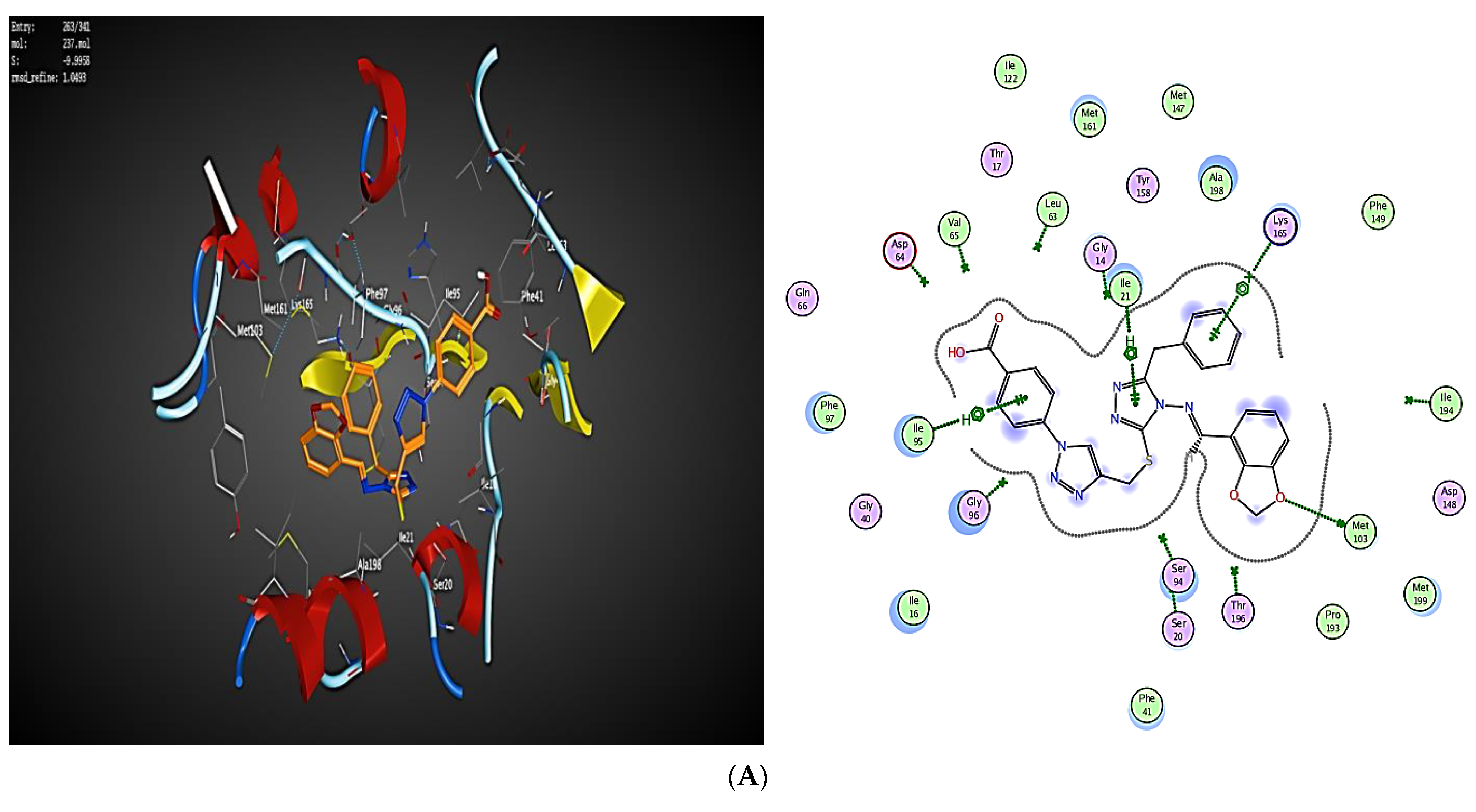
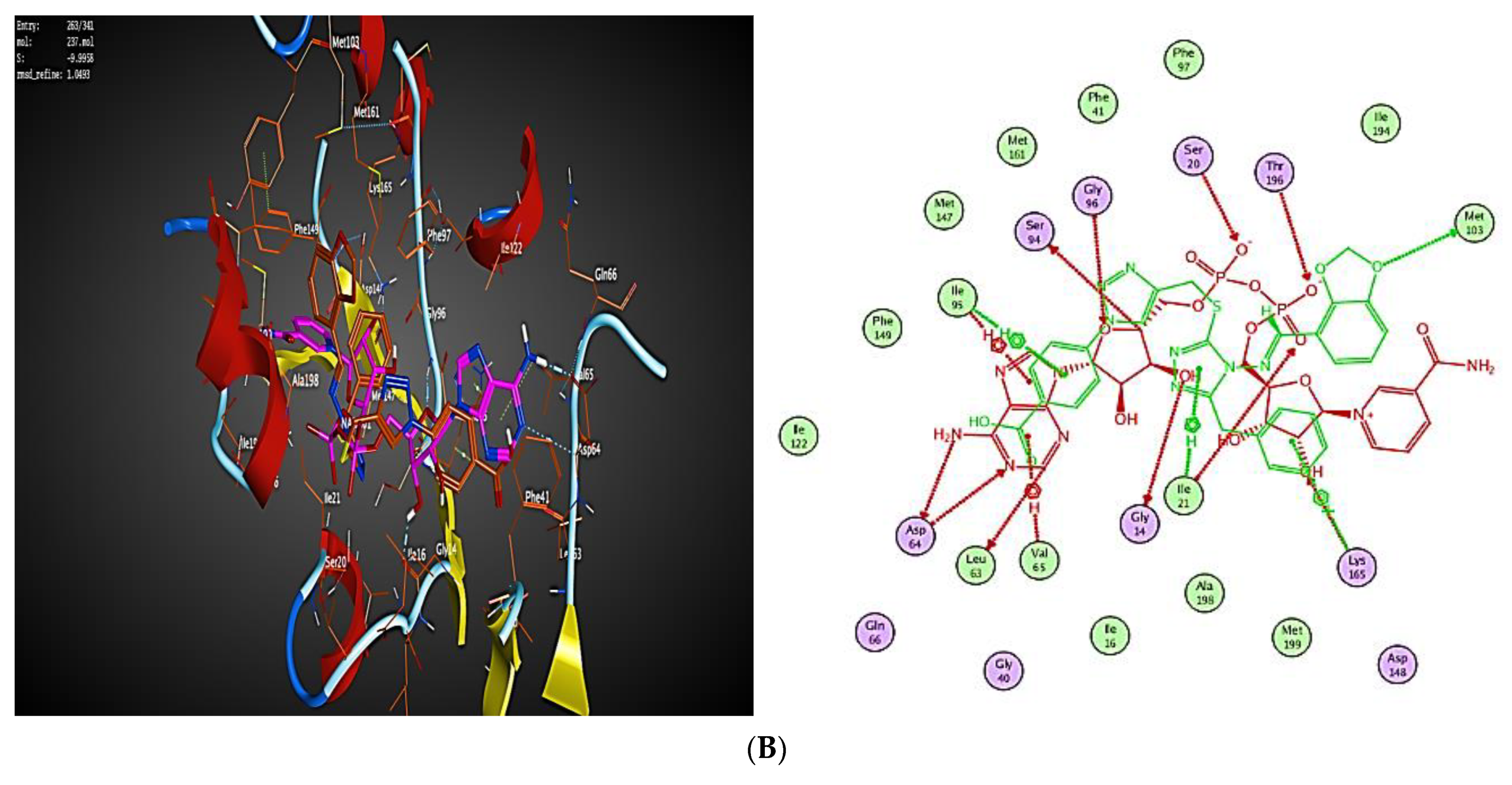
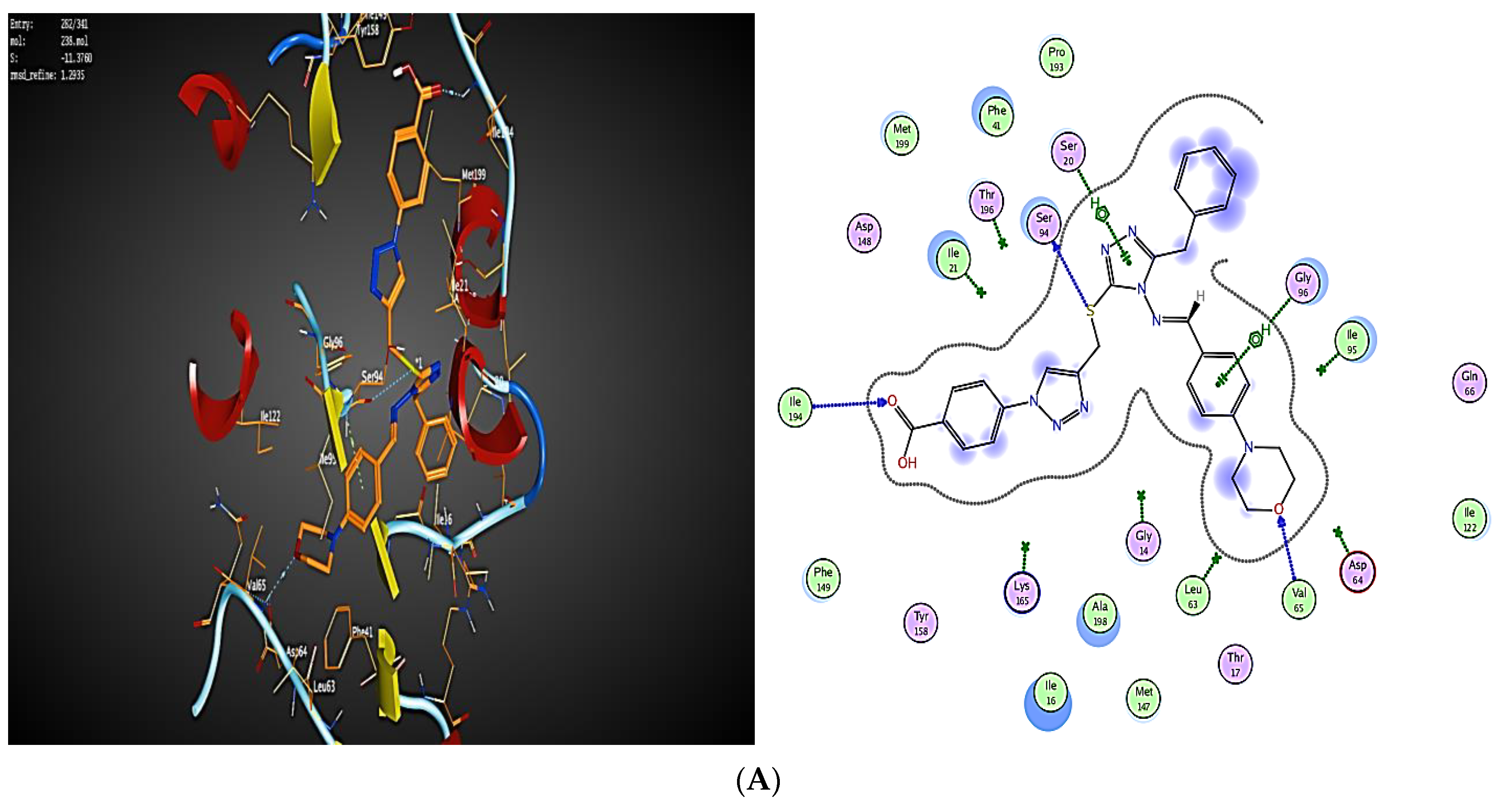
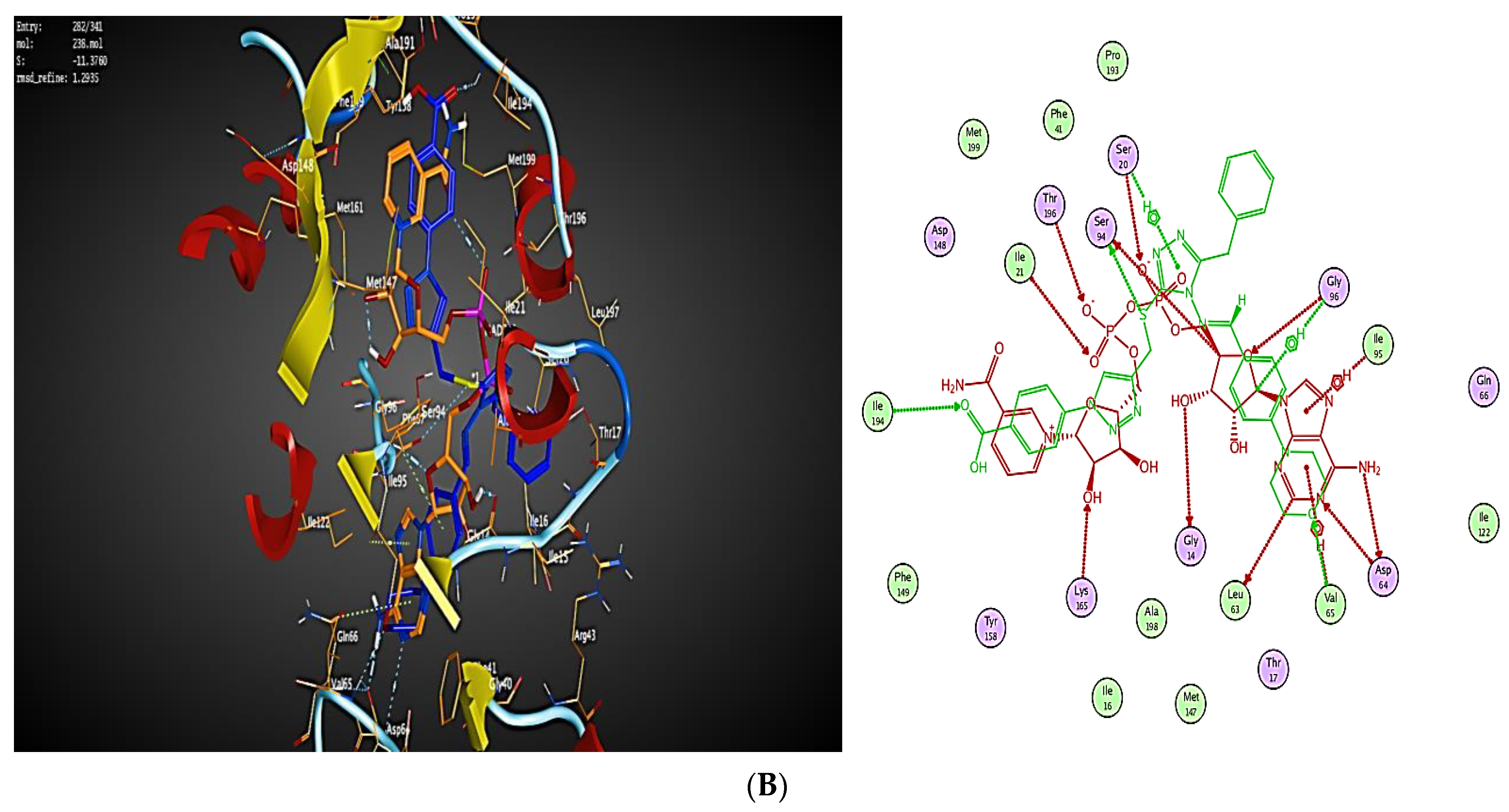
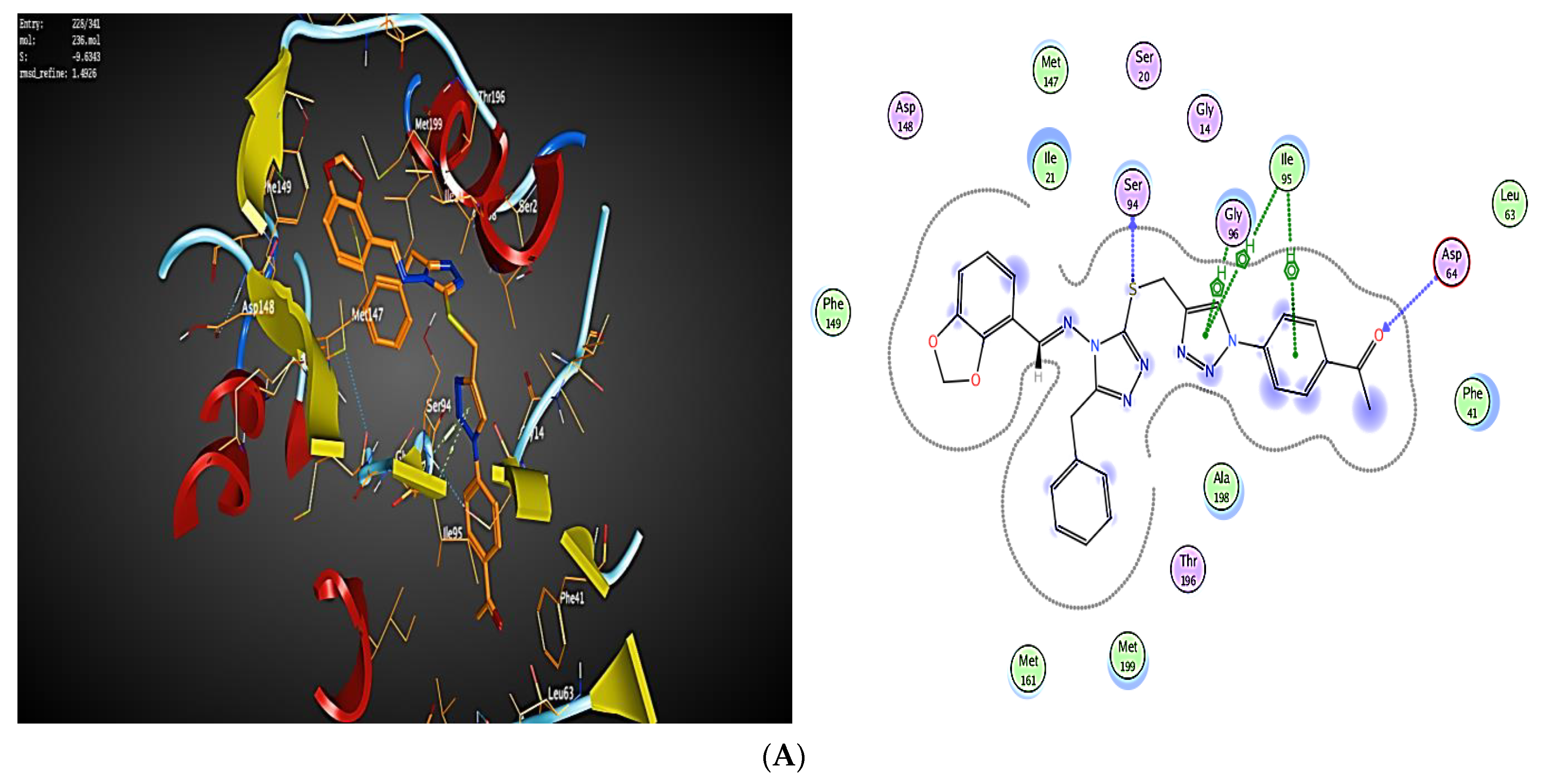
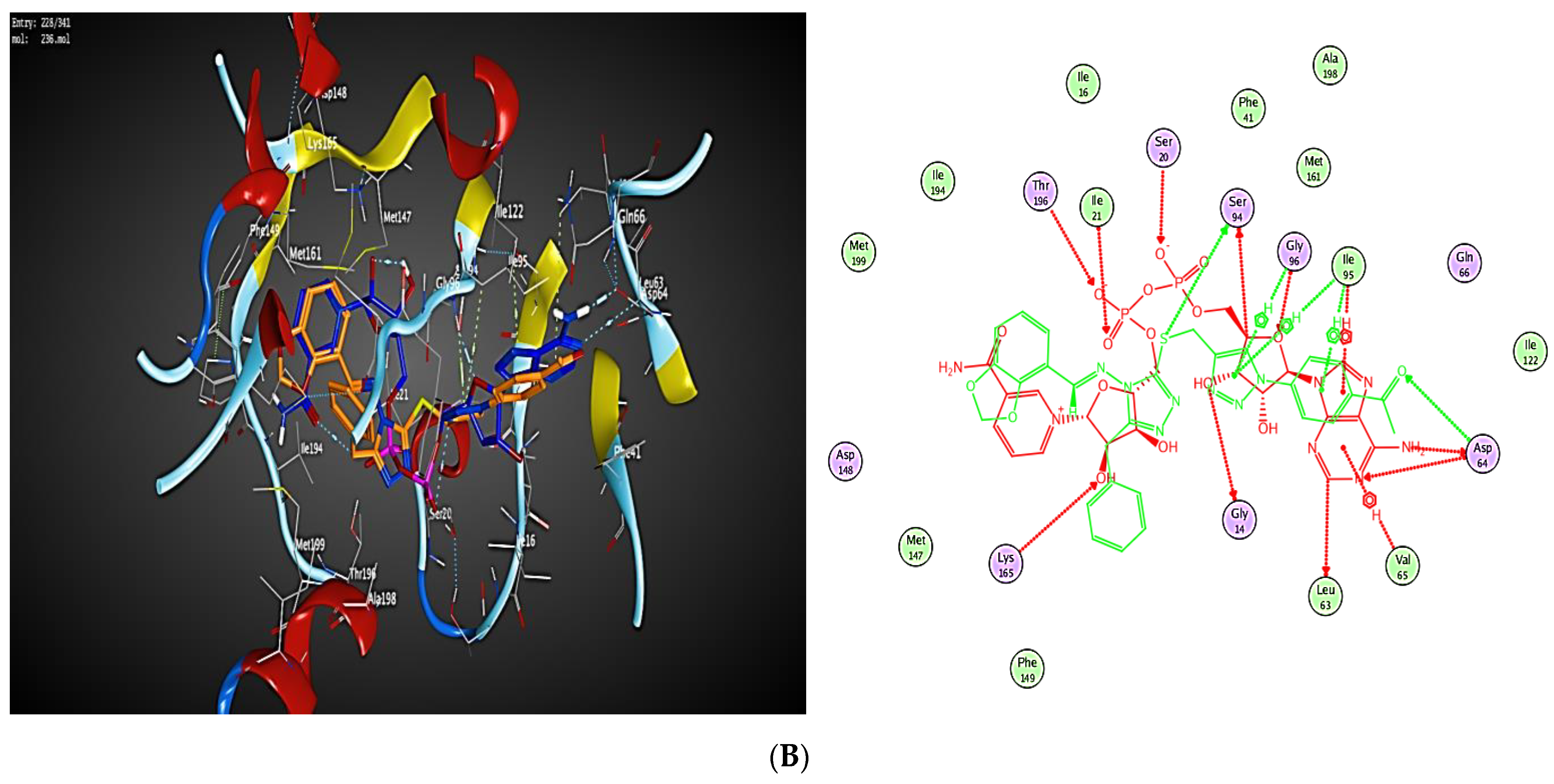
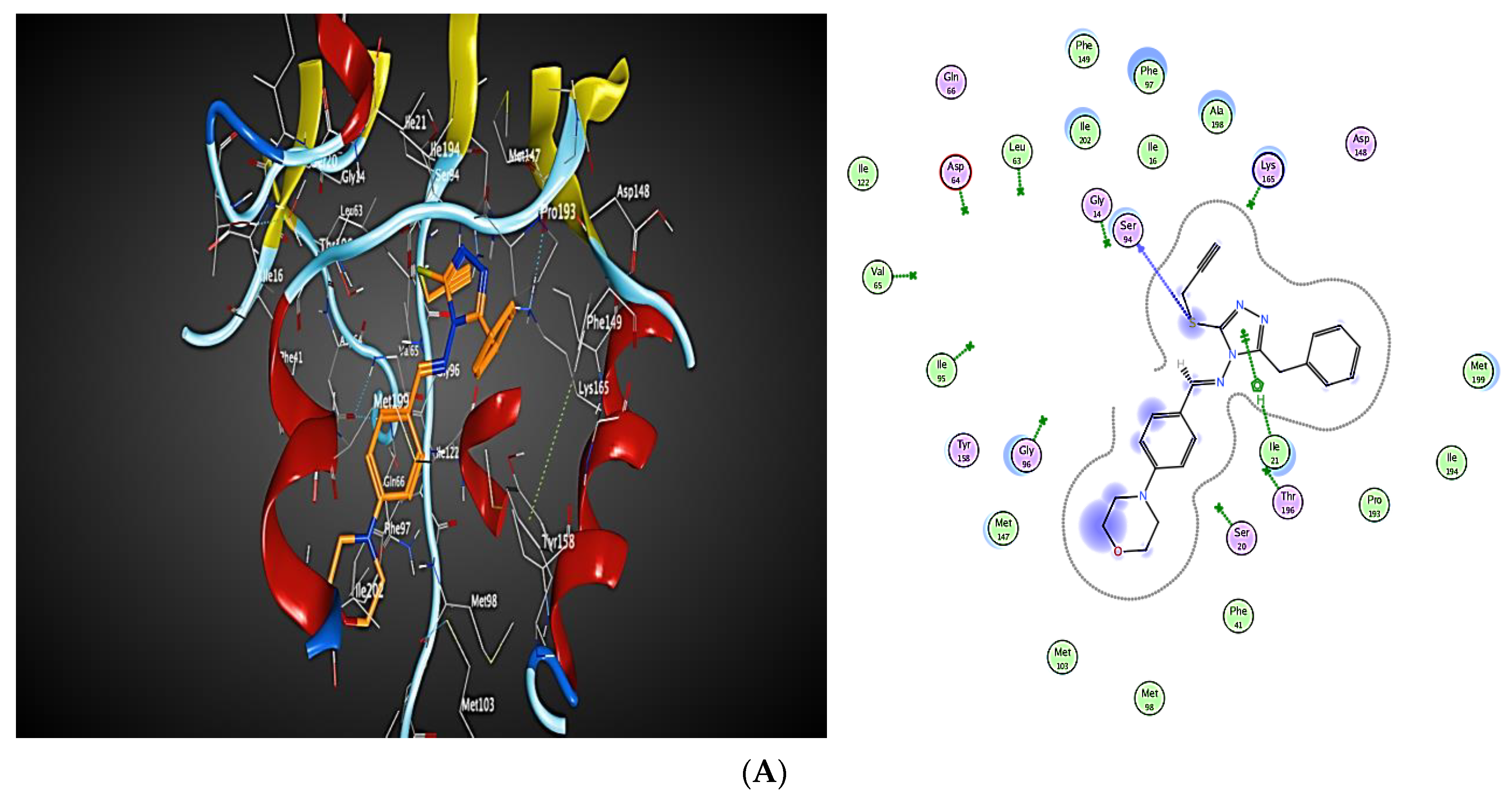
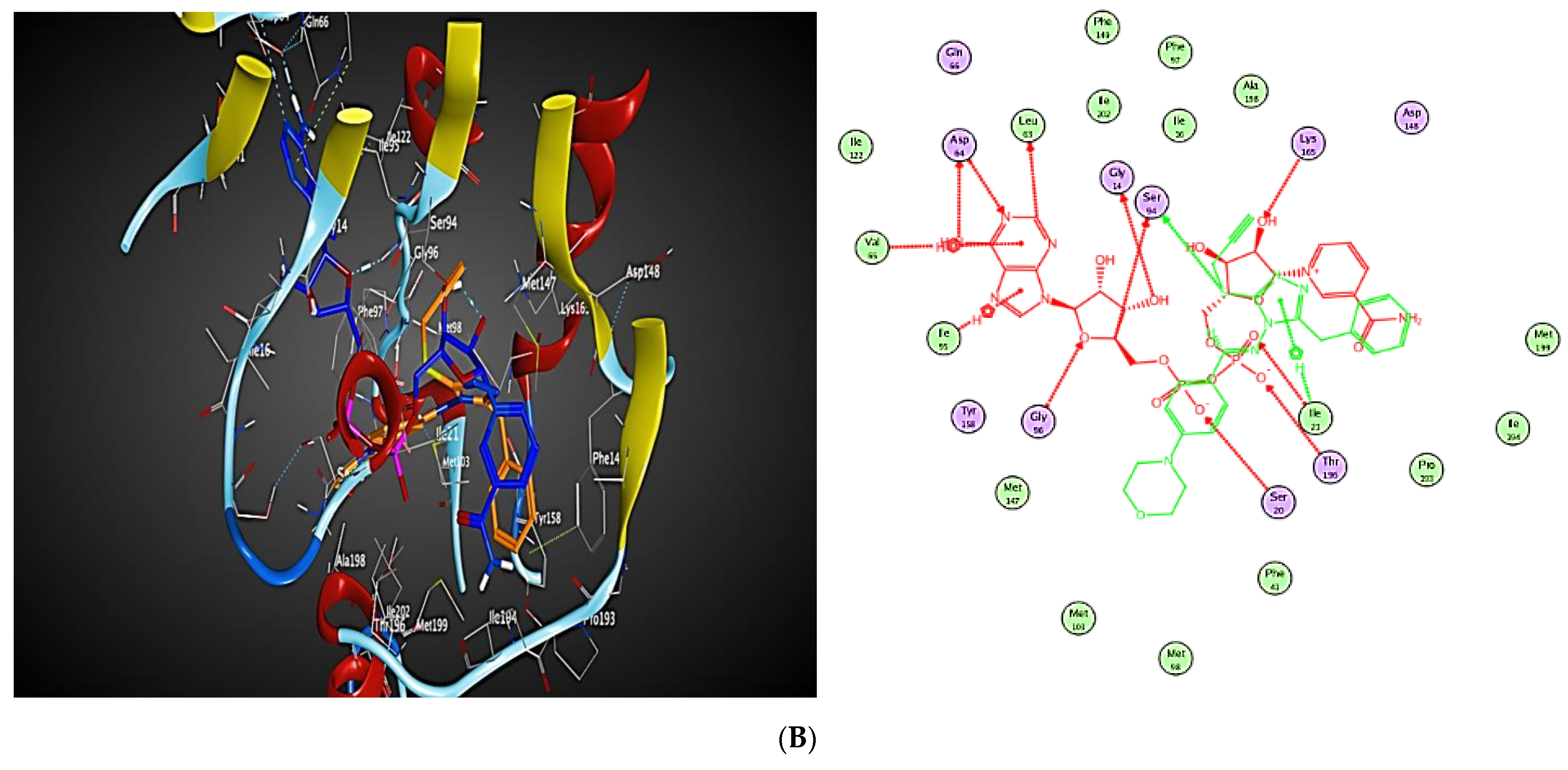
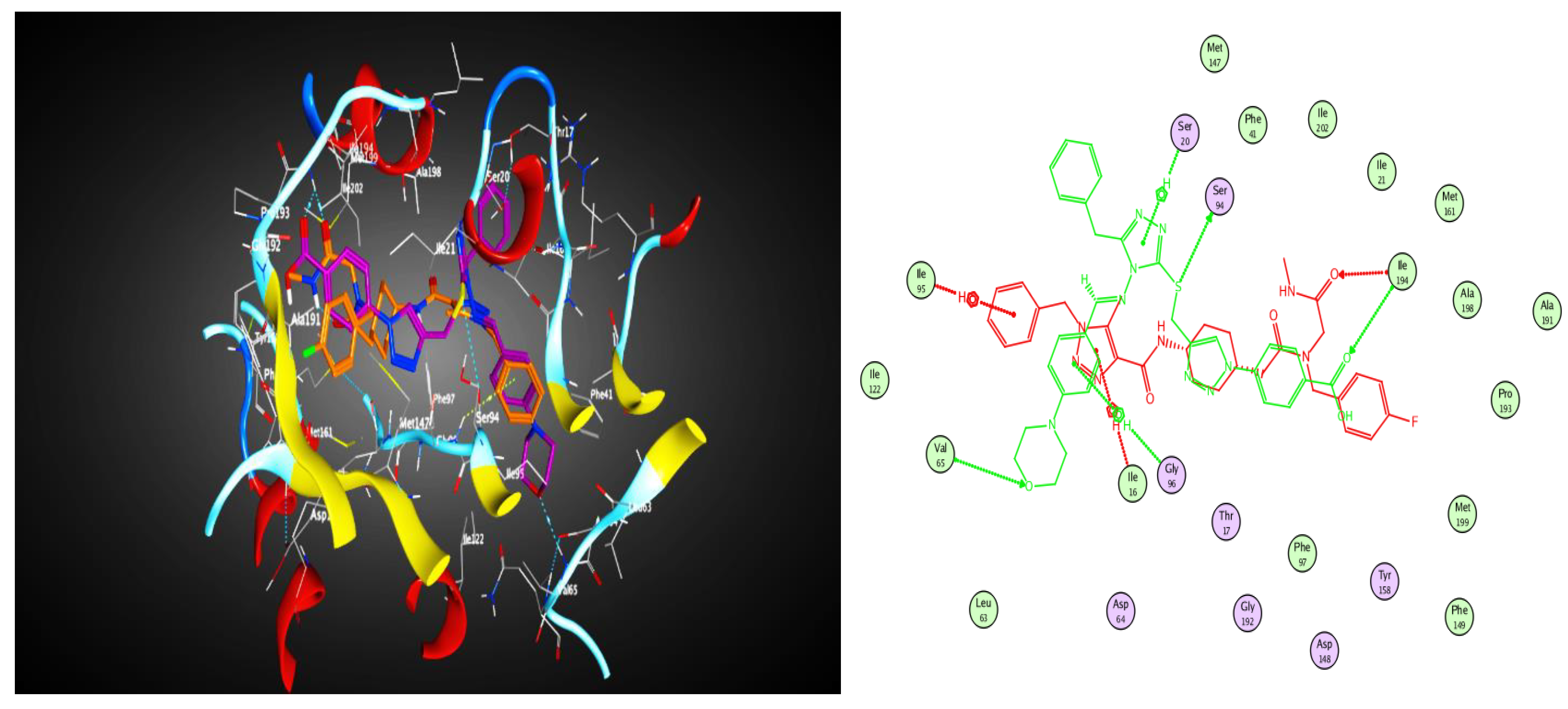
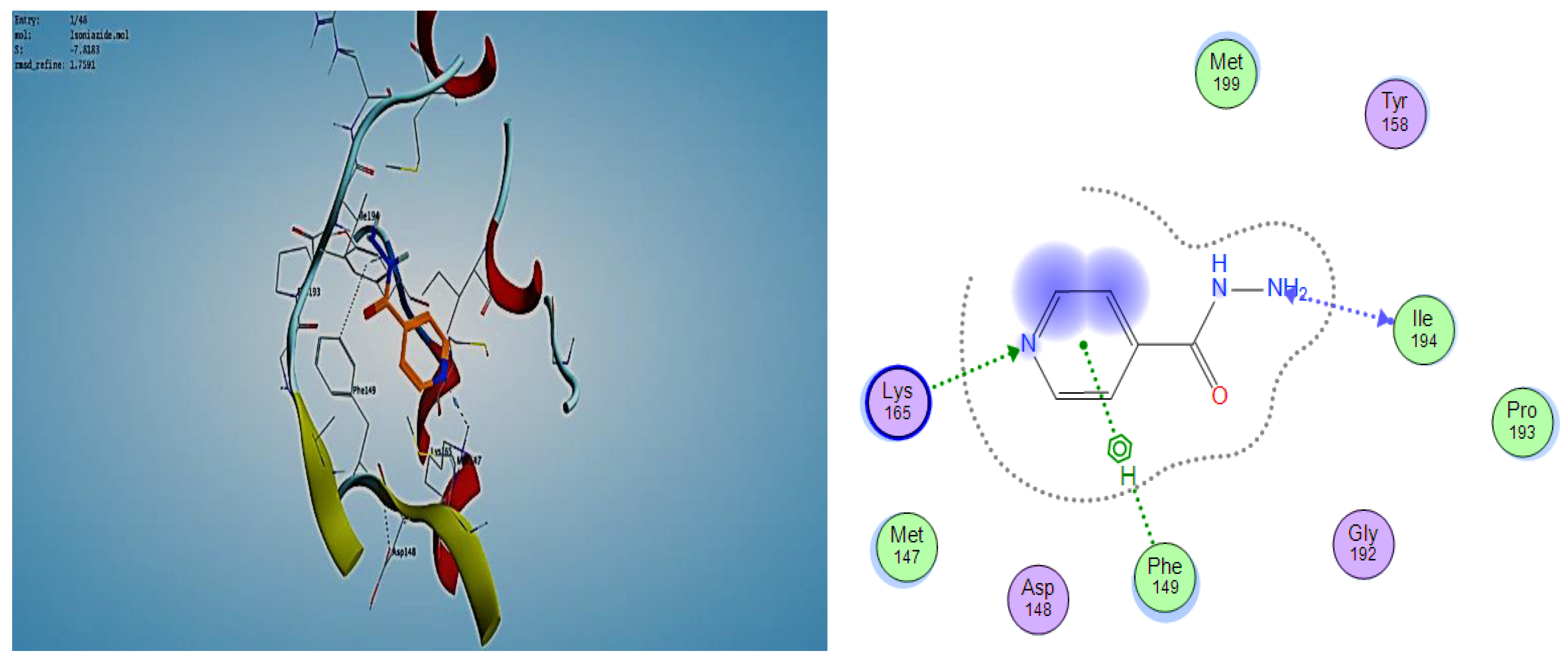
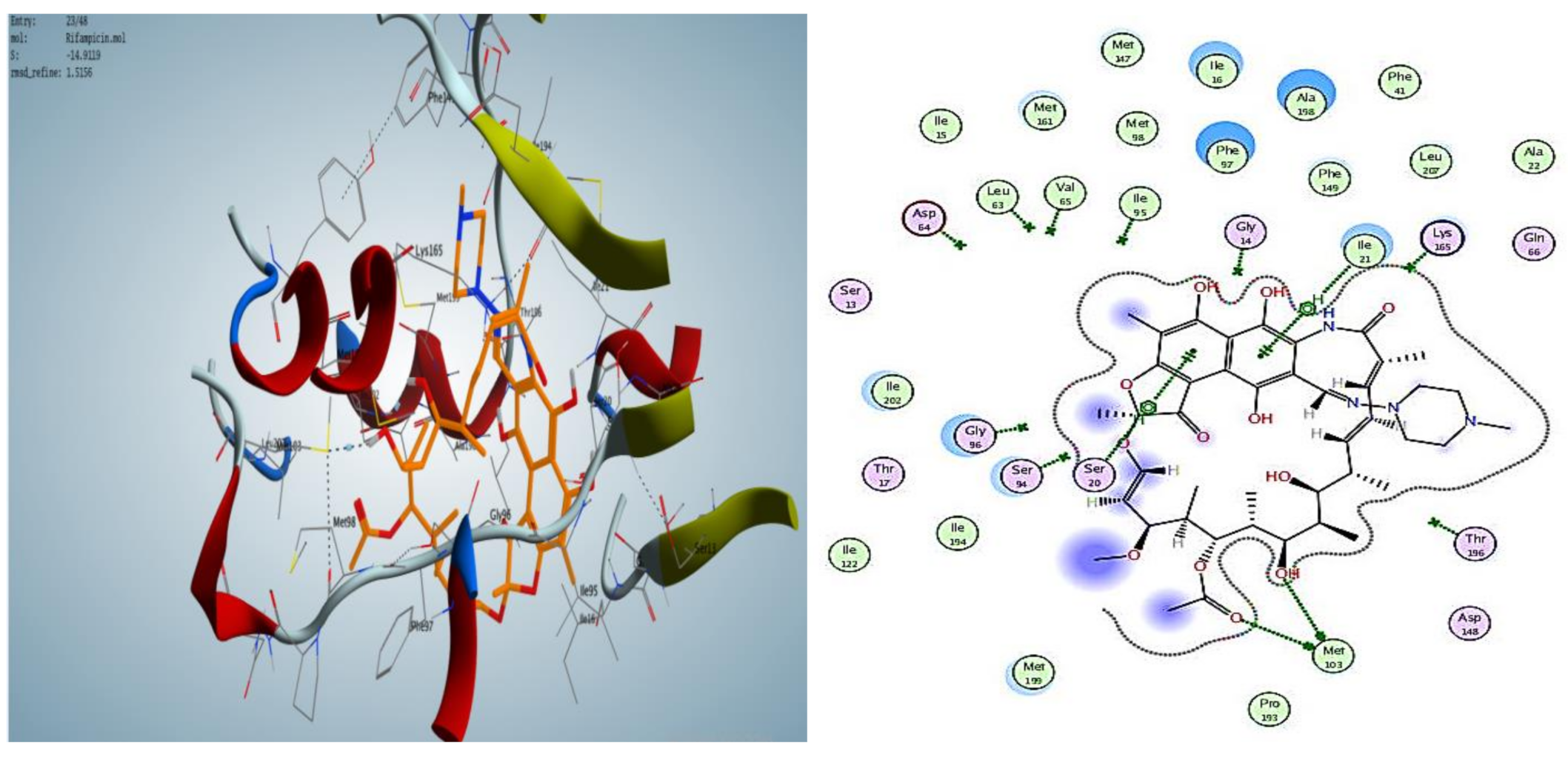
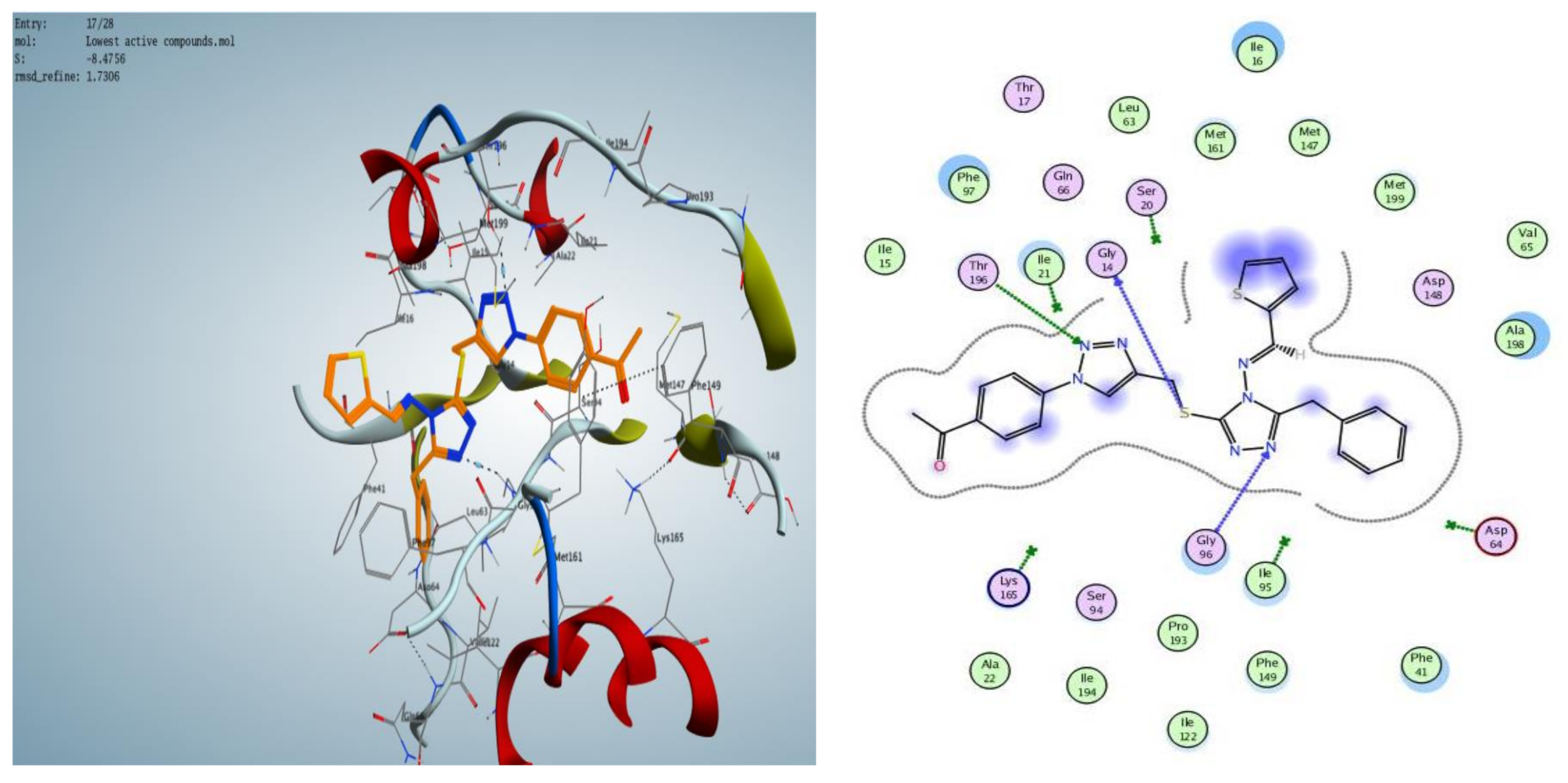
2.3. Molecular Modeling
2.3.1. Docking Simulations
2.3.2. Docking into inhA Active Site
3. Experimental
3.1. Chemistry
3.2. Synthesis of 4-Amino-5-Benzyl-2,4-Dihydro-1,2,4-Triazole-3-Thiol, 3
3.2.1. General Procedure
3.2.2. General Procedure
3.2.3. General Procedure
3.2.4. Enzymatic Inhibition Experiments
Docking Study
Preparation of the Protein Crystal Structures
Preparation of the Selected Compounds for Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Massarotti, A.; Aprile, S.; Mercalli, V.; Del Grosso, E.; Grosa, G.; Sorba, G.; Tron, G.C. Are 1,4-and 1,5-Disubstituted 1,2,3-Triazoles Good Pharmacophoric Groups? Chem. Med. Chem. 2014, 9, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.S.; Zheng, M.J.; Zhao, F.; Liu, D. 1,2,3-Triazole hybrids with anti-HIV-1 activity. Arch. Pharm. 2021, 354, 2000163. [Google Scholar] [CrossRef] [PubMed]
- Holanda, V.N.; Lima, E.M.d.A.; Silva, W.V.D.; Maia, R.T.; Medeiros, R.d.L.; Ghosh, A.; Lima, V.L.d.M.; Figueiredo, R.C.B.Q.D. Identification of 1,2,3-triazole-phthalimide derivatives as potential drugs against COVID-19: A virtual screening, docking and molecular dynamic study. J. Biomol. Struct. Dyn. 2021, 18, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Almehmadi, M.A.; Aljuhani, A.; Alraqa, S.Y.; Ali, I.; Rezki, N.; Aouad, M.R.; Hagar, M. Design, synthesis, DNA binding, modeling, anticancer studies and DFT calculations of Schiff bases tethering benzothiazole-1,2,3-triazole conjugates. J. Mol. Struct. 2021, 1225, 129148. [Google Scholar] [CrossRef]
- Said, M.A.; Khan, D.J.; Al-Blewi, F.F.; Al-Kaff, N.S.; Ali, A.A.; Rezki, N.; Aouad, M.R.; Hagar, M. New 1,2,3-Triazole Scaffold Schiff Bases as Potential Anti-COVID-19: Design, Synthesis, DFT-Molecular Docking, and Cytotoxicity Aspects. Vaccines 2021, 9, 1012. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.Y.; Shaaban, M.M.; Elwakil, B.H.; Hamed, M.T.; Rezki, N.; Aouad, M.R.; Zakaria, M.A.; Hagar, M. Anti-COVID-19 activity of some benzofused 1,2,3-triazolesulfonamide hybrids using in silico and in vitro analyses. Chemom. Intell. Lab. Syst. 2021, 217, 104421. [Google Scholar] [CrossRef]
- Sari, K.Ö.; Tan, O.Ü.; Sriram, D.; Balkan, A. Some new hydrazone derivatives bearing the 1,2,4-triazole moiety as potential antimycobacterial agents. Turk. J. Pharm. Sci. 2019, 16, 432. [Google Scholar] [CrossRef]
- Smit, F.J.; Seldon, R.; Aucamp, J.; Jordaan, A.; Warner, D.F.; N’Da, D.D. Synthesis and antimycobacterial activity of disubstituted benzyltriazoles. Med. Chem. Res. 2019, 28, 2279–2293. [Google Scholar] [CrossRef] [Green Version]
- Soutter, H.H.; Centrella, P.; Clark, M.A.; Cuozzo, J.W.; Dumelin, C.E.; Guie, M.-A.; Habeshian, S.; Keefe, A.D.; Kennedy, K.M.; Sigel, E.A. Discovery of cofactor-specific, bactericidal Mycobacterium tuberculosis InhA inhibitors using DNA-encoded library technology. Proc. Natl. Acad. Sci. USA 2016, 113, E7880–E7889. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; He, Y.; Zhang, X.; Xu, J.; Luo, Y.; Wang, Y.; Franzblau, S.G.; Yang, Z.; Chan, R.J.; Liu, Y. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl. Acad. Sci. USA 2010, 107, 4573–4578. [Google Scholar] [CrossRef] [Green Version]
- Karczmarzyk, Z.; Swatko-Ossor, M.; Wysocki, W.; Drozd, M.; Ginalska, G.; Pachuta-Stec, A.; Pitucha, M. New application of 1,2,4-triazole derivatives as antitubercular agents. Structure, in vitro screening and docking studies. Molecules 2020, 25, 6033. [Google Scholar] [CrossRef] [PubMed]
- Patpi, S.R.; Pulipati, L.; Yogeeswari, P.; Sriram, D.; Jain, N.; Sridhar, B.; Murthy, R.; Kalivendi, S.V.; Kantevari, S. Design, synthesis, and structure–activity correlations of novel dibenzo [b, d] furan, dibenzo [b, d] thiophene, and N-methylcarbazole clubbed 1,2,3-triazoles as potent inhibitors of mycobacterium tuberculosis. J. Med. Chem. 2012, 55, 3911–3922. [Google Scholar] [CrossRef] [PubMed]
- Badar, A.D.; Sulakhe, S.M.; Muluk, M.B.; Rehman, N.N.; Dixit, P.P.; Choudhari, P.B.; Rekha, E.M.; Sriram, D.; Haval, K.P. Synthesis of isoniazid-1, 2, 3-triazole conjugates: Antitubercular, antimicrobial evaluation and molecular docking study. J. Heterocycl. Chem. 2020, 57, 3544–3557. [Google Scholar] [CrossRef]
- Singh, G.; Devi, A.; Gupta, S. Tetrazole conjoined organosilane and organosilatrane via the ‘click approach’: A potent Mycobacterium tuberculosis enoyl ACP reductase inhibitor and a dual sensor for Fe (iii) and Cu (ii) ions. New J. Chem. 2022, 46, 2094. [Google Scholar] [CrossRef]
- Hervin, V.; Arora, R.; Rani, J.; Ramchandran, S.; Bajpai, U.; Agrofoglio, L.A.; Roy, V. Design and Synthesis of Various 5′-Deoxy-5′-(4-Substituted-1,2,3-Triazol-1-yl)-Uridine Analogues as Inhibitors of Mycobacterium tuberculosis Mur Ligases. Molecules 2020, 25, 4953. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Kandeel, M.; Pillay, M.; Deb, P.K.; Abdallah, H.H.; Mahomoodally, M.F.; Chopra, D. Anti-tubercular properties of 4-amino-5-(4-fluoro-3-phenoxyphenyl)-4h-1,2,4-triazole-3-thiol and its schiff bases: Computational input and molecular dynamics. Antibiotics 2020, 9, 559. [Google Scholar] [CrossRef]
- Chuprun, S.; Dar’in, D.; Rogacheva, E.; Kraeva, L.; Levin, O.; Manicheva, O.; Dogonadze, M.; Vinogradova, T.; Bakulina, O.; Krasavin, M. Mutually isomeric 2-and 4-(3-Nitro-1,2,4-Triazol-1-Yl) pyrimidines inspired by an antimycobacterial screening hit: Synthesis and biological activity against the eskape panel of pathogens. Antibiotics 2020, 9, 666. [Google Scholar] [CrossRef]
- Encinas, L.; O’Keefe, H.; Neu, M.; Remuinan, M.J.; Patel, A.M.; Guardia, A.; Davie, C.P.; Perez-Macias, N.; Yang, H.; Convery, M.A. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J. Med. Chem. 2014, 57, 1276–1288. [Google Scholar] [CrossRef] [Green Version]
- Kumari, A.; Singh, R.K. Morpholine as ubiquitous pharmacophore in medicinal chemistry: Deep insight into the structure-activity relationship (SAR). Bioorg. Chem. 2020, 96, 103578. [Google Scholar] [CrossRef]
- Lee, M.; Lee, J.; Carroll, M.W.; Choi, H.; Min, S.; Song, T.; Via, L.E.; Goldfeder, L.C.; Kang, E.; Jin, B. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med. 2012, 367, 1508–1518. [Google Scholar] [CrossRef] [Green Version]
- El-Ashry, E.S.H.; Rashed, N.; Awad, L.F.; Ramadan, E.S.; Abdel-Maggeed, S.M.; Rezki, N. Synthesis of 5-Aryl-3-Glycosylthio-4-Phenyl-4H-1,2,4-Triazoles and Their Acyclic Analogs Under Conventional and Microwave Conditions. J. Carbohydr. Chem. 2008, 27, 70–85. [Google Scholar] [CrossRef]
- Rostamizadeh, S.; Mollahoseini, K.; Moghadasi, S. A one-pot synthesis of 4,5-disubstituted-1,2,4-triazole-3-thiones on solid support under microwave irradiation. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 1839–1845. [Google Scholar] [CrossRef]
- Naik, S.K.; Mohanty, S.; Padhi, A.; Pati, R.; Sonawane, A. Evaluation of antibacterial and cytotoxic activity of Artemisia nilagirica and Murraya koenigii leaf extracts against mycobacteria and macrophages. BMC Complementary Altern. Med. 2014, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Chollet, A.; Mourey, L.; Lherbet, C.; Delbot, A.; Julien, S.; Baltas, M.; Bernadou, J.; Pratviel, G.; Maveyraud, L.; Bernardes-Génisson, V. Crystal structure of the enoyl-ACP reductase of Mycobacterium tuberculosis (InhA) in the apo-form and in complex with the active metabolite of isoniazid pre-formed by a biomimetic approach. J. Struct. Biol. 2015, 190, 328–337. [Google Scholar] [CrossRef]
- Husain, A.; Naseer, M.A. Studies on fused heterocyclic 3,6-disubstituted-1,2,4-triazolo-1,3,4-thiadiazoles: Synthesis and biological evaluation. Med. Chem. Res. 2011, 20, 47–54. [Google Scholar] [CrossRef]
- El Ashry, E.; Elshatanofy, M.; Badawy, M.; Kandeel, K.; Elhady, O.; Abdel-Sayed, M. Synthesis and Evaluation of Antioxidant, Antibacterial, and Target Protein-Molecular Docking of Novel 5-Phenyl-2,4-dihydro-3H-1,2,4-triazole Derivatives Hybridized with 1,2,3-Triazole via the Flexible SCH2-Bonding. Russ. J. Gen. Chem. 2020, 90, 2419–2434. [Google Scholar] [CrossRef]
- Huisgen, R. Centenary Lecture-1,3-dipolar cycloadditions; Royal Soc Chemistry Thomas Graham House, Science Park: Milton, RD, USA; Cambridge, UK, 1961; p. 357. [Google Scholar]
- Rodriguez, F.; Saffon, N.; Sammartino, J.C.; Degiacomi, G.; Pasca, M.R.; Lherbet, C. First triclosan-based macrocyclic inhibitors of InhA enzyme. Bioorg. Chem. 2020, 95, 103498. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested Compounds | IC50 (nM) |
|---|---|
| 4a | 53.2 |
| 4b | 9.16 |
| 4c | 8.16 |
| 5a | 5.61 |
| 5b | 0.66 |
| 5c | 0.23 |
| 7a | 6.18 |
| 7b | 12.5 |
| 7c | 0.074 |
| 7d | 0.18 |
| 7e | 0.13 |
| 7f | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Sawy, M.A.; Elshatanofy, M.M.; El Kilany, Y.; Kandeel, K.; Elwakil, B.H.; Hagar, M.; Aouad, M.R.; Albelwi, F.F.; Rezki, N.; Jaremko, M.; et al. Novel Hybrid 1,2,4- and 1,2,3-Triazoles Targeting Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase (InhA): Design, Synthesis, and Molecular Docking. Int. J. Mol. Sci. 2022, 23, 4706. https://doi.org/10.3390/ijms23094706
El Sawy MA, Elshatanofy MM, El Kilany Y, Kandeel K, Elwakil BH, Hagar M, Aouad MR, Albelwi FF, Rezki N, Jaremko M, et al. Novel Hybrid 1,2,4- and 1,2,3-Triazoles Targeting Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase (InhA): Design, Synthesis, and Molecular Docking. International Journal of Molecular Sciences. 2022; 23(9):4706. https://doi.org/10.3390/ijms23094706
Chicago/Turabian StyleEl Sawy, Maged A., Maram M. Elshatanofy, Yeldez El Kilany, Kamal Kandeel, Bassma H. Elwakil, Mohamed Hagar, Mohamed Reda Aouad, Fawzia Faleh Albelwi, Nadjet Rezki, Mariusz Jaremko, and et al. 2022. "Novel Hybrid 1,2,4- and 1,2,3-Triazoles Targeting Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase (InhA): Design, Synthesis, and Molecular Docking" International Journal of Molecular Sciences 23, no. 9: 4706. https://doi.org/10.3390/ijms23094706