
Bioinformatics Screening of Potential Biomarkers from mRNA Expression Profiles to Discover Drug Targets and Agents for Cervical Cancer

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Identification of cDEGs
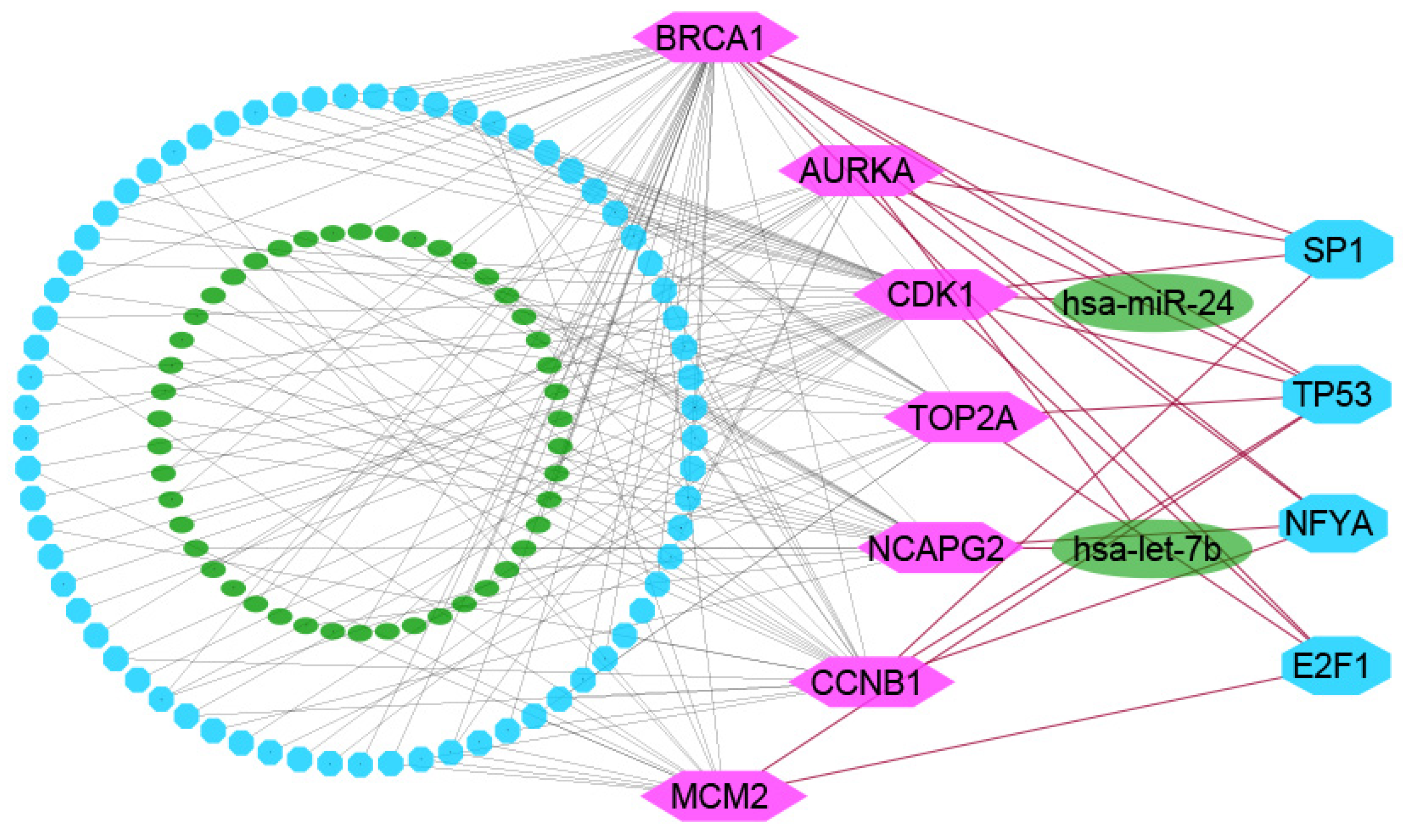
2.2. PPI Network Analysis of cDEGs for Identification of KGs
2.3. The Regulatory Network Analysis of KGs
2.4. GO Functions and KEGG Pathway Enrichment Analysis of cDEGs Highlighting KGs
2.5. Survival Analysis with KGs
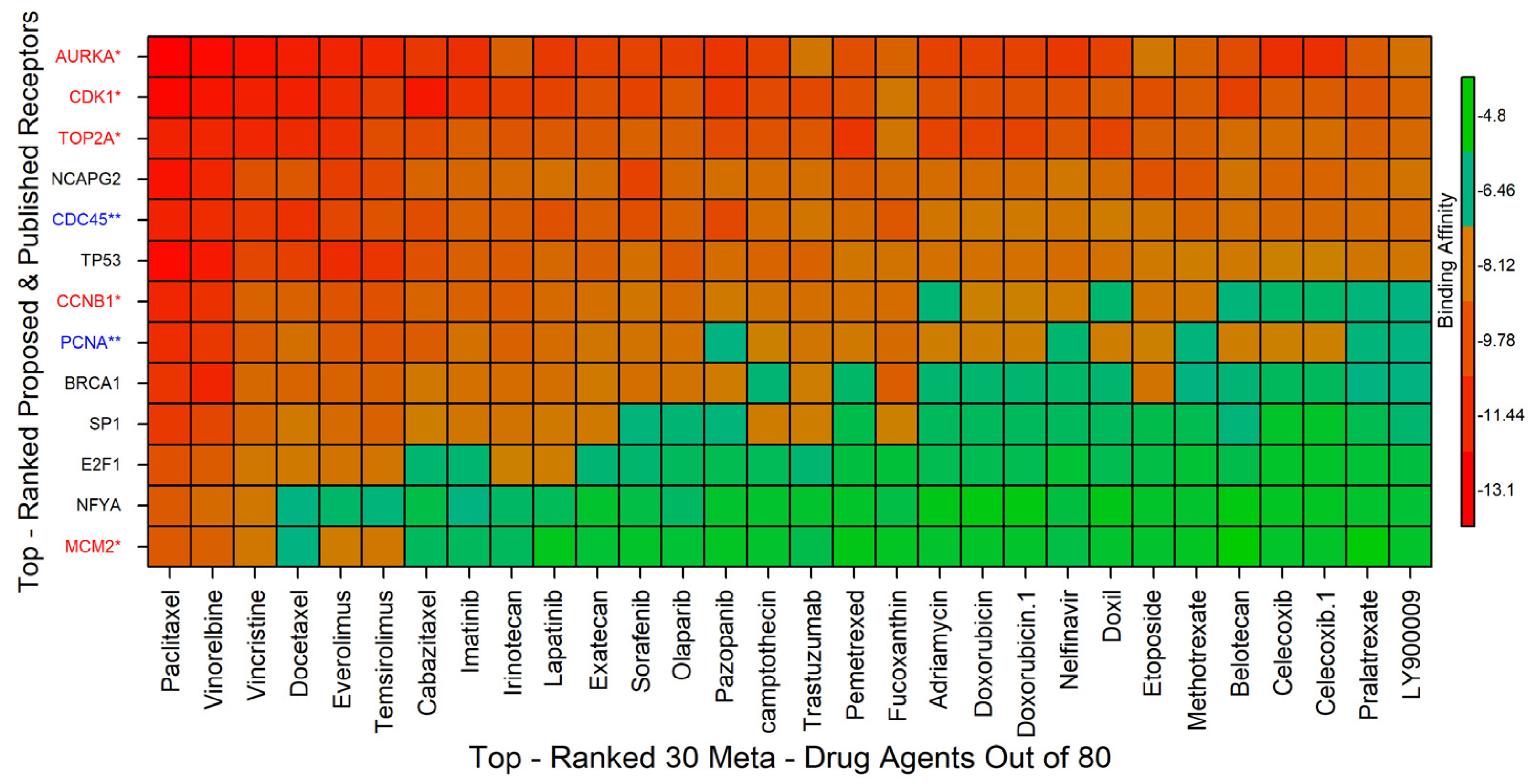
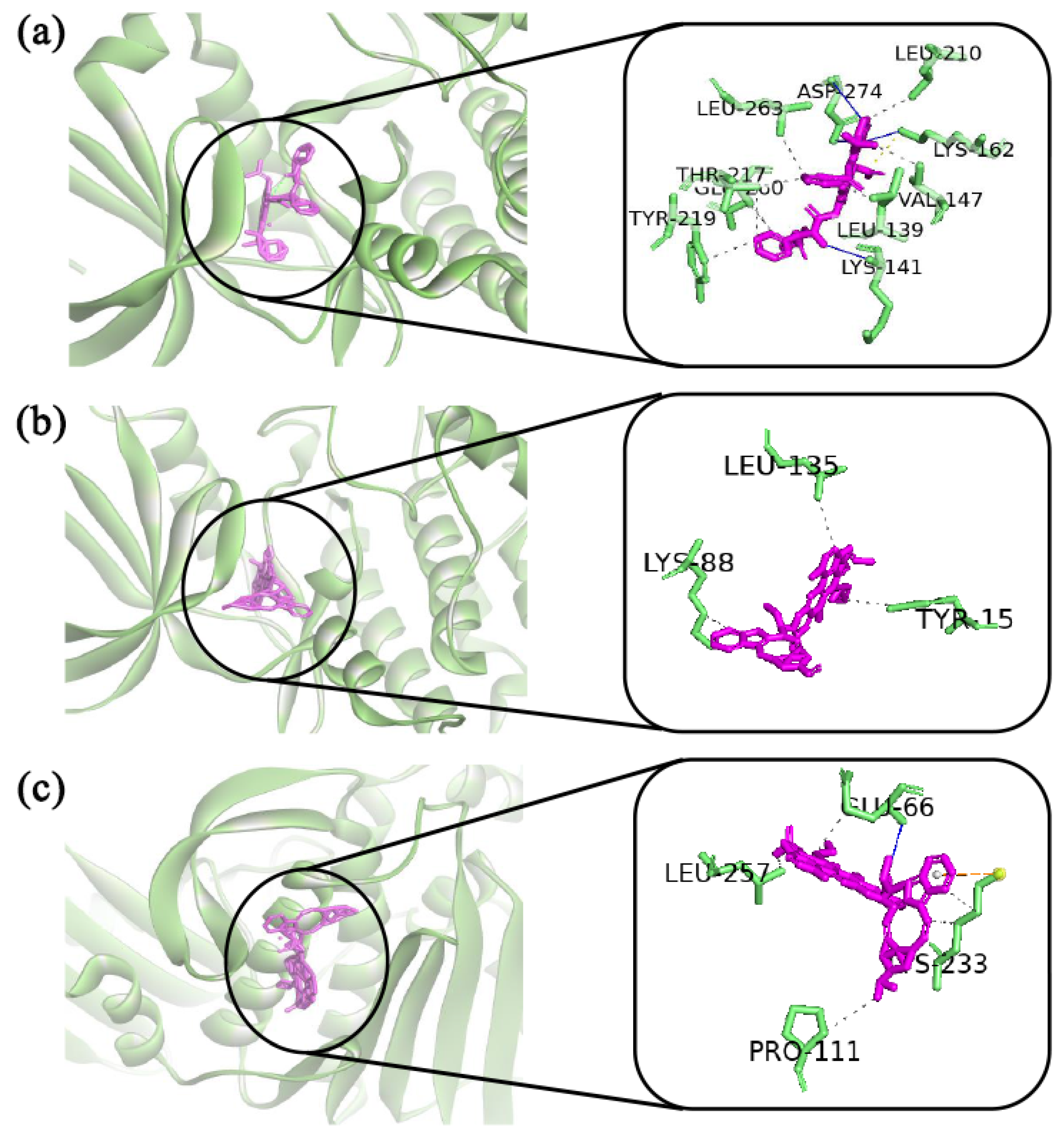
2.6. Drug Repurposing by Molecular Docking
2.7. MD Simulations
3. Discussion
4. Materials and Methods
4.1. Data Sources and Descriptions
4.2. Collection of Microarray Exploring Profiles for Genomic Biomarkers and Drug Target Receptors
4.3. Collection of Meta-Drug Agents for Exploring Candidate Drugs
4.4. Collection of Independent Meta-Receptors for Cross-Validation with the Proposed Drugs
4.5. Identification of cDEGs for CC Patients
4.6. Construction of Protein–Protein Interaction (PPI) Network for Identification of KGs
4.7. Regulatory Network Analysis of KGs
4.8. GO Terms and KEGG Pathway Enrichment Analysis of KGs
4.9. Survival Analysis
4.10. Drug Repurposing by Molecular Docking Study
4.11. Molecular Dynamic (MD) Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CC | Cervical Cancer |
| CSCC | Cervical Squamous Cell Carcinoma |
| HPV | Human Papillomavirus |
| (MLICs) | Middle- and Low-Income Countries |
| LIMMA | Linear Models for Microarray Data |
| PPI | Protein–Protein Interaction |
| ENCODE | Encyclopedia Of DNA Elements |
| MCODE | Molecular Complex Detection |
| DEGs | Differentially Expressed Genes |
| cDEGs | Common Differentially Expressed Genes |
| cHubGs | Common Hub Genes |
| cHubPs | Common Hub Proteins |
| KGs | Key Genes |
| KPs | Key Proteins |
| DR | Drug Repurposing |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| TFs | Transcription Factors |
| miRNAs | Micro-RNAs |
| MD | Molecular Dynamic |
| MM-PBSA | Molecular Mechanics Poisson–Boltzmann Surface Area |
| RMSD | Root Mean Square Deviation |
| 3D | Three-Dimensional |
| PDB | Protein Data Bank |
| PLIP | Protein–Ligand Interaction Profiler |
| YASARA | Yet Another Scientific Artificial Reality Application |
References
- Urasa, M.; Darj, E. Knowledge of cervical cancer and screening practices of nurses at a regional hospital in Tanzania. Afr. Health Sci. 2011, 11, 48–57. [Google Scholar] [PubMed]
- Small, W.; Bacon, M.A.; Bajaj, A.; Chuang, L.T.; Fisher, B.J.; Harkenrider, M.M.; Jhingran, A.; Kitchener, H.C.; Mileshkin, L.R.; Viswanathan, A.N.; et al. Cervical cancer: A global health crisis. Cancer 2017, 123, 2404–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- Bruni, L.; Alberto, G.; Serrano, B.; Mena, M.; Gómez, D.; Muñoz, J.; Fx, B.; De, S.S. ICO/IARC Information Centre on HPV and Cancer (HPV Information Centre). Human Papillomavirus and Related Diseases in India. Summ. Rep. 2018, 27. Available online: http://www.hpvcentre.net/statistics/reports/XEX.pdf (accessed on 12 December 2021).
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Canfell, K.; Kim, J.J.; Brisson, M.; Keane, A.; Simms, K.T.; Caruana, M.; Burger, E.A.; Martin, D.; Nguyen, D.T.N.; Bénard, É.; et al. Mortality impact of achieving WHO cervical cancer elimination targets: A comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet 2020, 395, 591–603. [Google Scholar] [CrossRef] [Green Version]
- Vaccarella, S.; Laversanne, M.; Ferlay, J.; Bray, F. Cervical cancer in Africa, Latin America and the Caribbean and Asia: Regional inequalities and changing trends. Int. J. Cancer 2017, 141, 1997–2001. [Google Scholar] [CrossRef] [Green Version]
- TCGA Integrated genomic and molecular characterization of cervical cancer The Cancer Genome Atlas Research Network. Nature 2017, 543, 378–384. [CrossRef]
- He, H.; Liu, X.; Liu, Y.; Zhang, M.; Lai, Y.; Hao, Y.; Wang, Q.; Shi, D.; Wang, N.; Luo, X.-G.; et al. Human Papillomavirus E6/E7 and Long Noncoding RNA TMPOP2 Mutually Upregulated Gene Expression in Cervical Cancer Cells. J. Virol. 2019, 93, e01808-18. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, T.; Lin, Z.; Gao, Y. Aberrant methylation of MEG3 functions as a potential plasma-based biomarker for cervical cancer. Sci. Rep. 2017, 7, 6271. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.H.; Qin, Y.E.; Tang, W.F.; Tao, J.; Song, H.M.; Zuo, M. MiR-34a and miR-206 act as novel prognostic and therapy biomarkers in cervical cancer. Cancer Cell Int. 2017, 17, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudrapal, M.; Khairnar, S.; Jadhav, A. Drug Repurposing (DR): An Emerging Approach in Drug Discovery. In Drug Repurposing—Hypothesis, Molecular Aspects and Therapeutic Applications; 2020; Available online: https://www.intechopen.com/chapters/72744 (accessed on 10 March 2022).
- Venkataramnan, S.; Binti Zainol Izam Khan, W.N. Cervical Cancer and Gene Expression Analysis with Key Genes Identification by Computational Method. J. Bio Innov. 2020, 9, 868–878. [Google Scholar] [CrossRef]
- Qiu, H.Z.; Huang, J.; Xiang, C.C.; Li, R.; Zuo, E.D.; Zhang, Y.; Shan, L.; Cheng, X. Screening and Discovery of New Potential Biomarkers and Small Molecule Drugs for Cervical Cancer: A Bioinformatics Analysis. Technol. Cancer Res. Treat. 2020, 19, 1533033820980112. [Google Scholar] [CrossRef]
- Wu, X.; Peng, L.; Zhang, Y.; Chen, S.; Lei, Q.; Li, G.; Zhang, C. Identification of key genes and pathways in cervical cancer by bioinformatics analysis. Int. J. Med. Sci. 2019, 16, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.; Fang, Y.; Wu, K.; Liu, Y.; Zhang, W. Comprehensive gene and pathway analysis of cervical cancer progression. Oncol. Lett. 2020, 19, 3316–3332. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.J.; Xue, J.-M.; Li, J.; Wan, L.-H.; Zhu, Y. xi Identification of key genes and pathways of diagnosis and prognosis in cervical cancer by bioinformatics analysis. Mol. Genet. Genomic Med. 2020, 8, e1200. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zheng, H.; Han, Y.; Wang, G.; Li, Y. A Novel Four-Gene Prognostic Signature as a Risk Biomarker in Cervical Cancer. Int. J. Genom. 2020, 2020, 4535820. [Google Scholar] [CrossRef]
- Xue, H.; Sun, Z.; Wu, W.; Du, D.; Liao, S. Identification of hub genes as potential prognostic biomarkers in cervical cancer using comprehensive bioinformatics analysis and validation studies. Cancer Manag. Res. 2021, 13, 117–131. [Google Scholar] [CrossRef]
- Samieefar, N.; Yari Boroujeni, R.; Jamee, M.; Lotfi, M.; Golabchi, M.R.; Afshar, A.; Miri, H.; Khazeei Tabari, M.A.; Darzi, P.; Abdullatif Khafaie, M.; et al. Country Quarantine during COVID-19: Critical or Not? Disaster Med. Public Health Prep. 2020, 15, e24–e25. [Google Scholar] [CrossRef]
- Wu, B.; Xi, S. Bioinformatics analysis of differentially expressed genes and pathways in the development of cervical cancer. BMC Cancer 2021, 21, 733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bai, J.; Yuan, C.; Long, L.; Zheng, Z.; Wang, Q.; Chen, F.; Zhou, Y. Bioinformatics analysis and identification of potential genes related to pathogenesis of cervical intraepithelial neoplasia. J. Cancer 2020, 11, 2150–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Han, L.; Cao, R.; Wang, H.; Jiang, J.; Deng, Y.; Yu, X. Prediction of a miRNA-mRNA functional synergistic network for cervical squamous cell carcinoma. FEBS Open Bio 2019, 9, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, H.; Zhu, L.; Hu, S.; Xi, X.; Liu, Y.; Liu, J.; Zhong, T. Bioinformatics analysis shows that top2a functions as a key candidate gene in the progression of cervical cancer. Biomed. Rep. 2020, 13, 21. [Google Scholar] [CrossRef]
- Xu, Z.; Zhou, Y.; Shi, F.; Cao, Y.; Dinh, T.L.A.; Wan, J.; Zhao, M. Investigation of differentially-expressed microRNAs and genes in cervical cancer using an integrated bioinformatics analysis. Oncol. Lett. 2017, 13, 2784–2790. [Google Scholar] [CrossRef]
- Mei, Y.; Jiang, P.; Shen, N.; Fu, S.; Zhang, J. Identification of miRNA-mRNA Regulatory Network and Construction of Prognostic Signature in Cervical Cancer. DNA Cell Biol. 2020, 39, 1023–1040. [Google Scholar] [CrossRef]
- Wei, J.; Wang, Y.; Shi, K.; Wang, Y. Identification of Core Prognosis-Related Candidate Genes in Cervical Cancer via Integrated Bioinformatical Analysis. Biomed Res. Int. 2020, 2020, 8959210. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, Q.; Qiao, L.; Fan, X.; Zhang, W.; Zhao, W.; Chen, J.J. Cdc6 contributes to abrogating the G1 checkpoint under hypoxic conditions in HPV E7 expressing cells. Sci. Rep. 2017, 7, 1629–1641. [Google Scholar] [CrossRef]
- Chen, Z.; Ling, K.; Zhu, Y.; Deng, L.; Li, Y.; Liang, Z. Rucaparib antagonize multidrug resistance in cervical cancer cells through blocking the function of ABC transporters. Gene 2020, 759, 145000. [Google Scholar] [CrossRef]
- Arruabarrena-Aristorena, A.; Maag, J.L.V.; Kittane, S.; Cai, Y.; Karthaus, W.R.; Ladewig, E.; Park, J.; Kannan, S.; Ferrando, L.; Cocco, E.; et al. FOXA1 Mutations Reveal Distinct Chromatin Profiles and Influence Therapeutic Response in Breast Cancer. Cancer Cell 2020, 38, 534–550.e9. [Google Scholar] [CrossRef]
- Murugesan, M.; Premkumar, K. Integrative miRNA-mRNA functional analysis identifies miR-182 as a potential prognostic biomarker in breast cancer. Mol. Omi. 2021, 17, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, L.; Bassi, Z.I.; Geymonat, M.; Todesca, S.; Copoiu, L.; Enright, A.J.; Callaini, G.; Riparbelli, M.G.; Yu, L.; Choudhary, J.S.; et al. The midbody interactome reveals unexpected roles for PP1 phosphatases in cytokinesis. Nat. Commun. 2019, 10, 4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear erk: Mechanism of translocation, substrates, and role in cancer. Int. J. Mol. Sci. 2019, 20, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, M. The MCM Complex: Its Role in DNA Replication and Implications for Cancer Therapy. Curr. Cancer Drug Targets 2005, 5, 365–380. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- The Uniprot Consortium. UniProt: A worldwide hub of protein knowledge The UniProt Consortium. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yi, Y.; Wu, W.; Wu, K.; Zhang, W. Bioinformatics prediction and analysis of hub genes and pathways of three types of gynecological cancer. Oncol. Lett. 2019, 18, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.M.; Liu, Y.; Wan, L.H.; Zhu, Y.X. Comprehensive analysis of differential gene expression to identify common gene signatures in multiple cancers. Med. Sci. Monit. 2020, 26, e919953-1. [Google Scholar] [CrossRef]
- Wang, M.; Li, L.; Liu, J.; Wang, J. A gene interaction network-based method to measure the common and heterogeneous mechanisms of gynecological cancer. Mol. Med. Rep. 2018, 18, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Shi, X.; Li, B.; Peng, M.; Zhu, T.; Lv, G.; Liu, L.; Jin, H.; Li, L.; Qin, D. Integrated analysis of key microRNAs /TFs /mRNAs/ in HPV-positive cervical cancer based on microRNA sequencing and bioinformatics analysis. Pathol. Res. Pract. 2020, 216, 152952. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.Z.; Poortahmasebi, V.; Mokhtari-Azad, T.; Shahmahmoodi, S.; Farahmand, M.; Farzanehpour, M.; Jalilvand, S. The dysregulation of microarray gene expression in cervical cancer is associated with overexpression of a unique messenger rna signature. Iran. J. Microbiol. 2020, 12, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Mishra, A. Network analysis revealed aurora kinase dysregulation in five gynecological types of cancer. Oncol. Lett. 2018, 15, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Li, Y.; Zhao, Y.; Chang, J.; Zhang, X.; Zou, B.; Gao, L.; Wang, W. Comprehensive Analysis of circRNA Expression Profiles During Cervical Carcinogenesis. Front. Oncol. 2021, 11, 676609. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yi, Y.; Liu, F.; Wu, W.; Chen, Y.; Zhang, W. Identification of key pathways and genes in the progression of cervical cancer using bioinformatics analysis. Oncol. Lett. 2018, 16, 1003–1009. [Google Scholar] [CrossRef]
- He, Z.; Wang, X.; Yang, Z.; Jiang, Y.; Li, L.; Wang, X.; Song, Z.; Wang, X.; Wan, J.; Jiang, S.; et al. Expression and prognosis of CDC45 in cervical cancer based on the GEO database. PeerJ 2021, 9, e12114. [Google Scholar] [CrossRef]
- Wu, B.; Xi, S. Bioinformatics analysis of the transcriptional expression of minichromosome maintenance proteins as potential indicators of survival in patients with cervical cancer. BMC Cancer 2021, 21, 928. [Google Scholar] [CrossRef]
- Wen, X.; Liu, S.; Cui, M. Effect of BRCA1 on the Concurrent Chemoradiotherapy Resistance of Cervical Squamous Cell Carcinoma Based on Transcriptome Sequencing Analysis. Biomed Res. Int. 2020, 2020, 3598417. [Google Scholar] [CrossRef]
- Li, S.; Liu, N.; Piao, J.; Meng, F.; Li, Y. Ccnb1 expedites the progression of cervical squamous cell carcinoma via the regulation by foxm1. Onco. Targets Ther. 2020, 13, 12383–12395. [Google Scholar] [CrossRef]
- Deng, Y.R.; Chen, X.J.; Chen, W.; Wu, L.F.; Jiang, H.P.; Lin, D.; Wang, L.J.; Wang, W.; Guo, S.Q. Sp1 contributes to radioresistance of cervical cancer through targeting g2/m cell cycle checkpoint CDK1. Cancer Manag. Res. 2019, 11, 5835–5844. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Tiwari, D.; Dongre, A.; Mustafa, S.S.; Das, C.R.; Massey, S.; Bose, P.D.; Bose, S.; Husain, S.A. Exploring the p53 connection of cervical cancer pathogenesis involving north-east Indian patients. PLoS ONE 2020, 15, e0238500. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.T.; Feng, Q.; Ma, H.M.; Lei, D.; Zheng, P.S. NF-YA promotes the cell proliferation and tumorigenic properties by transcriptional activation of SOX2 in cervical cancer. J. Cell. Mol. Med. 2020, 24, 12464–12475. [Google Scholar] [CrossRef]
- Wang, X.; Gao, P.; Wang, M.; Liu, J.; Lin, J.; Zhang, S.; Zhao, Y.; Zhang, J.; Pan, W.; Sun, Z.; et al. Feedback between E2F1 and CIP2A regulated by human papillomavirus E7 in cervical cancer: Implications for prognosis. Am. J. Transl. Res. 2017, 9, 2327–2339. [Google Scholar] [PubMed]
- Meneur, C.; Eswaran, S.; Adiga, D.; Sriharikrishnaa, S.; Nadeem, K.G.; Mallya, S.; Chakrabarty, S.; Kabekkodu, S.P. Analysis of Nuclear Encoded Mitochondrial Gene Networks in Cervical Cancer. Asian Pacific J. Cancer Prev. 2021, 22, 1799–1811. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Ushijima, K. Chemotherapy for advanced or recurrent cervical cancer. Taiwan. J. Obstet. Gynecol. 2013, 52, 161–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, X.; Hu, W.; Ramirez, P.T.; Kavanagh, J.J. Chemotherapy for recurrent and metastatic cervical cancer. Gynecol. Oncol. 2008, 110, S67–S71. [Google Scholar] [CrossRef]
- Barra, F.; Lorusso, D.; Leone Roberti Maggiore, U.; Ditto, A.; Bogani, G.; Raspagliesi, F.; Ferrero, S. Investigational drugs for the treatment of cervical cancer. Expert Opin. Investig. Drugs 2017, 26, 389–402. [Google Scholar] [CrossRef]
- Sharma, S.; Deep, A.; Sharma, A.K. Current Treatment for Cervical Cancer: An Update. Anticancer Agents Med. Chem. 2020, 20, 1768–1779. [Google Scholar] [CrossRef]
- Blatt, J.M.; Weisskopf, V.F.; Critchfield, C.L. Theoretical Nuclear Physics. Am. J. Phys. 1953, 21, 235–236. [Google Scholar] [CrossRef]
- Lovering, A.L.; Seung, S.L.; Kim, Y.W.; Withers, S.G.; Strynadka, N.C.J. Mechanistic and structural analysis of a family 31 α-glycosidase and its glycosyl-enzyme intermediate. J. Biol. Chem. 2005, 280, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Pyeon, D.; Newton, M.A.; Lambert, P.F.; Den Boon, J.A.; Sengupta, S.; Marsit, C.J.; Woodworth, C.D.; Connor, J.P.; Haugen, T.H.; Smith, E.M.; et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007, 67, 4605–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffarel, M.M.; Chattopadhyay, A.; Araujo, A.M.; Bauer, J.; Scarpini, C.G.; Coleman, N. Tissue transglutaminase mediates the pro-malignant effects of oncostatin M receptor over-expression in cervical squamous cell carcinoma. J. Pathol. 2013, 231, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Arias-Pulido, H.; Subramaniyam, S.; Schneider, A.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: Potential role in progression. Genes Chromosom. Cancer 2008, 47, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Mason, S.P.; Barabási, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [Green Version]
- Pržulj, N.; Wigle, D.A.; Jurisica, I. Functional topology in a network of protein interactions. Bioinformatics 2004, 20, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.C. A Set of Measures of Centrality Based on Betweenness. Sociometry 1977, 40, 39–54. [Google Scholar] [CrossRef]
- Shimbel, A. Structural parameters of communication networks. Bull. Math. Biophys. 1953, 15, 501–507. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Feingold, E.A.; Good, P.J.; Guyer, M.S.; Kamholz, S.; Liefer, L.; Wetterstrand, K.; Collins, F.S.; Gingeras, T.R.; Kampa, D.; Sekinger, E.A.; et al. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar]
- Liu, Z.P.; Wu, C.; Miao, H.; Wu, H. RegNetwork: An integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, 2015, bav095. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO:: TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Doms, A.; Schroeder, M. GoPubMed: Exploring PubMed with the gene ontology. Nucleic Acids Res. 2005, 33, W783–W786. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An Online Biomarker Validation Tool and Database for Cancer Gene Expression Data Using Survival Analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visualizer. Accelrys Software Inc. D Sv4. Vol. 0(100). 13345; Accelrys Software Inc.: San Diego, CA, USA, 2005. [Google Scholar]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Oleg, T.; Arthur, J.O. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Delano, W.L.; Bromberg, S. PyMOL User’s Guide; 2004. Available online: http://pymol.sourceforge.net/newman/userman.pdf (accessed on 10 March 2022).
- Krieger, E.; Vriend, G.; Spronk, C. YASARA—Yet Another Scientific Artificial Reality Application. YASARA org 2013, 993, 51–78. [Google Scholar]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided. Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Genet. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Dash, R. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP bromodomain inhibitors. J. Mol. Graph. Model. 2018, 83, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Rajasekaran, R. Computational investigation of curcumin, a natural polyphenol that inhibits the destabilization and the aggregation of human SOD1 mutant (Ala4Val). RSC Adv. 2016, 6, 102744–102753. [Google Scholar] [CrossRef]
- Marquina, G.; Manzano, A.; Casado, A. Targeted Agents in Cervical Cancer: Beyond Bevacizumab. Curr. Oncol. Rep. 2018, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verschraegen, C.F. Irinotecan for the treatment of cervical cancer. Oncology 2002, 16, 32–34. [Google Scholar] [PubMed]
- Su, J.; Zhang, F.; Li, X.; Liu, Z. Osthole promotes the suppressive effects of cisplatin on NRF2 expression to prevent drug-resistant cervical cancer progression. Biochem. Biophys. Res. Commun. 2019, 514, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, S.; Beckmann, M.W.; Thiel, F.; Bogenrieder, T. Topotecan in cervical cancer. Int. J. Gynecol. Cancer. 2007, 17, 6. [Google Scholar] [CrossRef]
- Chandimali, N.; Sun, H.N.; Park, Y.H.; Kwon, T. BRM270 suppresses cervical cancer stem cell characteristics and progression by inhibiting SOX2. In Vivo 2020, 34, 1085–1094. [Google Scholar] [CrossRef]
- Markman, M. Advances in cervical cancer pharmacotherapies. Expert Rev. Clin. Pharmacol. 2014, 7, 219–223. [Google Scholar] [CrossRef]
- Moga, M.A.; Dima, L.; Balan, A.; Blidaru, A.; Dimienescu, O.G.; Podasca, C.; Toma, S. Are bioactive molecules from seaweeds a novel and challenging option for the prevention of HPV infection and cervical cancer therapy?—a review. Int. J. Mol. Sci. 2021, 22, 629. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, Y.M.; Kim, M.B.; Kim, D.Y.; Kim, J.H.; Nam, J.H.; Kim, Y.T. Chemosensitivity of uterine cervical cancer demonstrated by the histoculture drug response assay. Tohoku J. Exp. Med. 2009, 219, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Small, W. Potential for use of amifostine in cervical cancer. Semin. Oncol. 2002, 29, 34–37. [Google Scholar] [CrossRef]
- Ai, Z.; Wang, J.; Xu, Y.; Teng, Y. Bioinformatics analysis reveals potential candidate drugs for cervical cancer. J. Obstet. Gynaecol. Res. 2013, 39, 1052–1058. [Google Scholar] [CrossRef]
- Ujhelyi, Z.; Kalantari, A.; Vecsernyés, M.; Róka, E.; Fenyvesi, F.; Póka, R.; Kozma, B.; Bácskay, I. The enhanced inhibitory effect of different antitumor agents in self-microemulsifying drug delivery systems on human cervical cancer HeLa cells. Molecules 2015, 20, 13226–13239. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Gonzalez, A.; Gonzalez-Fierro, A. Pharmacodynamics of current and emerging treatments for cervical cancer. Expert Opin. Drug Metab. Toxicol. 2019, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nie, S.; Gao, M.; Jiang, Y.; Wan, Y.; Ma, X.; Zhou, S.; Cheng, W. Identification of EPHX2 and RMI2 as two novel key genes in cervical squamous cell carcinoma by an integrated bioinformatic analysis. J. Cell. Physiol. 2019, 234, 21260–21273. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Olvera, A.; Cetina, L.; Coronel, J.; Dueñas-González, A. Emerging drugs for the treatment of cervical cancer. Expert Opin. Emerg. Drugs 2015, 20, 165–182. [Google Scholar] [CrossRef]
- Klopp, A.H.; Eifel, P.J. Chemoradiotherapy for cervical cancer in 2010. Curr. Oncol. Rep. 2011, 13, 77–85. [Google Scholar] [CrossRef]
- Diaz-Padilla, I.; Monk, B.J.; Mackay, H.J.; Oaknin, A. Treatment of metastatic cervical cancer: Future directions involving targeted agents. Crit. Rev. Oncol. Hematol. 2013, 85, 303–314. [Google Scholar] [CrossRef]
- Tierney, J.F.; Vale, C.; Symonds, P. Concomitant and Neoadjuvant Chemotherapy for Cervical Cancer. Clin. Oncol. 2008, 20, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Xing, Y.; Lv, J.; Gu, D.; Pan, J.; Zhang, Y.; Liu, J. Construction of an immune-related gene signature for prediction of prognosis in patients with cervical cancer. Int. Immunopharmacol. 2020, 88, 106882. [Google Scholar] [CrossRef]
- Deng, S.P.; Zhu, L.; Huang, D.S. Predicting Hub Genes Associated with Cervical Cancer through Gene CoExpression Networks. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2016, 13, 27–35. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.; Gao, F.; Li, S.; Nie, S.; Meng, H.; Sun, R.; Wan, Y.; Jiang, Y.; Ma, X.; et al. A microRNAMessenger RNA Regulatory Network and Its Prognostic Value in Cervical Cancer. DNA Cell Biol. 2020, 39, 1328–1346. [Google Scholar] [CrossRef]
- Ouyang, D.; Ouyang, D.; Yang, P.; Cai, J.; Sun, S.; Wang, Z. Comprehensive analysis of prognostic alternative splicing signature in cervical cancer. Cancer Cell Int. 2020, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, X.; Jia, H.; Tao, Y.; Zhou, H.; Wang, M.; Wang, X.; Fang, X. Bioinformatics analysis of key genes and pathways of cervical cancer. Onco. Targets. Ther. 2020, 13, 13275–13283. [Google Scholar] [CrossRef]
- Ma, X.; Liu, J.; Wang, H.; Jiang, Y.; Wan, Y.; Xia, Y.; Cheng, W. Identification of crucial aberrantly methylated and differentially expressed genes related to cervical cancer using an integrated bioinformatics analysis. Biosci. Rep. 2020, 40, BSR20194365. [Google Scholar] [CrossRef] [PubMed]
- Mallik, S.; Seth, S.; Bhadra, T.; Zhao, Z. A linear regression and deep learning approach for detecting reliable genetic alterations in cancer using dna methylation and gene expression data. Genes 2020, 11, 931. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Lin, L.; Jiang, Y.; Wan, Y.; Zhou, S.; Cheng, W. Co-expression network analysis identified atypical chemokine receptor 1 (ACKR1) association with lymph node metastasis and prognosis in cervical cancer. Cancer Biomarkers 2020, 27, 213–223. [Google Scholar] [CrossRef]
- Tu, S.; Zhang, H.; Yang, X.; Wen, W.; Song, K.; Yu, X.; Qu, X. Screening of cervical cancer-related hub genes based on comprehensive bioinformatics analysis. Cancer Biomarkers 2021, 32, 303–315. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Z.; Wang, Y.; Nie, S.; Sun, R.; Yang, J.; Cheng, W. A prognostic signature based on immunerelated genes for cervical squamous cell carcinoma and endocervical adenocarcinoma. Int. Immunopharmacol. 2020, 88, 106884. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, S.; Yang, X. Construction of Gene Modules and Analysis of Prognostic Biomarkers for Cervical Cancer by Weighted Gene Co-Expression Network Analysis. Front. Oncol. 2021, 11, 327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y. Identification of hub genes and key pathways associated with the progression of gynecological cancer. Oncol. Lett. 2019, 18, 6516–6524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Zeng, X.; Huang, D.; Qiu, X. Identification of differentially expressed miRNAs in early-stage cervical cancer with lymph node metastasis across the cancer genome atlas datasets. Cancer Manag. Res. 2018, 10, 6489. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Han, F.; Qi, N.; Wen, L.; Li, J.; Feng, C.; Wang, Q. Determination of a six-gene prognostic model for cervical cancer based on WGCNA combined with LASSO and Cox-PH analysis. World J. Surg. Oncol. 2021, 19, 1–11. [Google Scholar] [CrossRef]
- Fu, X.H.; Wu, Y.F.; Xue, F. Probing pathway-related modules in invasive squamous cervical cancer based on topological centrality of network strategy. J. Cancer Res. Ther. 2018, 14, 1638. [Google Scholar] [CrossRef]
- Meng, H.; Liu, J.; Qiu, J.; Nie, S.; Jiang, Y.; Wan, Y.; Cheng, W. Identification of Key Genes in Association with Progression and Prognosis in Cervical Squamous Cell Carcinoma. DNA Cell Biol. 2020, 39, 848–863. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, L.; Zhang, C.; Song, J.; Jiang, Y. Screening of Significant Biomarkers Related to Prognosis of Cervical Cancer and Functional Study Based on lncRNA-associated ceRNA Regulatory Network. Comb. Chem. High Throughput Screen. 2020, 24, 472–482. [Google Scholar] [CrossRef]
- Oany, A.R.; Mia, M.; Pervin, T.; Alyami, S.A.; Moni, M.A. Integrative systems biology approaches to identify potential biomarkers and pathways of cervical cancer. J. Pers. Med. 2021, 11, 363. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhang, S.; Zheng, Q.; Zhang, S. Dysregulation of KIF14 regulates the cell cycle and predicts poor prognosis in cervical cancer: A study based on integrated approaches. Brazilian J. Med. Biol. Res. 2021, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Li, Y.; Ming, Z.; Wang, H.; Dong, Z.; Qiu, L.; Wang, T. Comprehensive circular RNA expression profile in radiation-treated HeLa cells and analysis of radioresistance-related circRNAs. PeerJ 2018, 6, e5011. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, P.; Luo, X.; Su, C.; Chen, Y.; Zhao, L.; Wei, L.; Zeng, H.; Varghese, Z.; Moorhead, J.F.; et al. High olive oil diets enhance cervical tumour growth in mice: Transcriptome analysis for potential candidate genes and pathways. Lipids Health Dis. 2019, 18, 76. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Shen, J.; Xu, S. Multi-Omics Data Analyses Construct a Six Immune-Related Genes Prognostic Model for Cervical Cancer in Tumor Microenvironment. Front. Genet. 2021, 12, 663617. [Google Scholar] [CrossRef]
- Jiang, P.; Cao, Y.; Gao, F.; Sun, W.; Liu, J.; Ma, Z.; Xie, M.; Fu, S. SNX10 and PTGDS are associated with the progression and prognosis of cervical squamous cell carcinoma. BMC Cancer 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Yang, C.; Xu, X.; Jin, H. Identification of potential miRNAs and candidate genes of cervical intraepithelial neoplasia by bioinformatic analysis. Eur. J. Gynaecol. Oncol. 2016, 37, 469–473. [Google Scholar] [CrossRef]
- Tong, Y.; Sun, P.; Yong, J.; Zhang, H.; Huang, Y.; Guo, Y.; Yu, J.; Zhou, S.; Wang, Y.; Wang, Y.; et al. Radiogenomic Analysis of Papillary Thyroid Carcinoma for Prediction of Cervical Lymph Node Metastasis: A Preliminary Study. Front. Oncol. 2021, 11, 682998. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, S.; Wang, K.; Shang, M.; Chen, Z.; Yang, H.; Chen, Y.; Chen, B. Identification of biomarkers associated with cervical lymph node metastasis in papillary thyroid carcinoma: Evidence from an integrated bioinformatic analysis. Clin. Hemorheol. Microcirc. 2021, 78, 117–126. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO ID | GO Term | cDEGs (Counts) | p-Value | Associated KGs | |
| Biological Process (BPs) | GO:0006260 | DNA replication | 18 | 1.58 × 10−16 | BRCA1, CDK1, MCM2 |
| GO:0051301 | Cell division | 22 | 7.94 × 10−15 | NCAPG2, AURKA, CCNB1, CDK1 | |
| GO:0000082 | G1/S transition of mitotic cell cycle | 12 | 5.61 × 10−11 | CDK1, MCM2 | |
| GO:0006270 | DNA replication initiation | 8 | 1.18 × 10−9 | MCM2 | |
| GO:0007067 | Mitotic nuclear division | 13 | 7.29 × 10−8 | NCAPG2, AURKA, CDK1 | |
| Molecular Function (MFs) | GO:0005524 | ATP binding | 30 | 3.80 × 10−8 | TOP2A, AURKA, CDK1, MCM2 |
| GO:0005515 | Protein binding | 83 | 2.65 × 10−7 | TOP2A, NCAPG2, BRCA1, MCM2, AURKA, CCNB1, CDK1 | |
| GO:0003678 | DNA helicase activity | 6 | 3.77 × 10−7 | MCM2 | |
| GO:0003682 | Chromatin binding | 12 | 4.13 × 10−5 | TOP2A, CDK1 | |
| GO:0003677 | DNA binding | 25 | 1.20 × 10−4 | TOP2A, BRCA1, MCM2 | |
| Cellular Component | GO:0005654 | Nucleoplasm | 56 | 4.91 × 10−17 | TOP2A, NCAPG2, BRCA1, MCM2, AURKA, CCNB1, CDK1 |
| GO:0030496 | Midbody | 13 | 2.54 × 10−11 | AURKA, CDK1 | |
| GO:0042555 | MCM complex | 6 | 1.04 × 10−9 | MCM2 | |
| GO:0005634 | Nucleus | 65 | 2.18 × 10−9 | TOP2A, NCAPG2, BRCA1, MCM2, AURKA, CCNB1, CDK1 | |
| GO:0005819 | Spindle | 10 | 6.21 × 10−8 | AURKA | |
| hsa ID | Pathways | cDEGs (Counts) | p-Value | Associated cHubGs | |
| KEGG Pathway | hsa03030 | DNA replication | 9 | 7.97 × 10−11 | MCM2 |
| hsa04110 | Cell cycle | 12 | 5.37 × 10−10 | CCNB1, CDK1, MCM2 | |
| hsa04115 | p53 signaling pathway | 5 | 0.001158992 | CCNB1, CDK1 | |
| hsa04114 | Oocyte meiosis | 5 | 0.007240129 | CCNB1, CDK1, AURKA | |
| hsa03460 | Fanconianemia pathway | 3 | 0.0485697 | BRCA1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reza, M.S.; Harun-Or-Roshid, M.; Islam, M.A.; Hossen, M.A.; Hossain, M.T.; Feng, S.; Xi, W.; Mollah, M.N.H.; Wei, Y. Bioinformatics Screening of Potential Biomarkers from mRNA Expression Profiles to Discover Drug Targets and Agents for Cervical Cancer. Int. J. Mol. Sci. 2022, 23, 3968. https://doi.org/10.3390/ijms23073968
Reza MS, Harun-Or-Roshid M, Islam MA, Hossen MA, Hossain MT, Feng S, Xi W, Mollah MNH, Wei Y. Bioinformatics Screening of Potential Biomarkers from mRNA Expression Profiles to Discover Drug Targets and Agents for Cervical Cancer. International Journal of Molecular Sciences. 2022; 23(7):3968. https://doi.org/10.3390/ijms23073968
Chicago/Turabian StyleReza, Md. Selim, Md. Harun-Or-Roshid, Md. Ariful Islam, Md. Alim Hossen, Md. Tofazzal Hossain, Shengzhong Feng, Wenhui Xi, Md. Nurul Haque Mollah, and Yanjie Wei. 2022. "Bioinformatics Screening of Potential Biomarkers from mRNA Expression Profiles to Discover Drug Targets and Agents for Cervical Cancer" International Journal of Molecular Sciences 23, no. 7: 3968. https://doi.org/10.3390/ijms23073968