
The Reconstitution Dynamics of Cultivated Hematopoietic Stem Cells and Progenitors Is Independent of Age

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
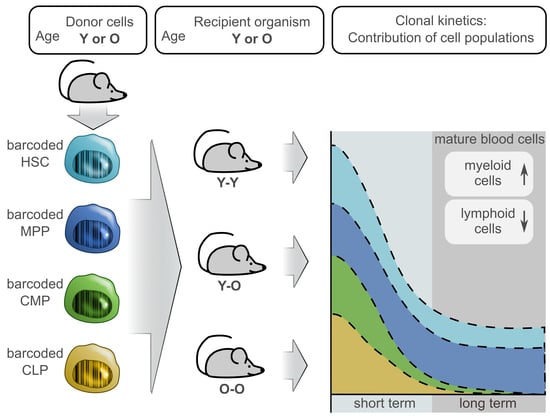
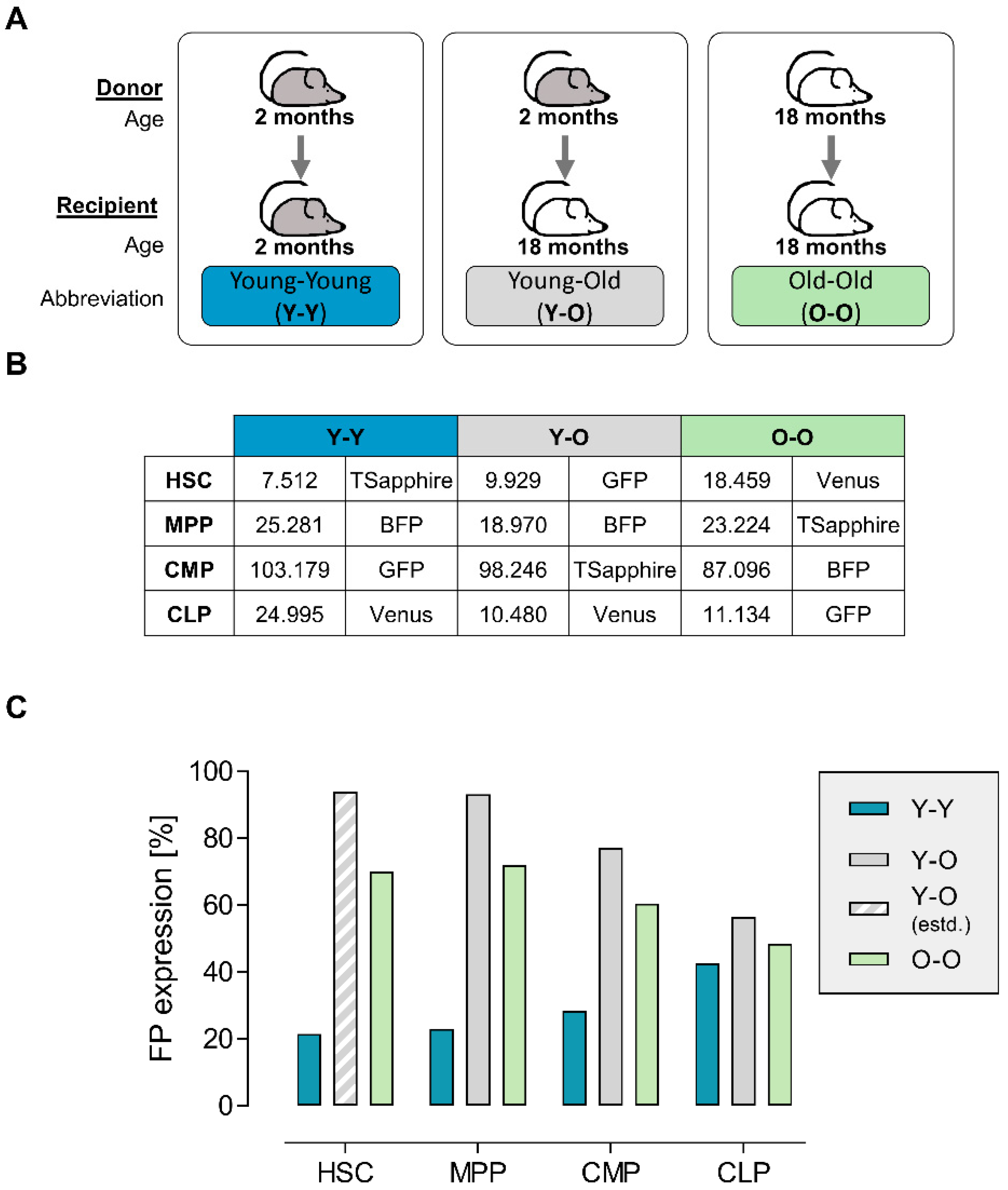
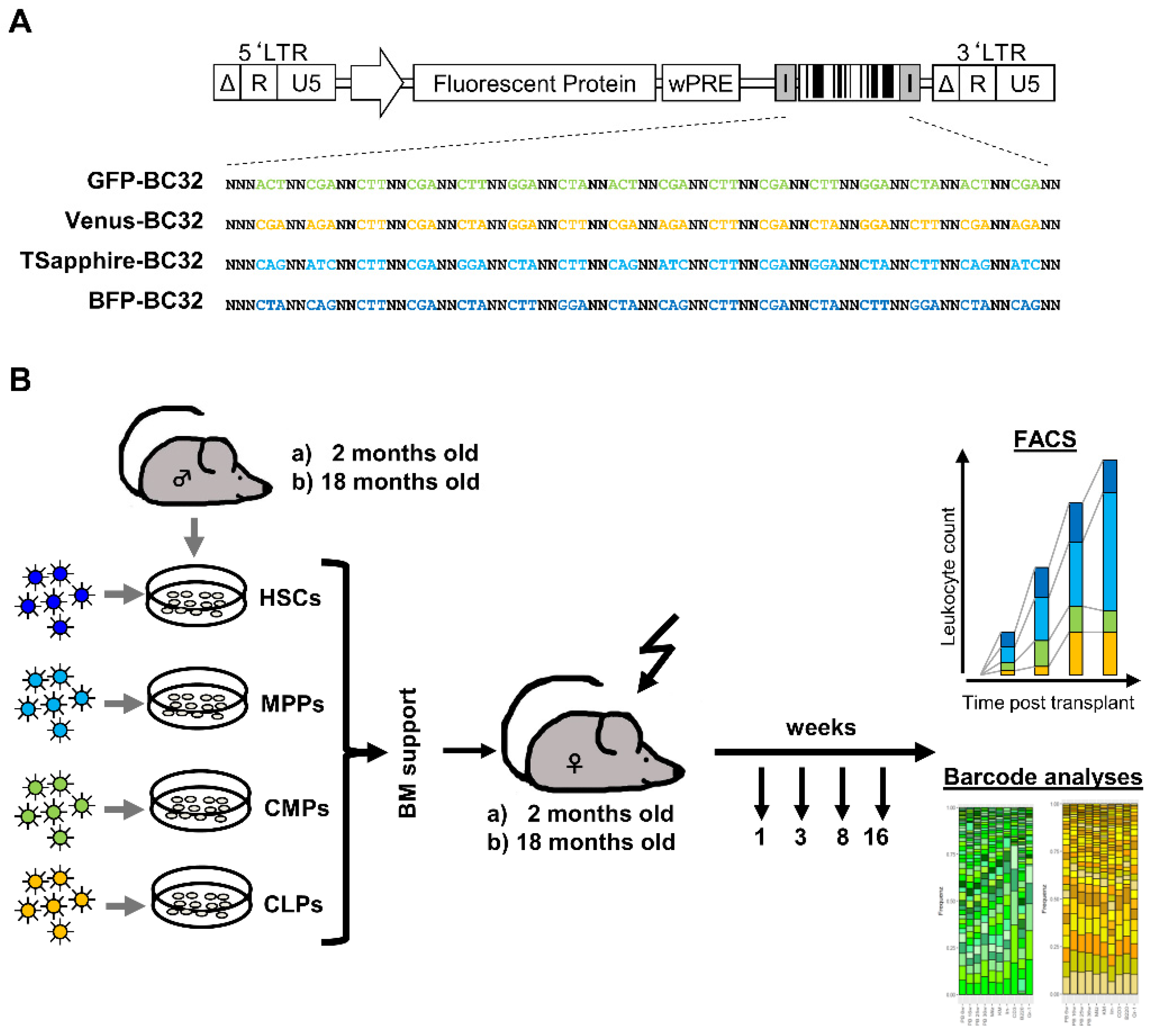
2.1. Graft Production and Quality Assessment
2.2. Dynamics of Blood Reconstitution from Old and Young Grafts
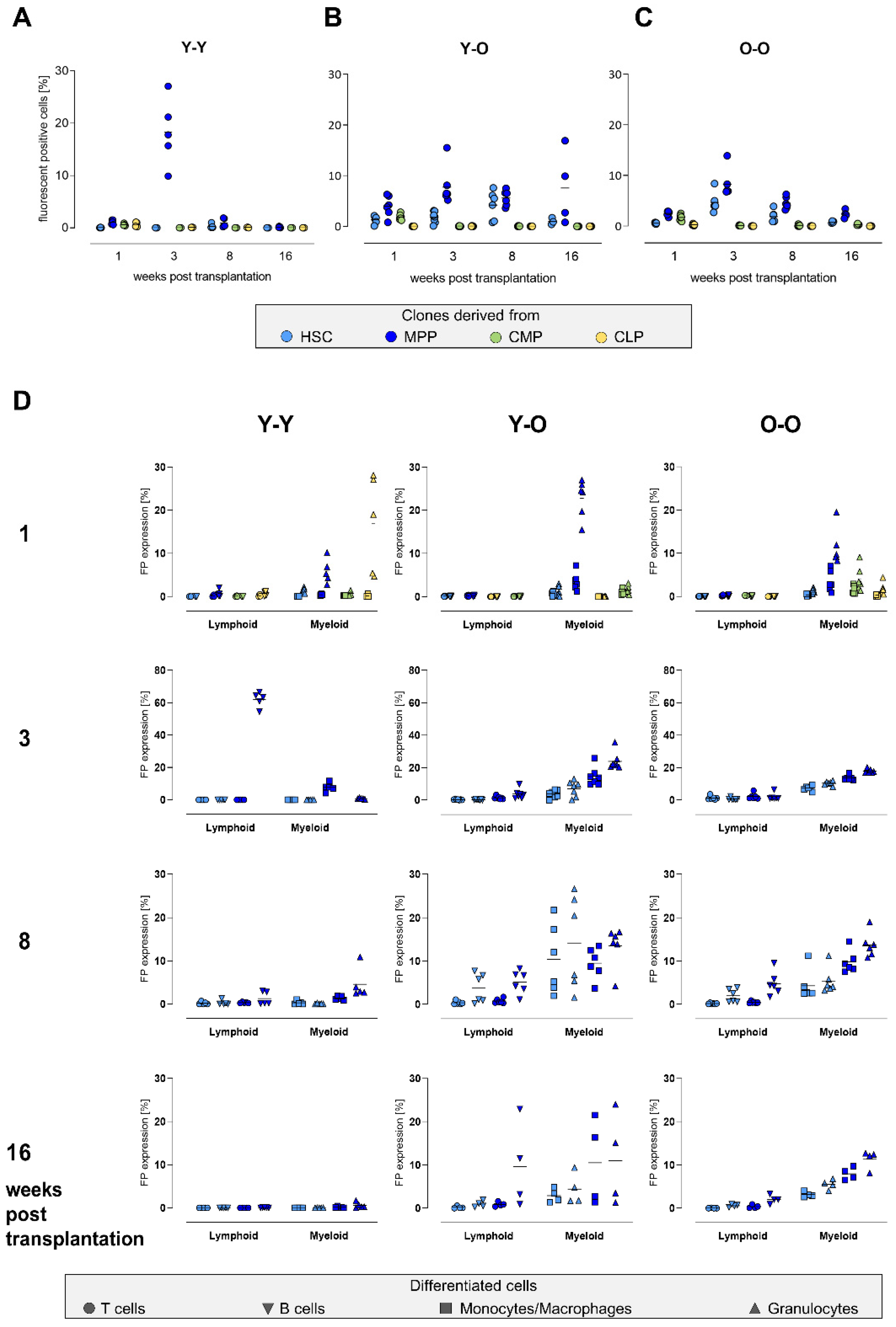
2.3. Contribution of the Transduced Cell Populations to Hematopoietic Reconstitution
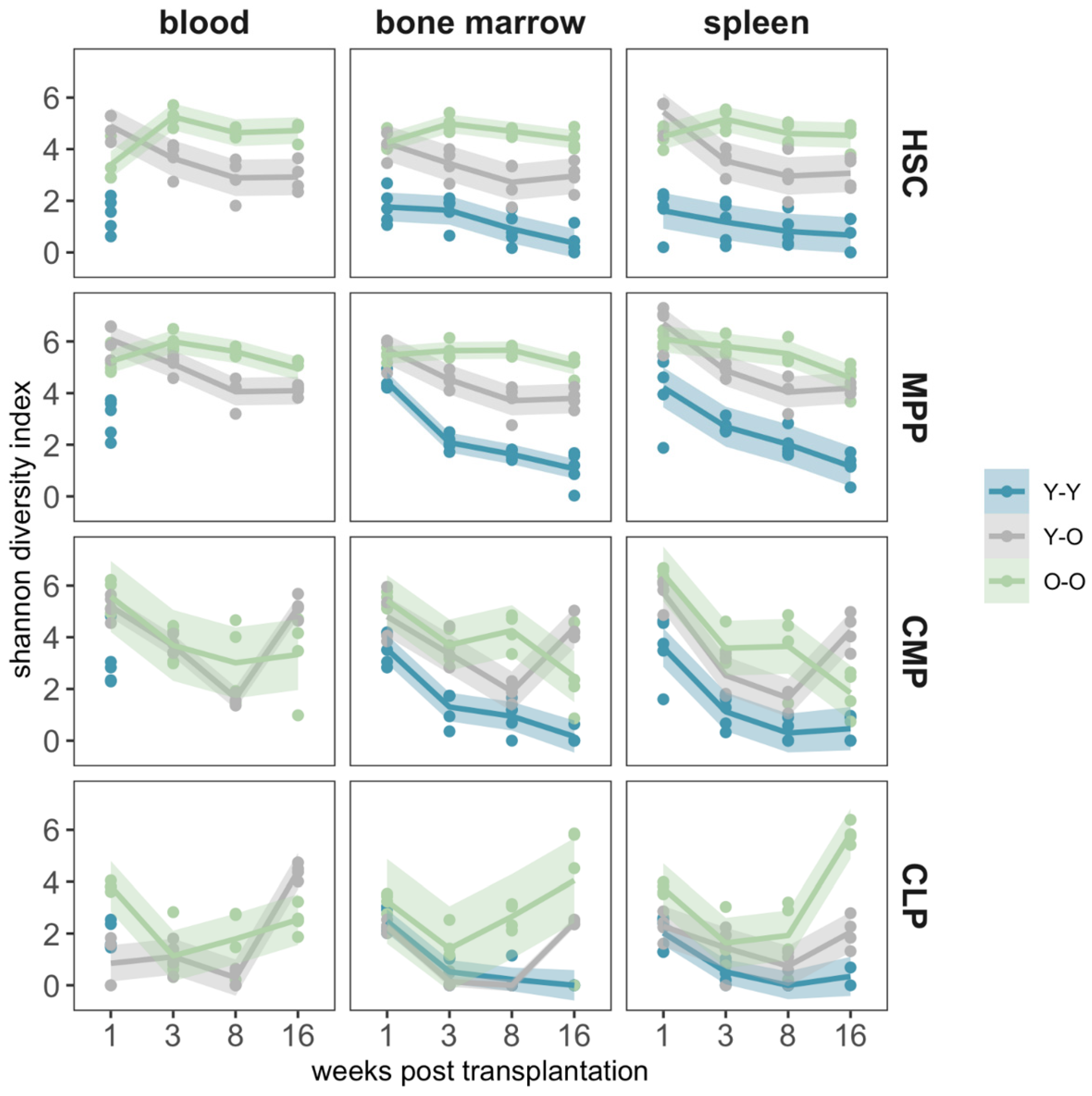
2.4. Clonal Composition of the Reconstituted Compartments
3. Discussion
Limitations
4. Materials and Methods
4.1. Generation and Production of Barcoded Lentiviral Vectors
4.2. Isolation and Transduction of the Target Cells
4.3. Murine Transplantation Experiment
4.4. Determination of Chimerism
4.5. Barcode Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Thomas, E.D.; Lochte, H.L., Jr.; Lu, W.C.; Ferrebee, J.W. Intravenous Infusion of Bone Marrow in Patients Receiving Radiation and Chemotherapy. N. Engl. J. Med. 1957, 257, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Aljurf, M.; Weisdorf, D.; Alfraih, F.; Szer, J.; Müller, C.; Confer, D.; Hashmi, S.; Kröger, N.; Shaw, B.E.; Greinix, H.; et al. Worldwide Network for Blood & Marrow Transplantation (WBMT) special article, challenges facing emerging al-ternate donor registries. Bone Marrow Transpl. 2019, 54, 1179–1188. [Google Scholar]
- Kollman, C.; Howe, C.W.S.; Anasetti, C.; Antin, J.H.; Davies, S.M.; Filipovich, A.H.; Hegland, J.; Kamani, N.; Kernan, N.; King, R.; et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: The effect of donor age. Blood 2001, 98, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, D.-P.; Liu, Q.-F.; Xu, L.-P.; Liu, K.-Y.; Zhang, X.-H.; Xu, Y.; Huang, F.; Huang, X. Donor and recipient age, gender and ABO incompatibility regardless of donor source: Validated criteria for donor selection for haematopoietic transplants. Leukemia 2018, 32, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Kollman, C.; Spellman, S.R.; Zhang, M.-J.; Hassebroek, A.; Anasetti, C.; Antin, J.H.; Champlin, R.E.; Confer, D.L.; Di Persio, J.F.; Fernandez-Viña, M.; et al. The effect of donor characteristics on survival after unrelated donor transplantation for hematologic malignancy. Blood 2016, 127, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artz, A.S. From Biology to Clinical Practice: Aging and Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2012, 18 (Suppl. S1), S40–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingemann, H.-G.; Storb, R.; Sanders, J.; Deeg, H.J.; Appelbaum, F.R.; Thomas, E.D. Acute lymphoblastic leukaemia after bone marrow transplantation for aplastic anaemia. Br. J. Haematol. 1986, 63, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Aschan, J.; Ringdén, O. Prognostic factors for long-term survival in leukemic marrow recipients with special emphasis on age and prophylaxis for graft-versus-host disease. Clin. Transplant. 1994, 8 Pt 1, 258–270. [Google Scholar] [PubMed]
- Bertz, H.; Auner, H.W.; Weissinger, F.; Salwender, H.J.; Einsele, H.; Egerer, G.; Sandherr, M.; Schüttrumpf, S.; Südhoff, T.; Maschmeyer, G. Antimicrobial therapy of febrile complications after high-dose chemo-/radiotherapy and autologous hematopoietic stem cell transplantation—Guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Oncology (DGHO). Ann. Hematol. 2003, 82 (Suppl. S2), S167–S174. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.; Brand, R.; Martino, R.; Van Biezen, A.; Finke, J.; Bacigalupo, A.; Beelen, D.; Devergie, A.; Alessandrino, E.; Willemze, R.; et al. Allogeneic Hematopoietic Stem-Cell Transplantation for Patients 50 Years or Older with Myelodysplastic Syndromes or Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Rezvani, A.R.; Storer, B.E.; Guthrie, K.A.; Schoch, H.G.; Maloney, D.G.; Sandmaier, B.M.; Storb, R. Impact of Donor Age on Outcome after Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2015, 21, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J. Clin. Oncol. 2022, 40, 189–201. [Google Scholar] [CrossRef]
- Oran, B.; Champlin, R.E.; Wang, F.; Tanaka, T.; Saliba, R.M.; Al-Atrash, G.; Garcia-Manero, G.; Kantarjian, H.; Cao, K.; Shpall, E.J.; et al. Donor clonal hematopoiesis increases risk of acute graft versus host disease after matched sibling transplantation. Leukemia 2022, 36, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J. Hematopoiesis: A Human Perspective. Cell Stem Cell 2012, 10, 120–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeti, R.; Park, C.Y.; Weissman, I.L. Identification of a Hierarchy of Multipotent Hematopoietic Progenitors in Human Cord Blood. Cell Stem Cell 2007, 1, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldini, L. Genetic engineering of hematopoiesis: Current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019, 11, e9958. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Swart, E.; Van Heijst, J.W.; Gerlach, C.; Castrucci, M.; Sie, D.; Heimerikx, M.; Velds, A.; Kerkhoven, R.M.; Arens, R.; et al. Dissecting T cell lineage relationships by cellular barcoding. J. Exp. Med. 2008, 205, 2309–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerrits, A.; Dykstra, B.; Kalmykowa, O.J.; Klauke, K.; Verovskaya, E.; Broekhuis, M.J.C.; de Haan, G.; Bystrykh, L.V. Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood 2010, 115, 2610–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyne, A.-M.; Kent, D.G.; Laurenti, E.; Cornils, K.; Glauche, I.; Perié, L. A track of the clones: New developments in cellular barcoding. Exp. Hematol. 2018, 68, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Pei, W.; Feyerabend, T.B.; Rössler, J.; Wang, X.; Postrach, D.; Busch, K.; Rode, I.; Klapproth, K.; Dietlein, N.; Quedenau, C.; et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 2017, 548, 456–460. [Google Scholar] [CrossRef] [Green Version]
- Pei, W.; Shang, F.; Wang, X.; Fanti, A.-K.; Greco, A.; Busch, K.; Klapproth, K.; Zhang, Q.; Quedenau, C.; Sauer, S.; et al. Resolving Fates and Single-Cell Transcriptomes of Hematopoietic Stem Cell Clones by Polylox Express Barcoding. Cell Stem Cell 2020, 27, 383–395.e8. [Google Scholar] [CrossRef]
- Rodriguez-Fraticelli, A.; Wolock, S.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef]
- Verovskaya, E.; Broekhuis, M.J.C.; Zwart, E.; Ritsema, M.; Van Os, R.; de Haan, G.; Bystrykh, L.V. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood 2013, 122, 523–532. [Google Scholar] [CrossRef]
- Verovskaya, E.; Broekhuis, M.J.C.; Zwart, E.; Weersing, E.; Ritsema, M.; Bosman, L.J.; Van Poele, T.; de Haan, G.; Bystrykh, L.V. Asymmetry in skeletal distribution of mouse hematopoietic stem cell clones and their equilibration by mobilizing cytokines. J. Exp. Med. 2014, 211, 487–497. [Google Scholar] [CrossRef]
- Lu, R.; Czechowicz, A.; Seita, J.; Jiang, D.; Weissman, I.L. Clonal-level lineage commitment pathways of hematopoietic stem cells in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Neff, N.F.; Quake, S.R.; Weissman, I.L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic bar-coding. Nat. Biotechnol. 2011, 29, 928–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.M.S.; Nguyen, L.V.; Carles, A.; Beer, P.; Miller, P.H.; Knapp, D.J.H.F.; Dhillon, K.; Hirst, M.; Eaves, C.J. Analysis of the clonal growth and differentiation dynamics of primitive barcoded human cord blood cells in NSG mice. Blood 2013, 122, 3129–3137. [Google Scholar] [CrossRef] [Green Version]
- Brugman, M.H.; Wiekmeijer, A.-S.; van Eggermond, M.; Wolvers-Tettero, I.; Langerak, A.W.; de Haas, E.F.E.; Bystrykh, L.V.; van Rood, J.J.; de Haan, G.; Fibbe, W.E.; et al. Development of a diverse human T-cell repertoire despite stringent restriction of hematopoietic clonality in the thymus. Proc. Natl. Acad. Sci. USA 2015, 112, E6020–E6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belderbos, M.E.; Jacobs, S.; Koster, T.K.; Ausema, A.; Weersing, E.; Zwart, E.; de Haan, G.; Bystrykh, L.V. Donor-to-Donor Heterogeneity in the Clonal Dynamics of Transplanted Human Cord Blood Stem Cellsin Murine Xenografts. Biol. Blood Marrow Transpl. 2020, 26, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Belderbos, M.E.; Koster, T.; Ausema, B.; Jacobs, S.; Sowdagar, S.; Zwart, E.; de Bont, E.; de Haan, G.; Bystrykh, L.V. Clonal selection and asymmetric distribution of human leukemia in murine xenografts revealed by cellular bar-coding. Blood 2017, 129, 3210–3220. [Google Scholar] [CrossRef]
- Wu, C.; Espinoza, D.A.; Koelle, S.J.; Yang, D.; Truitt, L.; Schlums, H.; Lafont, B.A.; Davidson-Moncada, J.K.; Lu, R.; Kaur, A.; et al. Clonal expansion and compartmentalized maintenance of rhesus macaque NK cell subsets. Sci. Immunol. 2018, 3, eaat9781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Li, B.; Lu, R.; Koelle, S.J.; Yang, Y.; Jares, A.; Krouse, A.E.; Metzger, M.; Liang, F.; Loré, K.; et al. Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell 2014, 14, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Koelle, S.J.; Espinoza, D.A.; Wu, C.; Xu, J.; Lu, R.; Li, B.; Donahue, R.E.; Dunbar, C.E. Quantitative stability of hematopoietic stem and progenitor cell clonal output in rhesus macaques receiving transplants. Blood 2017, 129, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, D.A.; Fan, X.; Yang, D.; Cordes, S.F.; Truitt, L.L.; Calvo, K.R.; Yabe, I.M.; Demirci, S.; Hope, K.J.; Hong, S.G.; et al. Aberrant Clonal Hematopoiesis following Lentiviral Vector Transduction of HSPCs in a Rhesus Macaque. Mol. Ther. 2019, 27, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-R.; Espinoza, D.A.; Wu, C.; Truitt, L.; Shin, T.-H.; Chen, S.; Fan, X.; Yabe, I.M.; Panch, S.; Hong, S.G.; et al. The impact of aging on primate hematopoiesis as interrogated by clonal tracking. Blood 2018, 131, 1195–1205. [Google Scholar] [CrossRef]
- Weber, K.; Thomaschewski, M.; Warlich, M.; Volz, T.; Cornils, K.; Niebuhr, B.; Täger, M.; Lütgehetmann, M.; Pollok, J.-M.; Stocking, C.; et al. RGB marking facilitates multicolor clonal cell tracking. Nat. Med. 2011, 17, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Cornils, K.; Thielecke, L.; Hüser, S.; Forgber, M.; Thomaschewski, M.; Kleist, N.; Hussein, K.; Riecken, K.; Volz, T.; Gerdes, S.; et al. Multiplexing clonality: Combining RGB marking and genetic barcoding. Nucleic Acids Res. 2014, 42, e56. [Google Scholar] [CrossRef]
- Thielecke, L.; Aranyossy, T.; Dahl, A.; Tiwari, R.; Roeder, I.; Geiger, H.; Fehse, B.; Glauche, I.; Cornils, K. Limitations and challenges of genetic barcode quantification. Sci. Rep. 2017, 7, 43249. [Google Scholar] [CrossRef] [Green Version]
- Aranyossy, T.; Thielecke, L.; Glauche, I.; Fehse, B.; Cornils, K. Genetic Barcodes Facilitate Competitive Clonal Analyses In Vivo. Hum. Gene Ther. 2017, 28, 926–937. [Google Scholar] [CrossRef]
- Fehse, B.; Kustikova, O.S.; Bubenheim, M.; Baum, C. Pois(s)on—It’s a question of dose. Gene Ther. 2004, 11, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Shemer, A.; Grozovski, J.; Tay, T.L.; Tao, J.; Volaski, A.; Süß, P.; Ardura-Fabregat, A.; Gross-Vered, M.; Kim, J.-S.; David, E.; et al. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat. Commun. 2018, 9, 5206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, S.N.; Baker, L.C.; Mittelman, D.; Porteus, M.H. Lentiviral and targeted cellular barcoding reveals ongoing clonal dynamics of cell lines in vitro and in vivo. Genome Biol. 2014, 15, R75. [Google Scholar] [CrossRef] [Green Version]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-Associated Characteristics of Murine Hematopoietic Stem Cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, H.; True, J.M.; de Haan, G.; Van Zant, G. Age- and stage-specific regulation patterns in the hematopoietic stem cell hierarchy. Blood 2001, 98, 2966–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Haan, G.; Nijhof, W.; Van Zant, G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: Correlation between lifespan and cycling activity. Blood 1997, 89, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Wandycz, A.M.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, S.; Adair, J.E.; Giese, M.A.; Chan, Y.-Y.; Norgaard, Z.K.; Enstrom, M.; Haworth, K.G.; Schefter, L.E.; Kiem, H.-P. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Weissman, I.L.; Akashi, K. Identification of Clonogenic Common Lymphoid Progenitors in Mouse Bone Marrow. Cell 1997, 91, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Akashi, K.; Traver, D.; Miyamoto, T.; Weissman, I.L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404, 193–197. [Google Scholar] [CrossRef]
- Liang, Y.; Van Zant, G.; Szilvassy, S.J. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood 2005, 106, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Moon, H.-B.; Spangrude, G.J. Major Age-Related Changes Of Mouse Hematopoietic Stem/Progenitor Cells. Ann. N. Y. Acad. Sci. 2003, 996, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.H.; Sieburg, H.B.; Muller-Sieburg, C.E. A new mechanism for the aging of hematopoietic stem cells: Aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood 2008, 111, 5553–5561. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.-J.; Klein, A.M.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Busch, K.; Klapproth, K.; Barile, M.; Flossdorf, M.; Holland-Letz, T.; Schlenner, S.; Reth, M.; Höfer, T.; Rodewald, H.-R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Borikar, S.; Bell, R.; Kuffler, L.; Philip, V.; Trowbridge, J.J. Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. J. Exp. Med. 2016, 213, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Oguro, H.; Ding, L.; Morrison, S.J. SLAM Family Markers Resolve Functionally Distinct Subpopulations of Hematopoietic Stem Cells and Multipotent Progenitors. Cell Stem Cell 2013, 13, 102–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Papa, E.F.; Dooner, M.S.; Machan, J.T.; Johnson, K.W.; Goldberg, L.R.; Quesenberry, P.J.; Colvin, G.A. Homing and Long-Term Engraftment of Long- and Short-Term Renewal Hematopoietic Stem Cells. PLoS ONE 2012, 7, e31300. [Google Scholar] [CrossRef]
- Wilson, A.; Laurenti, E.; Oser, G.; Van Der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezas-Wallscheid, N.; Klimmeck, D.; Hansson, J.; Lipka, D.B.; Reyes, A.; Wang, Q.; Weichenhan, D.; Lier, A.; von Paleske, L.; Renders, S.; et al. Identification of Regulatory Networks in HSCs and Their Immediate Progeny via Integrated Proteome, Transcriptome, and DNA Methylome Analysis. Cell Stem Cell 2014, 15, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, S.; Whiting-Theobald, N.L.; Linton, G.F.; Holmes, K.L.; Anderson-Cohen, M.; Kelly, P.F.; Vanin, E.F.; Pilon, A.M.; Bodine, D.M.; Horwitz, M.E.; et al. Concentrated RD114-pseudotyped MFGS-gp91phox vector achieves high levels of functional correction of the chronic granulomatous disease oxidase defect in NOD/SCID/beta -microglobulin-/- repopulating mobilized human peripheral blood CD34+ cells. Blood 2003, 102, 2789–2797. [Google Scholar] [CrossRef]
- Weber, K.; Thomaschewski, M.; Benten, D.; Fehse, B. RGB marking with lentiviral vectors for multicolor clonal cell tracking. Nat. Protoc. 2012, 7, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLOS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, Ö.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Bystrykh, L.V.; Belderbos, M.E. Clonal Analysis of Cells with Cellular Barcoding: When Numbers and Sizes Matter. Methods Mol. Biol. 2016, 1516, 57–89. [Google Scholar] [CrossRef]
- Lange, C.; Brunswig-Spickenheier, B.; Cappallo-Obermann, H.; Eggert, K.; Gehling, U.M.; Rudolph, C.; Schlegelberger, B.; Cornils, K.; Zustin, J.; Spiess, A.-N.; et al. Radiation Rescue: Mesenchymal Stromal Cells Protect from Lethal Irradiation. PLoS ONE 2011, 6, e14486. [Google Scholar] [CrossRef] [PubMed]
- Thielecke, L.; Cornils, K.; Glauche, I. genBaRcode: A comprehensive R-package for genetic barcode analysis. Bioinformatics 2020, 36, 2189–2194. [Google Scholar] [CrossRef]
- Biasco, L.; Pellin, D.; Scala, S.; Dionisio, F.; Basso-Ricci, L.; Leonardelli, L.; Scaramuzza, S.; Baricordi, C.; Ferrua, F.; Cicalese, M.P.; et al. In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Re-constitution Phases. Cell Stem Cell 2016, 19, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, S.; Basso-Ricci, L.; Dionisio, F.; Pellin, D.; Giannelli, S.; Salerio, F.A.; Leonardelli, L.; Cicalese, M.P.; Ferrua, F.; Aiuti, A.; et al. Dynamics of genetically engineered hematopoietic stem and progenitor cells after autologous transplantation in humans. Nat. Med. 2018, 24, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Bonner, M.; Pierciey, J.F.J.; Uchida, N.; Rottman, J.; Demopoulos, L.; Schmidt, M.; Kanter, J.; Walters, M.C.; Thompson, A.A.; et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020, 4, 2058–2063. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gotzhein, F.; Aranyossy, T.; Thielecke, L.; Sonntag, T.; Thaden, V.; Fehse, B.; Müller, I.; Glauche, I.; Cornils, K. The Reconstitution Dynamics of Cultivated Hematopoietic Stem Cells and Progenitors Is Independent of Age. Int. J. Mol. Sci. 2022, 23, 3160. https://doi.org/10.3390/ijms23063160
Gotzhein F, Aranyossy T, Thielecke L, Sonntag T, Thaden V, Fehse B, Müller I, Glauche I, Cornils K. The Reconstitution Dynamics of Cultivated Hematopoietic Stem Cells and Progenitors Is Independent of Age. International Journal of Molecular Sciences. 2022; 23(6):3160. https://doi.org/10.3390/ijms23063160
Chicago/Turabian StyleGotzhein, Frauke, Tim Aranyossy, Lars Thielecke, Tanja Sonntag, Vanessa Thaden, Boris Fehse, Ingo Müller, Ingmar Glauche, and Kerstin Cornils. 2022. "The Reconstitution Dynamics of Cultivated Hematopoietic Stem Cells and Progenitors Is Independent of Age" International Journal of Molecular Sciences 23, no. 6: 3160. https://doi.org/10.3390/ijms23063160