
Structure Elucidation of Fucan Sulfate from Sea Cucumber Holothuria fuscopunctata through a Bottom-Up Strategy and the Antioxidant Activity Analysis

Abstract
:1. Introduction
2. Results and Discussion
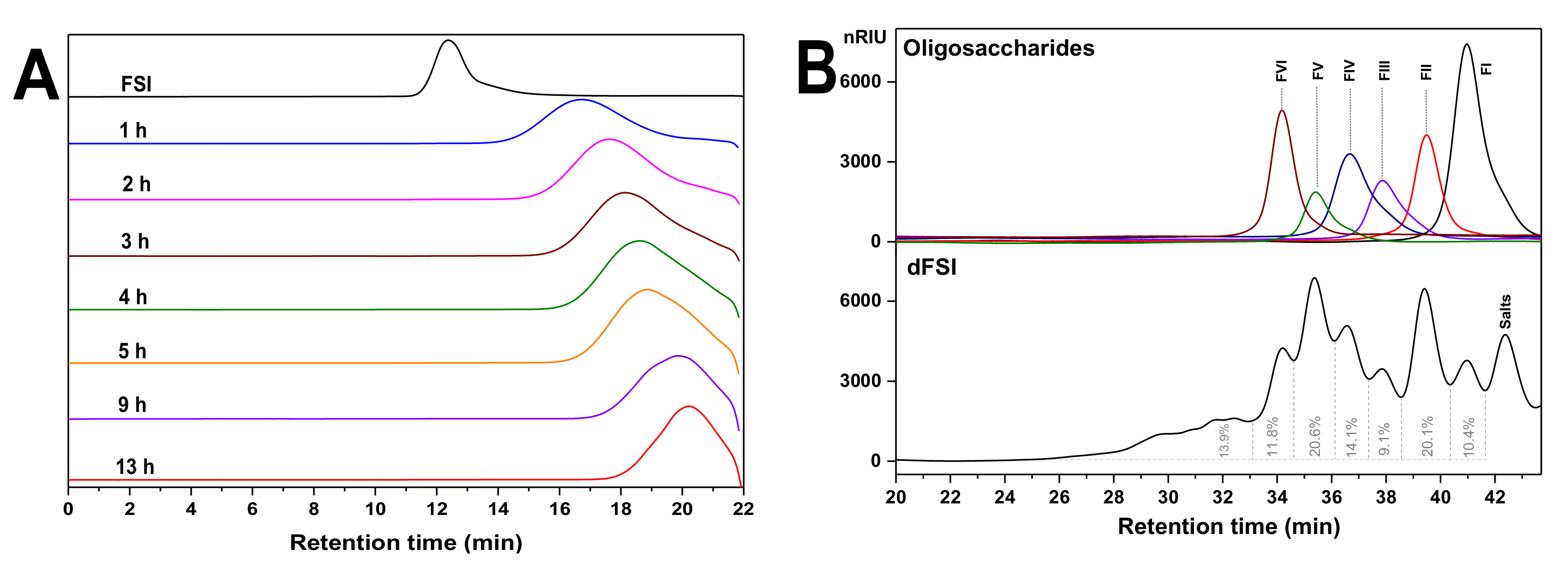
2.1. Optimization of Reaction Temperature and Preparation of Mild Acid Hydrolyzed Product (dFSI)
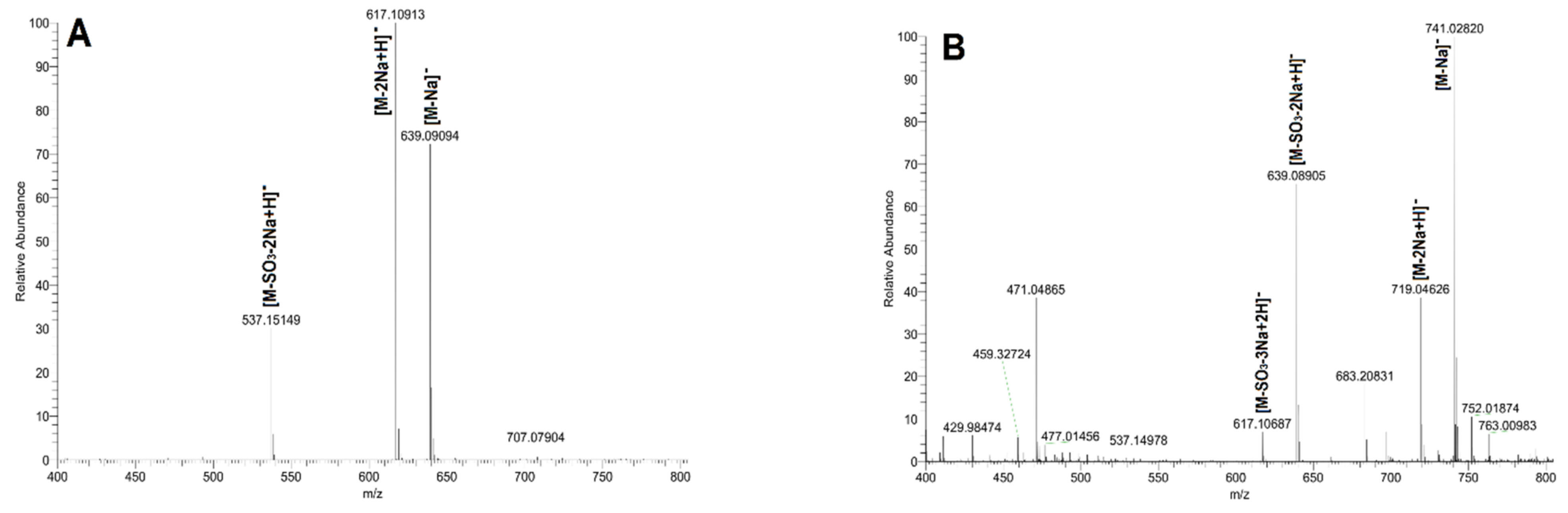
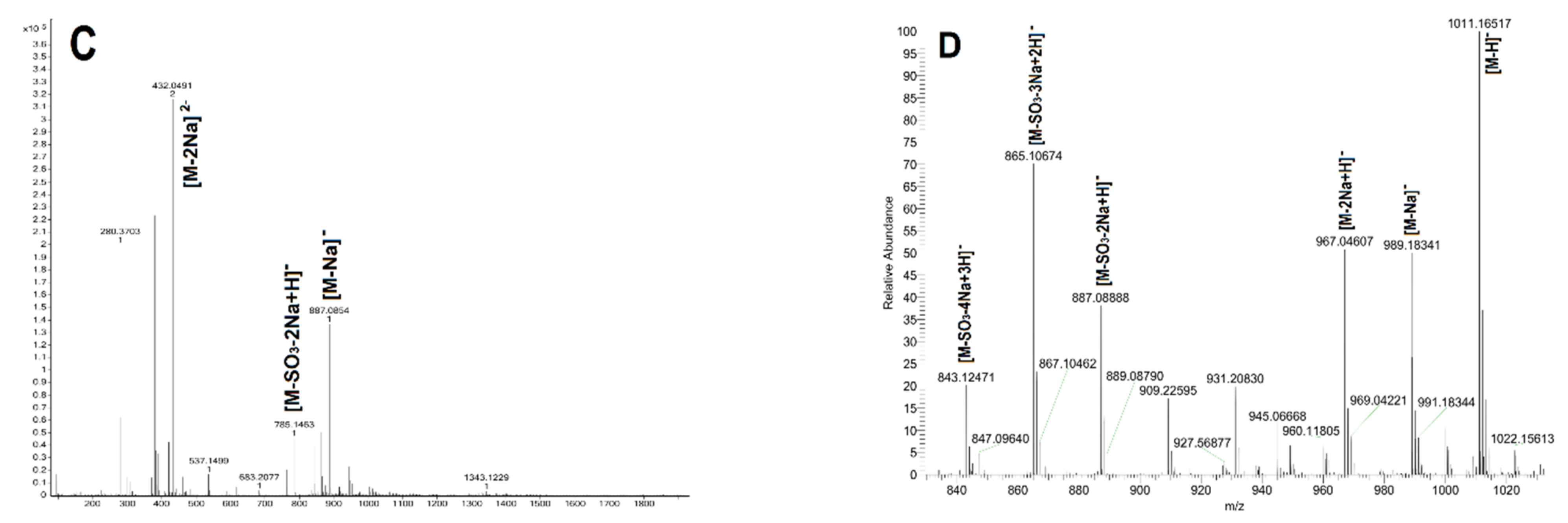
2.2. Purification of Size-Homogeneous Oligosaccharides from dFSI
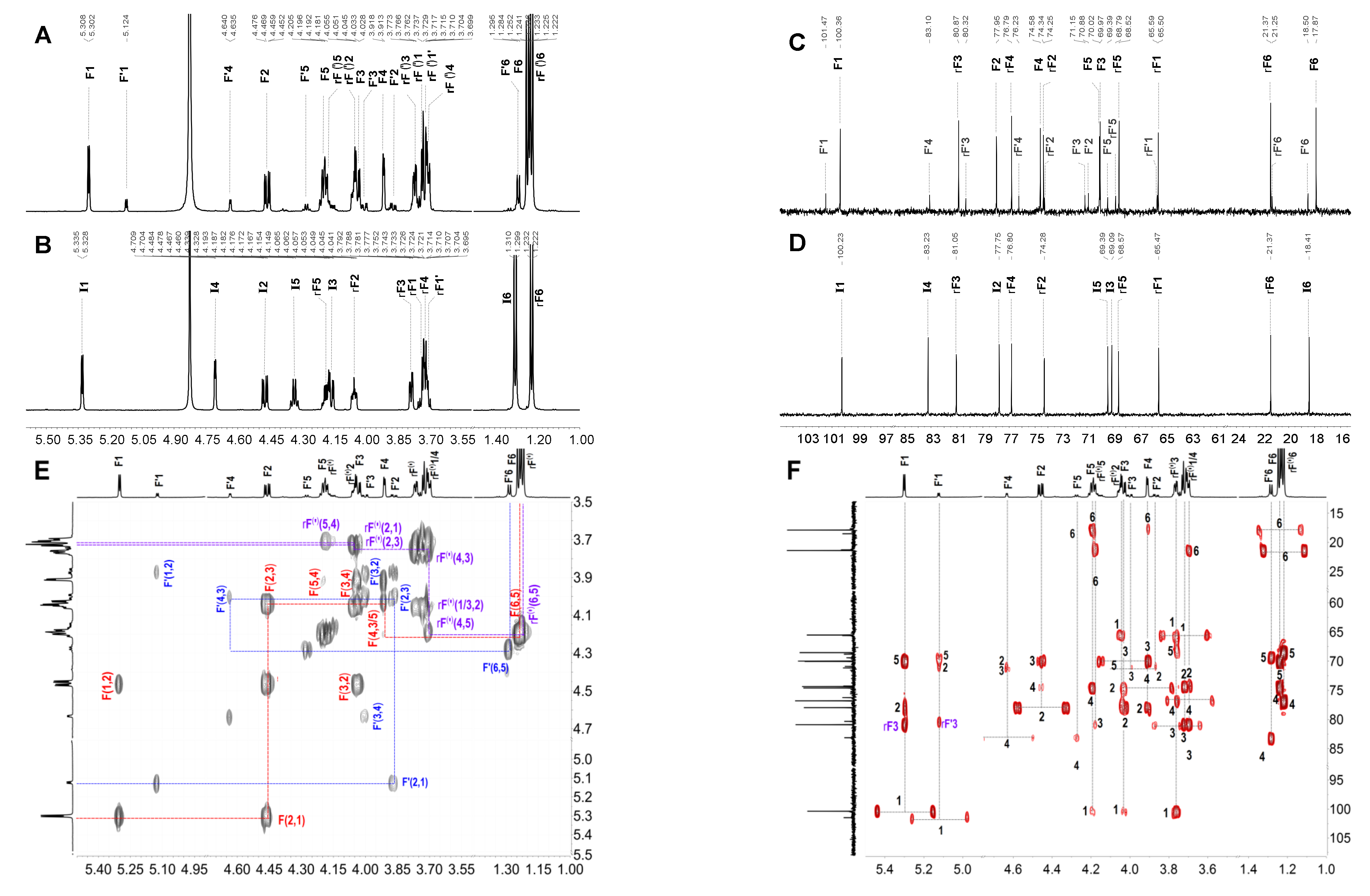
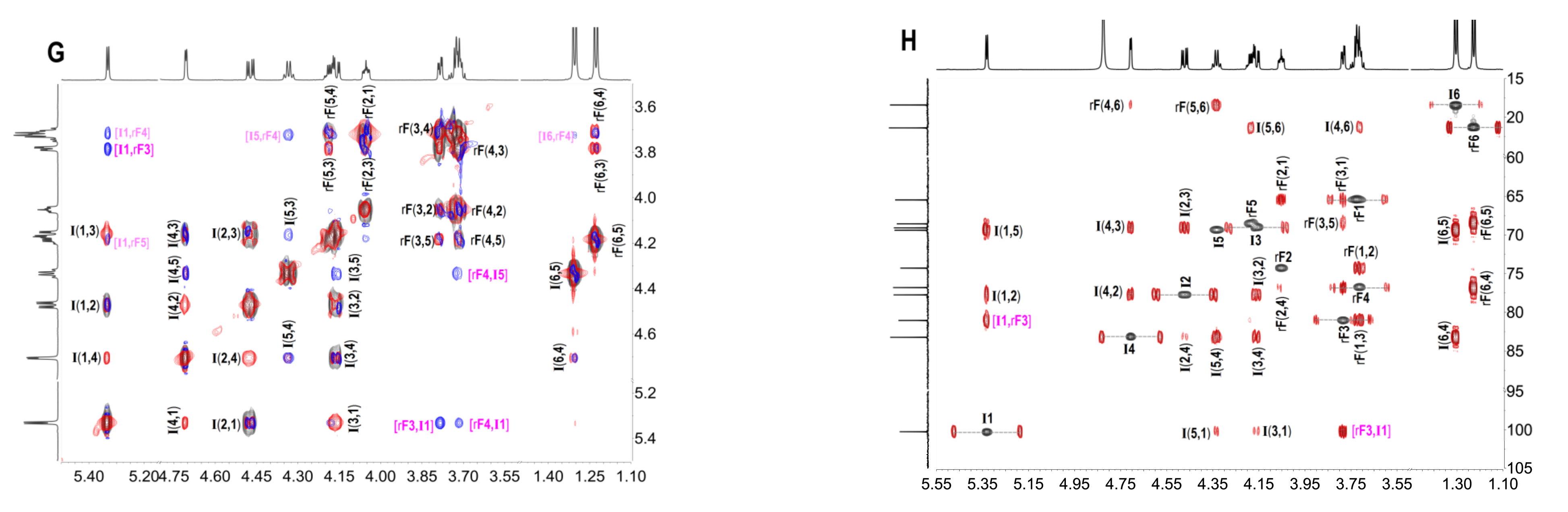
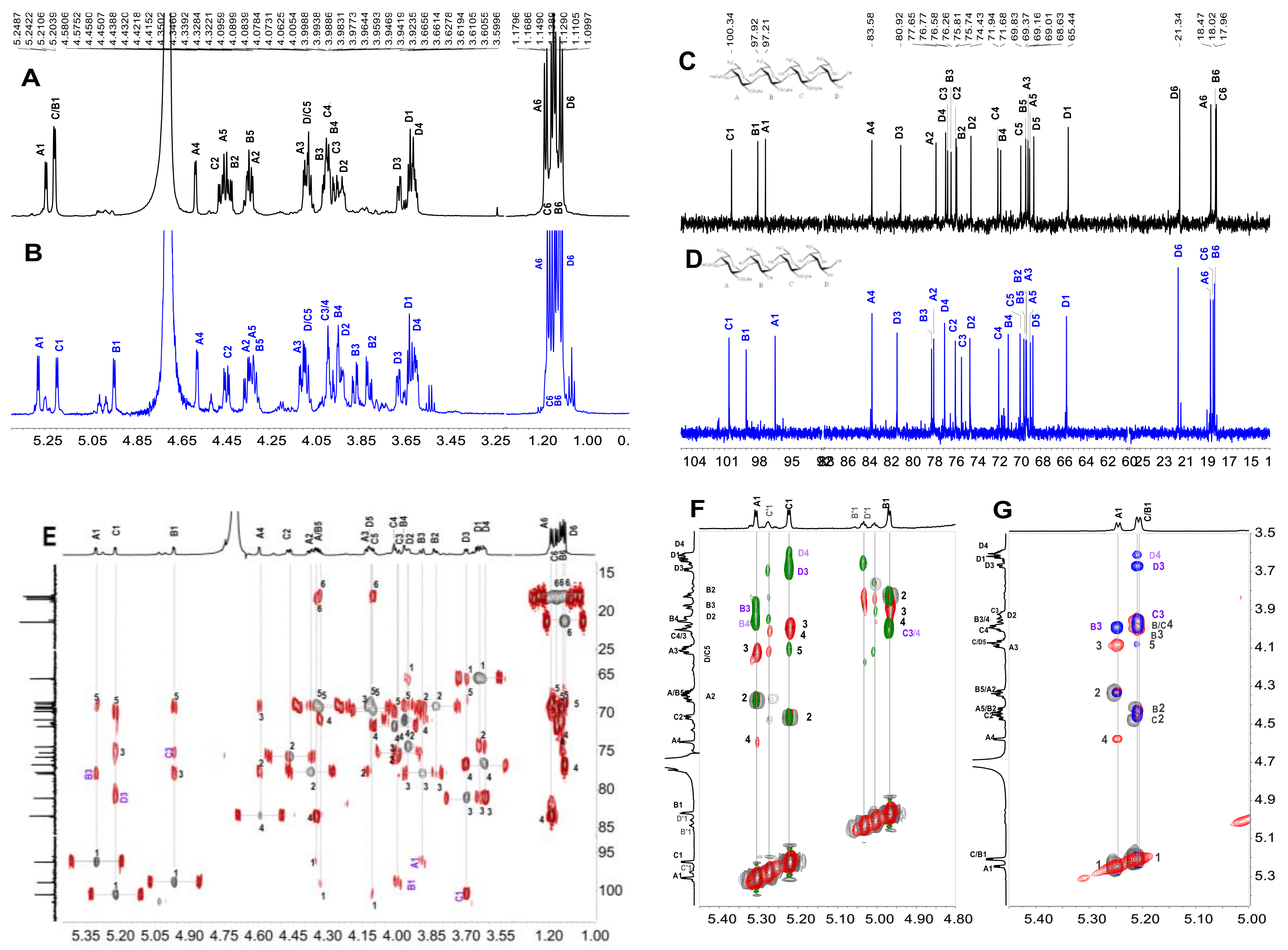
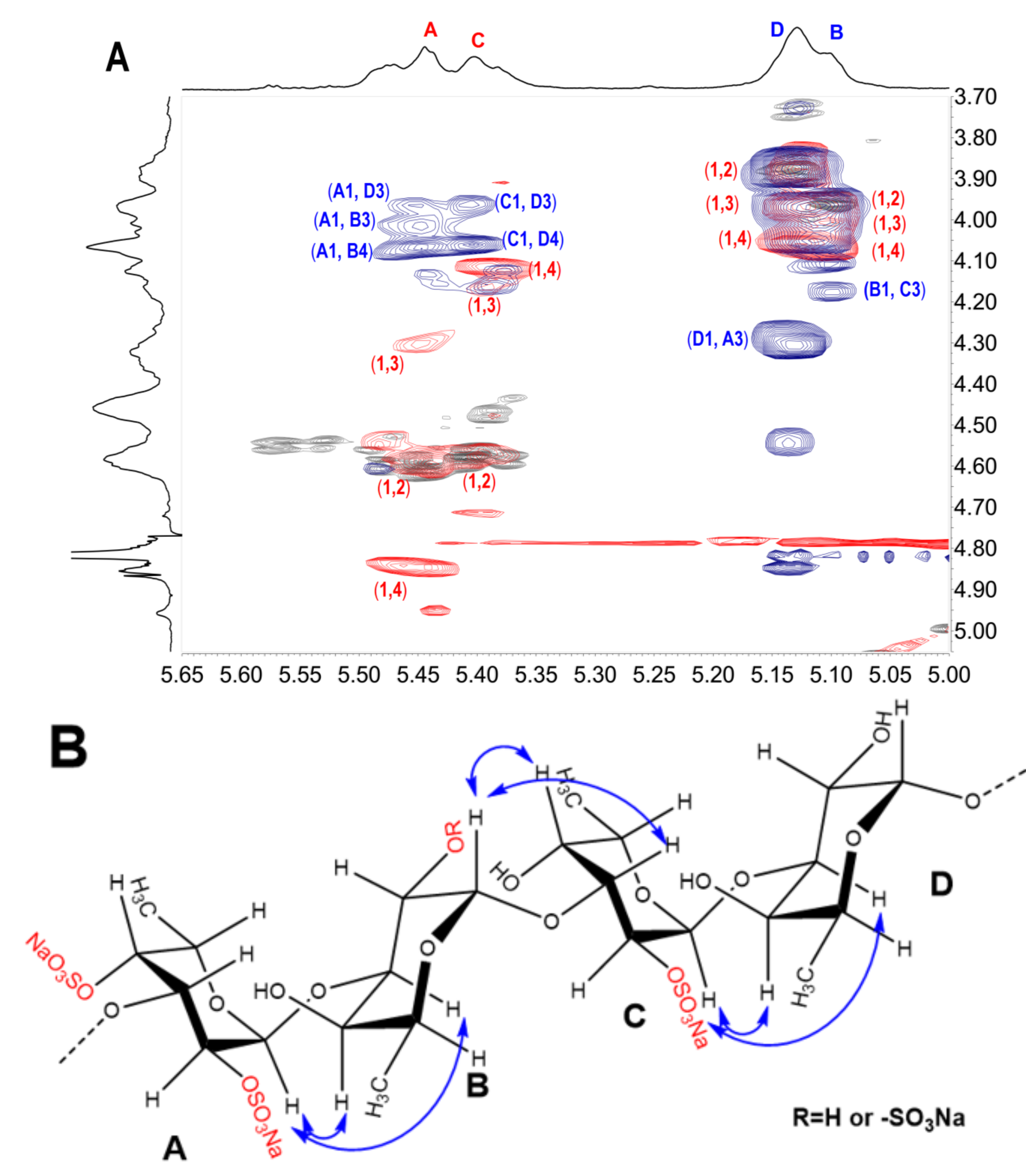
2.3. Structural Determination of Oligosaccharides by NMR
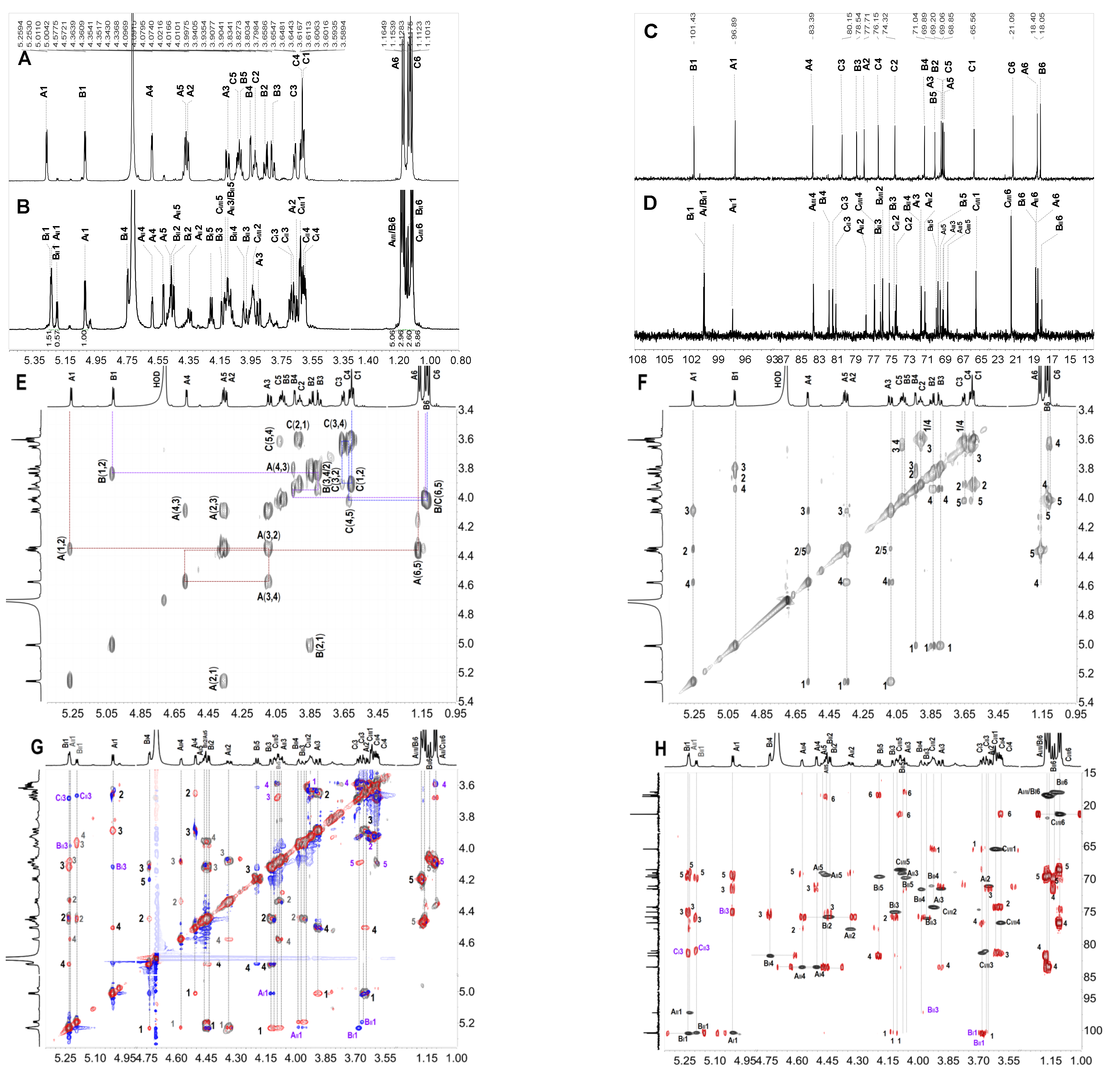
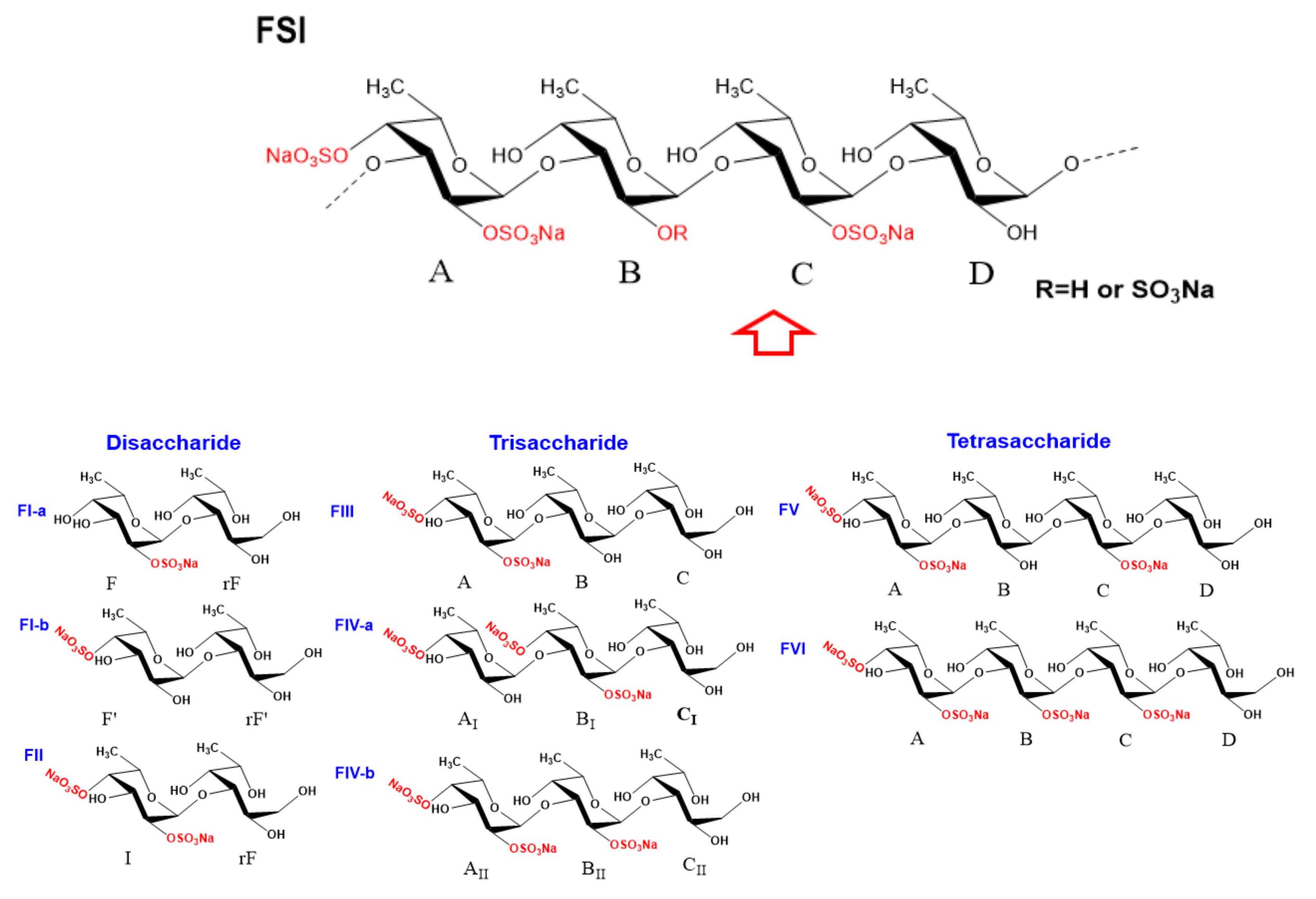
2.4. Structural Characteristics of Fucan Sulfate I (FSI) through a “Bottom-Up” Strategy
2.5. Antioxidant Activities of FSI and Its Low-Molecular-Weight Derivative dFSI′
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Extraction and Purification of Native FSI
4.3. Mild Acid Hydrolysis of FSI
4.3.1. Optimization of Acid Hydrolysis Conditions of FSI
4.3.2. Mild Acid Hydrolysis of FSI by Trifluoroacetic Acid
4.4. Purification of Oligosaccharides by Gel Permeation Chromatography
4.5. One-Dimensional (1D) and Two-Dimensional (2D) NMR Analysis of Oligosaccharides
4.6. Mass Spectrometry Analysis
4.7. Antioxidant Activity Assays of FSI and dFSI′ In Vitro
4.7.1. DPPH Radical Scavenging Assay
4.7.2. Hydroxyl Radical Scavenging Assay
4.7.3. Superoxide Radical Scavenging Assay
4.7.4. ABTS Radical Scavenging Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tian, M.; Xue, C.; Chang, Y.; Shen, J.; Zhang, Y.; Li, Z.; Wang, Y. Collagen fibrils of sea cucumber (Apostichopus japonicus) are heterotypic. Food Chem. 2020, 316, 126272. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wang, L.; Cavalcante, M.C.M.; Cardilo-Reis, L.; Ferreira, P.L.; Mourão, P.A.S.; Esko, J.D.; Pavão, M.S.G. Selectin blocking activity of a fucosylated chondroitin sulfate glycosaminoglycan from sea cucumber: Effect on tumor metastasis and neutrophil recruitment. J. Biol. Chem. 2007, 282, 14984–14991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhou, Q.; Liu, B.; Chen, F.; Wang, M. Holothurian fucosylated chondroitin sulfates and their potential benefits for human health: Structures and biological activities. Carbohydr. Polym. 2022, 275, 118691. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.J.C.; Mourão, P.A.S. Pharmacological activities of sulfated fucose-rich polysaccharides after oral administration: Perspectives for the development of new carbohydrate-based drugs. Mar. Drugs 2021, 19, 425. [Google Scholar] [CrossRef]
- Wijesinghe, W.; Jeon, Y.J. Biological activities and potential industrial applications of fucose rich sulfated polysaccharides and fucoidans isolated from brown seaweeds: A review. Carbohydr. Polym. 2012, 88, 13–20. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, X. A critical review of the abilities, determinants, and possible molecular mechanisms of seaweed polysaccharides antioxidants. Int. J. Mol. Sci. 2020, 21, 7774. [Google Scholar] [CrossRef]
- Dwivedi, R.; Pomin, V.H. Marine antithrombotics. Mar. Drugs 2020, 18, 514. [Google Scholar] [CrossRef]
- Shang, F.; Mou, R.; Zhang, Z.; Gao, N.; Lin, L.; Li, Z.; Wu, M.; Zhao, J. Structural analysis and anticoagulant activities of three highly regular fucan sulfates as novel intrinsic factor Xase inhibitors. Carbohydr. Polym. 2018, 195, 257–266. [Google Scholar] [CrossRef]
- Pereira, M.S.; Mulloy, B.; Mourão, P.A. Structure and anticoagulant activity of sulfated fucans. Comparison between the regular, repetitive, and linear fucans from echinoderms with the more heterogeneous and branched polymers from brown algae. J. Biol. Chem. 1999, 274, 7656–7667. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Hu, Y.; Ye, X.; Li, G.; Yu, G.; Xue, C.; Chai, W. Sequence determination and anticoagulant and antithrombotic activities of a novel sulfated fucan isolated from the sea cucumber Isostichopus badionotus. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 989–1000. [Google Scholar] [CrossRef]
- Yu, L.; Xu, X.; Xue, C.; Chang, Y.; Ge, L.; Wang, Y.; Zhang, C.; Liu, G.; He, C. Enzymatic preparation and structural determination of oligosaccharides derived from sea cucumber (Acaudina molpadioides) fucoidan. Food Chem. 2013, 139, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Pan, R.; Deng, X.; Chen, Y.; Zhao, G.; Wang, C. Separation, purification, anticoagulant activity and preliminary structural characterization of two sulfated polysaccharides from sea cucumber Acaudina molpadioidea and Holothuria nobilis. Process Biochem. 2014, 49, 1352–1361. [Google Scholar] [CrossRef]
- Chang, Y.; Hu, Y.; Yu, L.; McClements, D.J.; Xu, X.; Liu, G.; Xue, C. Primary structure and chain conformation of fucoidan extracted from sea cucumber Holothuria tubulosa. Carbohydr. Polym. 2016, 136, 1091–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Xue, C.; Chang, Y.; Xu, X.; Ge, L.; Liu, G.; Wang, Y. Structure elucidation of fucoidan composed of a novel tetrafucose repeating unit from sea cucumber Thelenota ananas. Food Chem. 2014, 146, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Chen, R.; Mou, R.; Xiang, J.; Zhou, K.; Li, Z.; Zhao, J. Purification, structural characterization and anticoagulant activities of four sulfated polysaccharides from sea cucumber Holothuria fuscopunctata. Int. J. Biol. Macromol. 2020, 164, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yu, L.; Zhang, Y.; Chang, Y.; Liu, Y.; Shen, J.; Xue, C. Utilizing heterologously overexpressed endo-1,3-fucanase to investigate the structure of sulfated fucan from sea cucumber (Holothuria hilla). Carbohydr. Polym. 2021, 272, 118480. [Google Scholar] [CrossRef]
- Pielesz, A.; Biniaś, W.; Paluch, J. Mild acid hydrolysis of fucoidan: Characterization by electrophoresis and FT-Raman spectroscopy. Carbohydr. Res. 2011, 346, 1937–1944. [Google Scholar] [CrossRef]
- Li, X.; Li, S.; Liu, J.; Lin, L.; Sun, H.; Yang, W.; Cai, Y.; Gao, N.; Zhou, L.; Qin, H.; et al. A regular fucan sulfate from Stichopus herrmanni and its peroxide depolymerization: Structure and anticoagulant activity. Carbohydr. Polym. 2021, 256, 117513. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Naumenko, O.I.; Senchenkova, S.N.; Perepelov, A.V. Chemical methods for selective cleavage of glycosidic bonds in the structural analysis of bacterial polysaccharides. Russ. Chem. Rev. 2019, 88, 406–424. [Google Scholar] [CrossRef]
- Koh, H.S.A.; Lu, J.; Zhou, W. Structure characterization and antioxidant activity of fucoidan isolated from Undaria pinnatifida grown in New Zealand. Carbohydr. Polym. 2019, 212, 178–185. [Google Scholar] [CrossRef]
- Pomin, V.H.; Valente, A.P.; Pereira, M.S.; Mourão, P.A.S. Mild acid hydrolysis of sulfated fucans: A selective 2-desulfation reaction and an alternative approach for preparing tailored sulfated oligosaccharides. Glycobiology 2005, 15, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xu, J.; Wang, Y.; Hu, S.; Wang, Y.; Wang, J.; Xue, C. Fucoidan isolated from the sea cucumber Acaudina molpadioides improves insulin resistance in adipocytes via activating PKB/GLUT4 pathway. Eur. Food Res. Technol. 2015, 240, 753–761. [Google Scholar] [CrossRef]
- Bordbar, S.; Anwar, F.; Saari, N. High-value components and bioactives from sea cucumbers for functional foods—A review. Mar. Drugs 2011, 9, 1761–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, P.A.G.; Queiroz, I.N.L.; Santos, G.R.C.; Mourão, P.A.S.; Pomin, V.H. NMR-based conformation and dynamics of a tetrasaccharide-repeating sulfated fucan substituted by different counterions. Biopolymers 2016, 105, 840–851. [Google Scholar] [CrossRef]
- Queiroz, I.N.L.; Wang, X.C.; Glushka, J.N.; Santos, G.R.C.; Valente, A.P.; Prestegard, J.H.; Woods, R.J.; Mourão, P.A.S.; Pomin, V.H. Impact of sulfation pattern on the conformation and dynamics of sulfated fucan oligosaccharides as revealed by NMR and MD. Glycobiology 2015, 25, 535–547. [Google Scholar] [CrossRef] [Green Version]
- An, Z.; Zhang, Z.; Zhang, X.; Yang, H.; Lu, H.; Liu, M.; Shao, Y.; Zhao, X.; Zhang, H. Oligosaccharide mapping analysis by HILIC-ESI-HCD-MS/MS for structural elucidation of fucoidan from sea cucumber Holothuria floridana. Carbohydr. Polym. 2022, 275, 118694. [Google Scholar] [CrossRef] [PubMed]
- Menshova, R.V.; Shevchenko, N.M.; Imbs, T.I.; Zvyagintseva, T.N.; Malyarenko, O.S.; Zaporoshets, T.S.; Besednova, N.N.; Ermakova, S.P. Fucoidans from brown alga Fucus evanescens: Structure and biological activity. Front. Mar. Sci. 2016, 3, 129. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.J.; Yuan, Q.X.; Zhang, Y.X.; Li, J.L.; Zhu, X.J.; Zhao, L.L.; Wen, J.; Liu, J.K.; Zhao, L.Y.; Zhao, J.H. Enzyme-assisted extraction optimization, characterization and antioxidant activity of polysaccharides from sea cucumber Phyllophorus proteus. Molecules 2018, 23, 590. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Wu, M.; Xu, L.; Lian, W.; Xiang, J.; Lu, F.; Gao, N.; Xiao, C.; Wang, S.; Zhao, J. Comparison of physicochemical characteristics and anticoagulant activities of polysaccharides from three sea cucumbers. Mar. Drugs 2013, 11, 399–417. [Google Scholar] [CrossRef] [Green Version]
- Albalasmeh, A.A.; Berhe, A.A.; Ghezzehei, T.A. A new method for rapid determination of carbohydrate and total carbon concentrations using UV spectrophotometry. Carbohydr. Polym. 2013, 97, 253–261. [Google Scholar] [CrossRef]
- Wang, J.; Liu, L.; Zhang, Q.B.; Zhang, Z.S.; Qi, H.M.; Li, P.C. Synthesized oversulphated, acetylated and benzoylated derivatives of fucoidan extracted from Laminaria japonica and their potential antioxidant activity in vitro. Food Chem. 2009, 114, 1285–1290. [Google Scholar] [CrossRef]
- Chen, S.; Huang, H.; Huang, G. Extraction, derivatization and antioxidant activity of cucumber polysaccharide. Int. J. Biol. Macromol. 2019, 140, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zou, G.; Li, C. Purification, characterization and in vitro antioxidant activities of polysaccharide from Chaenomeles speciosa. Int. J. Biol. Macromol. 2016, 92, 702–707. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | Residue | Chemical Shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| FI-a | F: L-Fuc2S-α1- | H | 5.305 (J1,2 = 4.05) | 4.463 | 4.039 | 3.915 | 4.199 | 1.247 |
| C | 100.37 | 77.96 | 69.98 | 74.59 | 70.03 | 17.87 | ||
| rF: -3-L-Fuc-ol | H | 3.742 (J1,1′ = 11.46) | 4.055 | 3.769 | 3.707 | 4.189 | 1.227 | |
| 3.713 (J1,2 = 4.96) | ||||||||
| C | 65.51 | 74.35 | 80.88 | 76.80 | 68.53 | 21.37 | ||
| FI-b | F′: L-Fuc4S-α1- | H | 5.124 (J1,2 = 4.20) | 3.870 | 4.006 | 4.638 | 4.279 | 1.290 |
| C | 101.48 | 70.89 | 71.15 | 83.11 | 69.40 | 18.50 | ||
| rF′: -3-L-Fuc-ol | H | 3.742 (J1,1′ = 11.46) | 4.043 | 3.776 | 3.707 | 4.155 | 1.229 | |
| 3.713 (J1,2 = 4.96) | ||||||||
| C | 65.60 | 74.26 | 80.32 | 76.24 | 68.79 | 21.25 | ||
| FII | I: L-Fuc2S4S-α1- | H | 5.332 (J1,1 = 3.90) | 4.472 | 4.160 | 4.706 | 4.333 | 1.304 |
| C | 100.24 | 77.76 | 69.09 | 83.24 | 69.40 | 18.41 | ||
| rF: -3-L-Fuc-ol | H | 3.739 (J1,1′ = 11.40) | 4.053 | 3.784 | 3.713 | 4.184 | 1.227 | |
| 3.710 (J1,2 = 5.04) | ||||||||
| C | 65.48 | 74.29 | 81.06 | 76.81 | 68.57 | 21.37 | ||
| Fraction | Residue | Chemical Shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| FIII | A: L-Fuc2S4S-α1- | H | 5.256 (J1,2 = 3.84) | 4.348 | 4.086 | 4.575 | 4.357 | 1.159 |
| C | 96.90 | 77.71 | 69.21 | 83.40 | 68.86 | 18.41 | ||
| B: -3-L-Fuc-α1- | H | 5.008 (J1,2 = 4.08) | 3.839 | 3.792 | 3.938 | 4.003 | 1.123 | |
| C | 101.44 | 69.07 | 78.55 | 71.50 | 69.90 | 18.05 | ||
| C: -3-L-Fuc-ol | H | 3.596 (J1,2 = 5.40) (J1,2 = 7.20) | 3.908 | 3.652 | 3.608 | 4.019 | 1.108 | |
| C | 65.57 | 74.33 | 80.16 | 76.16 | 69.00 | 21.09 | ||
| FIV-a | AI: L-Fuc4S-α1- | H | 5.011 (J1,2 = 3.90) | 3.641 | 3.896 | 4.504 | 4.462 | 1.170 |
| C | 100.37 | 71.17 | 71.53 | 83.52 | 69.41 | 18.37 | ||
| BI: -3-L-Fuc2S4S-α1- | H | 5.232 (J1,2 = 3.48) | 4.438 | 4.127 | 4.736 | 4.195 | 1.164 | |
| C | 100.46 | 75.72 | 75.07 | 81.74 | 69.63 | 18.62 | ||
| CI: -3-L-Fuc-ol | H | 3.622 (J1,1′ = 11.48) (J1,2 = 4.62) | 3.931 | 3.678 | 3.584 | 4.105 | 1.096 | |
| C | 65.39 | 74.22 | 81.28 | 76.71 | 68.52 | 21.35 | ||
| FIV-b | AII: L-Fuc2S4S-α1- | H | 5.226 (J1,2 = 3.82) | 4.352 | 4.089 | 4.578 | 4.458 | 1.167 |
| C | 97.18 | 77.62 | 69.03 | 83.48 | 69.11 | 18.47 | ||
| BII: -3-L-Fuc2S-α1- | H | 5.192 (J1,2 = 4.02) | 4.456 | 3.966 | 3.988 | 4.062 | 1.137 | |
| C | 100.31 | 74.41 | 75.99 | 71.66 | 69.83 | 17.94 | ||
| CII: -3-L-Fuc-ol | H | 3.612 (J1,1′ = 11.48) (J1,2 = 4.62) | 3.943 | 3.661 | 3.602 | 4.100 | 1.093 | |
| C | 65.42 | 74.48 | 81.02 | 76.64 | 68.46 | 21.34 | ||
| Fraction | Residue | Chemical Shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| FV | A: L-Fuc2S4S-α1- | H | 5.306 (J1,2 = 3.84) | 4.378 | 4.129 | 4.596 | 4.353 | 1.188 |
| C | 96.32 | 77.87 | 69.31 | 83.57 | 68.95 | 18.50 | ||
| B: -3-L-Fuc-α1- | H | 4.968 (J1,2 = 4.00) | 3.832 | 3.892 | 3.968 | 4.336 | 1.147 | |
| C | 98.99 | 69.36 | 78.07 | 70.99 | 69.54 | 18.12 | ||
| C: -3-L-Fuc2S -α1- | H | 5.223 (J1,2 = 3.92) | 4.466 | 3.997 | 4.014 | 4.105 | 1.166 | |
| C | 100.58 | 75.87 | 75.30 | 71.86 | 69.89 | 18.25 | ||
| D: -3-L-Fuc-ol | H | 3.660 (J1, 1′ = 11.60) 3.636 (J1,2 = 4.88) | 3.954 | 3.699 | 3.620 | 4.115 | 1.133 | |
| C | 65.61 | 74.53 | 81.25 | 76.86 | 68.71 | 21.49 | ||
| FVI | A: L-Fuc2S4S-α1- | H | 5.245 (J1,2 = 3.90) | 4.335 | 4.083 | 4.578 | 4.444 | 1.174 |
| C | 97.21 | 77.65 | 69.16 | 83.58 | 69.01 | 18.47 | ||
| B: -3-L-Fuc2S-α1- | H | 5.206 (J1,2 = 3.96) | 4.427 | 3.999 | 3.980 | 4.344 | 1.133 | |
| C | 97.92 | 75.74 | 76.26 | 71.68 | 69.37 | 18.02 | ||
| C: -3-L-Fuc2S-α1- | H | 5.208 (J1,2 = 3.92) | 4.464 | 3.996 | 3.953 | 4.068 | 1.143 | |
| C | 100.34 | 75.81 | 76.58 | 71.94 | 69.83 | 17.96 | ||
| D: -3-L-Fuc-ol | H | 3.633 (J1,1′ = 11.52) 3.603 (J1,2 = 7.44) | 3.923 | 3.669 | 3.602 | 4.079 | 1.105 | |
| C | 65.44 | 74.43 | 80.92 | 76.77 | 68.63 | 21.34 | ||
| Sample | Actual Peak | Charge | Negative Ion | Predicted Peak | |
|---|---|---|---|---|---|
| Structure | Formula | ||||
| FI | 413.0748 | 1- | [M-H]− | C12H22NaO12S− | 413.0730 |
| FII | 493.0324 | 1- | [M-Na]− | C12H22NaO15S2− | 493.0298 |
| 471.0502 | 1- | [M-2Na+H]− | C12H23O15S2− | 471.0478 | |
| 391.0933 | 1- | [M-SO3-2Na+H]− | C12H23O12S− | 391.0910 | |
| FIII | 537.1515 | 1- | [M-SO3-2Na+H]− | C18H33O16S− | 537.1489 |
| 617.1092 | 1- | [M-2Na+H]− | C18H33O19S2− | 617.1057 | |
| 639.0910 | 1- | [M-Na]− | C18H32NaO19S2− | 639.0877 | |
| FIV | 741.0282 | 1- | [M-Na]− | C18H31Na2O22S3− | 741.0264 |
| 719.0463 | 1- | [M-2Na+H]− | C18H32NaO22S3− | 719.0445 | |
| 639.0890 | 1- | [M-SO3-2Na+H]− | C18H32NaO19S2− | 639.0877 | |
| 617.1069 | 1- | [M-SO3-3Na+2H]− | C18H33O19S2− | 617.1057 | |
| FV | 432.0491 | 2- | [M-2Na]2− | C24H41NaO26S32− | 432.0473 |
| 785.1463 | 1- | [M-SO3-2Na+H]− | C24H42NaO23S2− | 785.1456 | |
| 887.0854 | 1- | [M-Na]− | C24H41Na2O26S3− | 887.0844 | |
| FVI | 1011.1652 | 1- | [M-H]− | C24H39Na4O29S4− | 1011.0051 |
| 989.1834 | 1- | [M-Na]− | C24H40Na3O29S4− | 989.0231 | |
| 967.0461 | 1- | [M-2Na+H]− | C24H41Na2O29S4− | 967.0412 | |
| 887.0888 | 1- | [M-SO3-2Na+H]− | C24H41Na2O26S3− | 887.0844 | |
| 865.1067 | 1- | [M-SO3-3Na+2H]− | C24H42NaO26S3− | 865.1024 | |
| 843.1247 | 1- | [M-SO3-4Na+3H]− | C24H43O26S3− | 843.1205 | |
Publisher′s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, L.; Xu, C.; Tao, X.; Zuo, Z.; Ning, Z.; Wang, L.; Gao, N.; Zhao, J. Structure Elucidation of Fucan Sulfate from Sea Cucumber Holothuria fuscopunctata through a Bottom-Up Strategy and the Antioxidant Activity Analysis. Int. J. Mol. Sci. 2022, 23, 4488. https://doi.org/10.3390/ijms23094488
Gao L, Xu C, Tao X, Zuo Z, Ning Z, Wang L, Gao N, Zhao J. Structure Elucidation of Fucan Sulfate from Sea Cucumber Holothuria fuscopunctata through a Bottom-Up Strategy and the Antioxidant Activity Analysis. International Journal of Molecular Sciences. 2022; 23(9):4488. https://doi.org/10.3390/ijms23094488
Chicago/Turabian StyleGao, Li, Chen Xu, Xuelin Tao, Zhichuang Zuo, Zimo Ning, Linghui Wang, Na Gao, and Jinhua Zhao. 2022. "Structure Elucidation of Fucan Sulfate from Sea Cucumber Holothuria fuscopunctata through a Bottom-Up Strategy and the Antioxidant Activity Analysis" International Journal of Molecular Sciences 23, no. 9: 4488. https://doi.org/10.3390/ijms23094488