
More than Addiction—The Nucleus Accumbens Contribution to Development of Mental Disorders and Neurodegenerative Diseases

 , ,
, , {kind=link}
Abstract
:1. Introduction
2. Addiction-Induced Short- and Long-Term Changes in Synaptic Plasticity Affect Molecular Signaling and Spino-Dendritic Morphology of the Accumbal Neurons
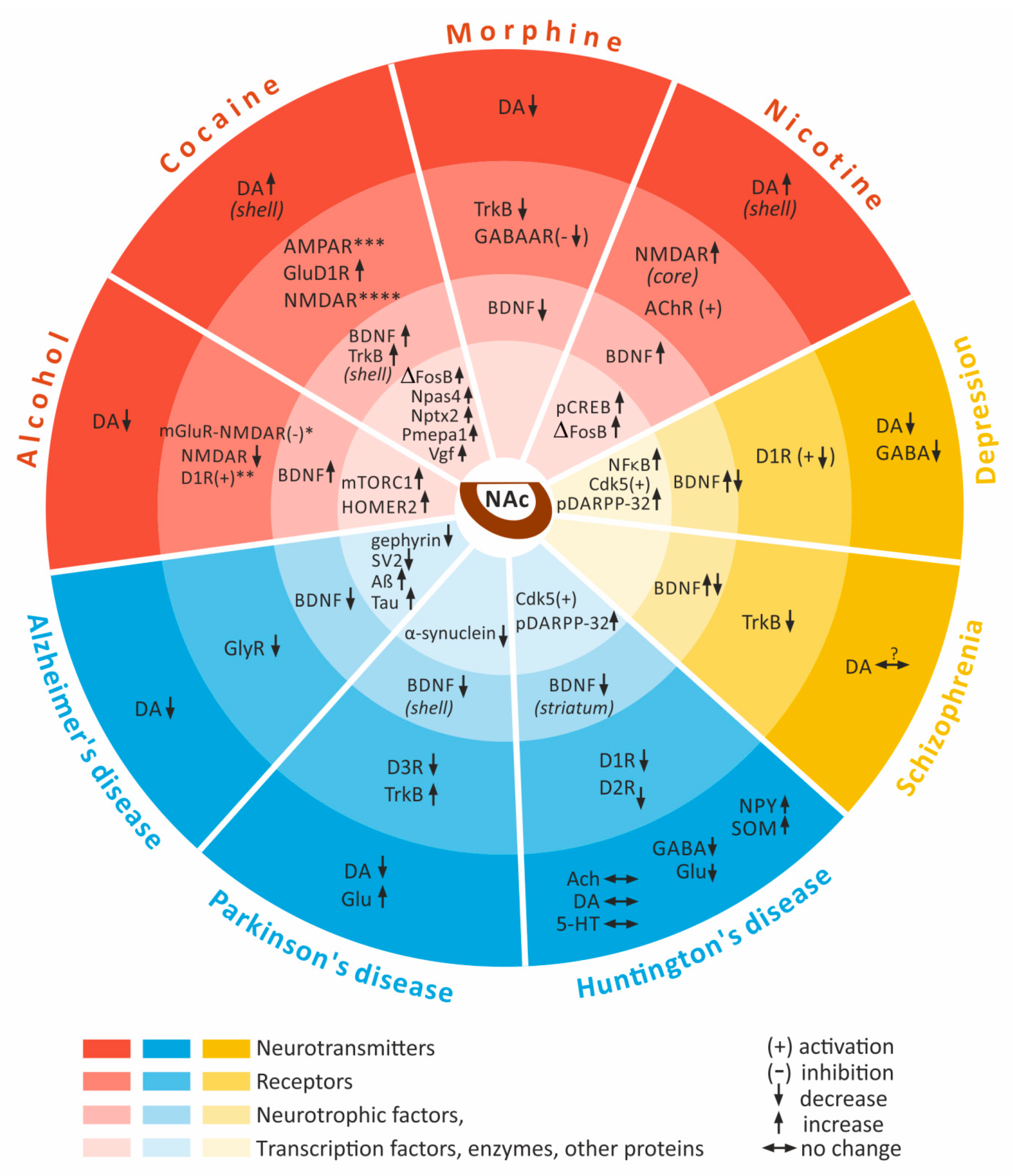
2.1. Alcohol
2.2. Cocaine
2.3. Morphine
2.4. Nicotine
3. Impairment of the Synaptic Plasticity and Dysregulation of Signaling Pathways Contribute to the Brain Reward System Dysfunction in Mental Illnesses
3.1. Depression
3.2. Schizophrenia
4. The Nucleus Accumbens Contribution to Neurodegeneration Results Not Only from Changes Occurring within the Nucleus Itself but Also within the Functionally Related Brain Areas
4.1. Alzheimer’s Disease
4.2. Huntington’s Disease
4.3. Parkinson Disease (PD)
5. Conclusions and Perspectives for Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aß | amyloid ß |
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| Amg | amygdala |
| AMPARs | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptors |
| BDNF | brain-derived neurotrophic factor |
| BRS | brain reward system |
| Cdk5 | cyclin-dependent kinase 5 |
| CNS | central nervous system |
| CUMS | chronic unpredictable mild stress |
| DA | dopamine |
| DARPP-32 | cAMP-regulated phosphoprotein 32 |
| DBS | deep brain stimulation |
| D1R | D1 dopamine receptor |
| GABA | γ-aminobutyric acid |
| GABAARs | γ-aminobutyric acid A receptors |
| Glu | glutamate |
| GluD1R | glutamate delta-1 receptor |
| HD | Huntington’s disease |
| Hip | hippocampus |
| 5-HT | serotonin |
| LTP | long-term potentiation |
| MDD | major depressive disorder |
| MGluRs | metabotropic glutamatergic receptors |
| MSNs | medium spiny neurons |
| mPFC | medial prefrontal cortex |
| NAc | nucleus accumbens |
| NADPH-d | dihydronicotinamide adenine dinucleotide phosphate diaphorase |
| NMDARs | N-methyl-D-aspartate receptors |
| NPY | neuropeptide Y |
| pCREB | phosphorylated cAMP (adenosine 3′5′ cyclic monophosphate)-Response Element Binding protein |
| PD | Parkinson’s disease |
| PFC | prefrontal cortex |
| SOM | somatostatin |
| TPD | tangle-predominant dementia |
| TrkB | tropomyosin receptor kinase B |
| vSub | ventral subiculum |
| VTA | ventral tegmental area |
| Enk | enkephalin |
| SP | substance P |
References
- Salgado, S.; Kaplitt, M.G. The nucleus accumbens: A comprehensive review. Stereotact. Funct. Neurosurg. 2015, 93, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Grueter, B.A.; Rothwell, P.E.; Malenka, R.C. Integrating synaptic plasticity and striatal circuit function in addiction. Curr. Opin. Neurobiol. 2012, 22, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.; Robison, A.J.; Mazei-Robison, M.S. Reward circuitry in addiction. Neurotherapeutics 2017, 14, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsden, W.N. Synaptic plasticity in depression: Molecular, cellular and functional correlates. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2013, 43, 168–184. [Google Scholar] [CrossRef]
- Russo, S.J.; Dietz, D.M.; Dumitriu, D.; Morrison, J.H.; Malenka, R.C.; Nestler, E.J. The addicted synapse: Mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010, 33, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.O.; Ip, N.Y. Structural plasticity of dendritic spines: The underlying mechanisms and its dysregulation in brain disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2257–2263. [Google Scholar] [CrossRef] [Green Version]
- Bayassi-Jakowicka, M.; Lietzau, G.; Czuba, E.; Steliga, A.; Waśkow, M.; Kowiański, P. Neuroplasticity and multilevel system of connections determine the integrative role of nucleus accumbens in the brain reward system. Int. J. Mol. Sci. 2021, 22, 9806. [Google Scholar]
- Dydyk, A.M.; Sizemore, D.C.; Trachsel, L.A.; Dulebohn, S.C.; Porter, B.R. Tennessee controlled substance prescribing for acute and chronic pain. StatPearls 2021. [Google Scholar]
- Malenka, R.C.; Nestler, E.J.; Hyman, S.E.; Sydor, A.; Brown, R.Y. Chapter 15: Reinforcement and Addictive Disorders. In Molecular Neuropharmacology: A Foundation for Clinical Neuroscience, 2nd ed.; McGraw-Hill Medical: New York, NY, USA, 2009; Volume 375, pp. 364–365. ISBN 978-0-07-148127-4. [Google Scholar]
- Volkow, N.D.; Koob, G.F.; McLellan, A.T. Neurobiologic advances from the brain disease model of addiction. N. Engl. J. Med. 2016, 374, 363–371. [Google Scholar] [CrossRef]
- Hasin, D.S.; O’Brien, C.P.; Auriacombe, M.; Borges, G.; Bucholz, K.; Budney, A.; Compton, W.M.; Crowley, T.; Ling, W.; Petry, N.M.; et al. DSM-5 criteria for substance use disorders: Recommendations and rationale. Am. J. Psychiatry 2013, 170, 834–851. [Google Scholar] [CrossRef] [Green Version]
- Guha, M. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. In Reference Reviews, 5th ed.; Emerald Group Publishing Limited: Bingley, UK, 2014; Volume 28, pp. 36–37. ISBN 0950-4125. [Google Scholar]
- Wallner, M.; Olsen, R.W. Physiology and pharmacology of alcohol: The imidazobenzodiazepine alcohol antagonist site on subtypes of GABA A receptors as an opportunity for drug development? Br. J. Pharmacol. 2008, 154, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Mishra, D.; Zhang, X.; Chergui, K. Ethanol disrupts the mechanisms of induction of long-term potentiation in the mouse nucleus accumbens. Alcohol. Clin. Exp. Res. 2012, 36, 2117–2125. [Google Scholar] [CrossRef] [Green Version]
- Abrahao, K.P.; Ariwodola, O.J.; Butler, T.R.; Rau, A.R.; Skelly, M.J.; Carter, E.; Alexander, N.P.; McCool, B.A.; Souza-Formigoni, M.L.O.; Weiner, J.L. Locomotor sensitization to ethanol impairs NMDA receptor-dependent synaptic plasticity in the nucleus accumbens and increases ethanol self-administration. J. Neurosci. 2013, 33, 4834–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckley, J.T.; Laguesse, S.; Phamluong, K.; Morisot, N.; Wegner, S.A.; Ron, D. The first alcohol drink triggers mTORC1-dependent synaptic plasticity in nucleus accumbens dopamine D1 receptor neurons. J. Neurosci. 2016, 36, 701–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggio, G.M.; di Marco, R.; Gulisano, W.; D’Ascenzo, M.; Torrisi, S.A.; Geraci, F.; Lavanco, G.; Dahl, K.; Giurdanella, G.; Castorina, A.; et al. Dopaminergic-GABAergic interplay and alcohol binge drinking. Pharmacol. Res. 2019, 141, 384–391. [Google Scholar] [CrossRef]
- Castelli, V.; Brancato, A.; Cavallaro, A.; Lavanco, G.; Cannizzaro, C. Homer2 and alcohol: A mutual interaction. Front. Psychiatry 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raivio, N.; Miettinen, P.; Kiianmaa, K. Innate BDNF expression is associated with ethanol intake in alcohol-preferring AA and alcohol-avoiding ANA rats. Brain Res. 2014, 1579, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Keller, C.; Martinez, S.A.; Esparza, M.A.; Bollati, F.; Kalivas, P.W.; Cancela, L.M. Cross-sensitization between cocaine and acute restraint stress is associated with sensitized dopamine but not glutamate release in the nucleus accumbens. Eur. J. Neurosci. 2013, 37, 982–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wolf, M.E. Multiple faces of BDNF in cocaine addiction. Behav. Brain Res. 2015, 279, 240–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Gandhi, P.J.; Pavuluri, R.; Shelkar, G.P.; Dravid, S.M. Glutamate delta-1 receptor regulates cocaine-induced plasticity in the nucleus accumbens. Transl. Psychiatry 2018, 8, 219. [Google Scholar] [CrossRef]
- D’Ascenzo, M.; Podda, M.V.; Grassi, C. The role of D-serine as co-agonist of NMDA receptors in the nucleus accumbens: Relevance to cocaine addiction. Front. Synaptic Neurosci. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, M.E.; Fant, B.; Swinford-Jackson, S.E.; Testino, A.; van Nest, D.; Abel, T.; Pierce, R.C. H3.3 barcoding of nucleus accumbens transcriptional activity identifies novel molecular cascades associated with cocaine self-administration in mice. J. Neurosci. 2019, 39, 5247–5254. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.M.; Wissman, A.M.; Chemplanikal, J.; Buzin, N.; Guzman, D.; Larson, E.B.; Neve, R.L.; Nestler, E.J.; Cowan, C.W.; Self, D.W. BDNF-TrkB controls cocaine-induced dendritic spines in rodent nucleus accumbens dissociated from increases in addictive behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, 9469–9474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, J.C.; Abel, J.M.; Lacy, R.T.; Beckmann, J.S.; Witte, M.A.; Lynch, W.J.; Smith, M.A. The effects of resistance exercise on cocaine self-administration, muscle hypertrophy, and BDNF expression in the nucleus accumbens. Drug Alcohol. Depend. 2016, 163, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Marie, N.; Canestrelli, C.; Noble, F. Transfer of neuroplasticity from nucleus accumbens core to shell is required for cocaine reward. PLoS ONE 2012, 7, e30241. [Google Scholar] [CrossRef]
- Koo, J.W.; Lobo, M.K.; Chaudhury, D.; Labonté, B.; Friedman, A.; Heller, E.; Peña, C.J.; Han, M.H.; Nestler, E.J. Loss of BDNF signaling in D1R-expressing NAc neurons enhances morphine reward by reducing GABA inhibition. Neuropsychopharmacology 2014, 39, 2646–2653. [Google Scholar] [CrossRef] [Green Version]
- Nestler, E.J. Cellular basis of memory for addiction. Dialogues Clin. Neurosci. 2013, 15, 431–443. [Google Scholar]
- Ghodrati-Jaldbakhan, S.; Ahmadalipour, A.; Rashidy-Pour, A.; Vafaei, A.A.; Miladi-Gorji, H.; Alizadeh, M. Low- and high-intensity treadmill exercise attenuates chronic morphine-induced anxiogenesis and memory impairment but not reductions in hippocampal BDNF in female rats. Brain Res. 2017, 1663, 20–28. [Google Scholar] [CrossRef]
- Ávila-Ruiz, T.; Carranza, V.; Gustavo, L.L.; Limón, D.I.; Martínez, I.; Flores, G.; Flores-Hernández, J. Chronic administration of nicotine enhances NMDA-activated currents in the prefrontal cortex and core part of the nucleus accumbens of rats. Synapse 2014, 68, 248–256. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Steliga, A.; Czuba, E.; Ludkiewicz, B.; Waśkow, M.; Spodnik, J.H.; Moryś, J. Nicotine-induced CREB and DeltaFosB activity is modified by caffeine in the brain reward system of the rat. J. Chem. Neuroanat. 2018, 88, 1–12. [Google Scholar] [CrossRef]
- Tanda, G.; Goldberg, S.R. Alteration of the behavioral effects of nicotine by chronic caffeine exposure. Pharmacol. Biochem. Behav. 2000, 66, 47–64. [Google Scholar] [CrossRef]
- Liu, X.; Jernigan, C. Effects of caffeine on persistence and reinstatement of nicotine-seeking behavior in rats: Interaction with nicotine-associated cues. Psychopharmacology 2012, 220, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestler, E.J. Transcriptional Mechanisms of Addiction: Role of ΔFosB. In Proceedings of the Philosophical Transactions of the Royal Society B: Biological Sciences; Royal Society: London, UK, 2008; Volume 363, pp. 3245–3255. [Google Scholar]
- Kivinummi, T.; Kaste, K.; Rantamäki, T.; Castrén, E.; Ahtee, L. Alterations in BDNF and phospho-CREB levels following chronic oral nicotine treatment and its withdrawal in dopaminergic brain areas of mice. Neurosci. Lett. 2011, 491, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Birdogan, A.; Salur, E.; Tuzcu, F.; Gokmen, R.C.; Ozturk Bintepe, M.; Aypar, B.; Keser, A.; Balkan, B.; Koylu, E.O.; Kanit, L.; et al. Chronic oral nicotine administration and withdrawal regulate the expression of neuropeptide Y and its receptors in the mesocorticolimbic system. Neuropeptides 2021, 90, 102184. [Google Scholar] [CrossRef] [PubMed]
- Shelton, R.C. The molecular neurobiology of depression. Psychiatr. Clin. N. Am. 2007, 30, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Song, Z.; Sun, X.; Wang, J.H. microRNA and mRNA profiles in nucleus accumbens underlying depression versus resilience in response to chronic stress. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Zhang, P.; Li, T.; Li, M.; Zhang, Q.; He, F.; Zhang, L.; Cai, H.; Lv, X.; Qiao, H.; et al. NAc-VTA circuit underlies emotional stress-induced anxiety-like behavior in the three-chamber vicarious social defeat stress mouse model. Nat. Commun. 2022, 13, 1–19. [Google Scholar] [CrossRef]
- Pittenger, C.; Duman, R.S. Stress, depression, and neuroplasticity: A convergence of mechanisms. Neuropsychopharmacology 2008, 33, 88–109. [Google Scholar] [CrossRef]
- Bagot, R.C.; Parise, E.M.; Peña, C.J.; Zhang, H.X.; Maze, I.; Chaudhury, D.; Persaud, B.; Cachope, R.; Bolaños-Guzmán, C.A.; Cheer, J.; et al. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Russo, S.J.; Nestler, E.J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013, 14, 609–625. [Google Scholar] [CrossRef] [Green Version]
- Lorenzetti, V.; Allen, N.B.; Fornito, A.; Yücel, M. Structural brain abnormalities in major depressive disorder: A selective review of recent MRI studies. J. Affect. Disord. 2009, 117, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pizzagalli, D.A.; Holmes, A.J.; Dillon, D.G.; Goetz, E.L.; Birk, J.L.; Bogdan, R.; Dougherty, D.D.; Iosifescu, D.V.; Rauch, S.L.; Fava, M. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am. J. Psychiatry 2009, 166, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, C.S.; Goswami, D.B.; Austin, M.C.; Iyo, A.H.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. NeuroPsychopharmacol. Biol. Psychiatry 2011, 35, 1774–1779. [Google Scholar] [CrossRef] [Green Version]
- Avnioglu, S.; Velioglu, H.A.; Cankaya, S.; Yulug, B. Quantitative evaluation of brain volumes in drug-free major depressive disorder using MRI-Cloud method. Neuroreport 2021, 32, 1027–1034. [Google Scholar] [CrossRef]
- Ho, T.C. Editorial: Toward neurobiological-based treatments of depression and anxiety: A potential case for the nucleus accumbens. J. Am. Acad. Child Adolesc. Psychiatry 2022, 61, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Gallay, C.C.; Forsyth, G.; Can, A.T.; Dutton, M.; Jamieson, D.; Jensen, E.; Hermens, D.F.; Bennett, M.R.; Lagopoulos, J. Six-week oral ketamine treatment for chronic suicidality is associated with increased grey matter volume. Psychiatry Res. Neuroimaging 2021, 317, 111369. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Li, M.X.; Xu, C.; Chen, H.B.; An, S.C.; Ma, X.M. Dendritic spines in depression: What we learned from animal models. Neural Plast. 2016, 2016, 20–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, T.C.; Chandra, R.; Friend, D.M.; Finkel, E.; Dayrit, G.; Miranda, J.; Brooks, J.M.; Iñiguez, S.D.; O’Donnell, P.; Kravitz, A.; et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol. Psychiatry 2015, 77, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, M.E.; Chandra, R.; Menken, M.S.; Larkin, E.J.; Nam, H.; Engeln, M.; Francis, T.C.; Lobo, M.K. Dendritic remodeling of D1 neurons by RhoA/Rho-kinase mediates depression-like behavior. Mol. Psychiatry 2018, 25, 1022–1034. [Google Scholar] [CrossRef]
- Martinowich, K.; Manji, H.; Lu, B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 2007, 10, 1089–1093. [Google Scholar] [CrossRef]
- McEwen, B.S.; Magarinos, A.M. Stress Effects on Morphology and Function of the Hippocampus. Ann. N. Y. Acad. Sci. 1997, 821, 271–284. [Google Scholar] [CrossRef]
- Willner, P. Chronic mild stress (CMS) revisited: Consistency and behavioural- neurobiological concordance in the effects of CMS. Neuropsychobiology 2005, 52, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.C.; Cao, X.; Das, M.; Zhu, X.H.; Gao, T.M. Behavioral animal models of depression. Neurosci. Bull. 2010, 26, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrén, E.; Võikar, V.; Rantamäki, T. Role of neurotrophic factors in depression. Curr. Opin. Pharmacol. 2007, 7, 18–21. [Google Scholar] [CrossRef]
- Castrén, E.; Rantamäki, T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev. Neurobiol. 2010, 70, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.T.; Weickert, C.S.; Wyatt, E.; Webster, M.J. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry Neurosci. 2011, 36, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal volume reduction in major depression. Am. J. Psychiatry 2000, 157, 115–117. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, H.; Wei, G.; Dong, Z.; Zhao, H.; Han, X.; Song, X.; Zhang, H.; Zong, X.; Baloch, Z.; et al. Identification of key genes, pathways, and miRNA/mRNA regulatory networks of CUMS-induced depression in nucleus accumbens by integrated bioinformatics analysis. Neuropsychiatr. Dis. Treat. 2019, 15, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lai, S.; Wang, R.; Zhou, T.; Dong, N.; Zhu, L.; Chen, T.; Zhang, X.; Chen, Y. Dopamine D3 receptor in the nucleus accumbens alleviates neuroinflammation in a mouse model of depressive-like behavior. Brain. Behav. Immun. 2022, 101, 165–179. [Google Scholar] [CrossRef]
- Caviedes, A.; Lafourcade, C.; Soto, C.; Wyneken, U. BDNF/NF-κB signaling in the neurobiology of depression. Curr. Pharm. Des. 2017, 23, 3154–3163. [Google Scholar] [CrossRef]
- Weiss, A.P.; Goff, D.; Schacter, D.L.; Ditman, T.; Freudenreich, O.; Henderson, D.; Heckers, S. Fronto-hippocampal function during temporal context monitoring in schizophrenia. Biol. Psychiatry 2006, 60, 1268–1277. [Google Scholar] [CrossRef]
- O’Donnell, P.; Greene, J.; Pabello, N.; Lewis, B.L.; Grace, A.A. Modulation of Cell Firing in the Nucleus Accumbens. Ann. N. Y. Acad. Sci. 1999, 877, 157–175. [Google Scholar] [CrossRef]
- Lodge, D.J.; Grace, A.A. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J. Neurosci. 2007, 27, 11424–11430. [Google Scholar] [CrossRef] [PubMed]
- Silbersweig, D.A.; Stern, E.; Frith, C.; Cahill, C.; Holmes, A.; Grootoonk, S.; Seaward, J.; Mc Kenna, P.; Chua, S.E.; Schnorr, L.; et al. A functional neuroanatomy of hallucinations in schizophrenia. Nature 1995, 378, 176–179. [Google Scholar] [CrossRef]
- Belujon, P.; Patton, M.H.; Grace, A.A. Role of the prefrontal cortex in altered hippocampal-accumbens synaptic plasticity in a developmental animal model of schizophrenia. Cereb. Cortex 2014, 24, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Molina, V.; Sanz, J.; Sarramea, F.; Benito, C.; Palomo, T. Prefrontal atrophy in first episodes of schizophrenia associated with limbic metabolic hyperactivity. J. Psychiatr. Res. 2005, 39, 117–127. [Google Scholar] [CrossRef]
- Grace, A.A.; Floresco, S.B.; Goto, Y.; Lodge, D.J. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007, 30, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Mogenson, G.J.; Yang, C.R.; Yim, C.Y. Influence of dopamine on limbic inputs to the nucleus accumbens. Ann. N. Y. Acad. Sci. 1988, 537, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Bird, E.D.; Spokes, E.G.S.; Iversen, L.L. Increased dopamine concentration in limbic areas of brain from patients dying with schizophrenia. Brain 1979, 102, 347–360. [Google Scholar] [CrossRef]
- Mackay, A.V.P.; Iversen, L.L.; Rossor, M.; Spokes, E.; Bird, E.; Arregui, A.; Creese, I.; Snyder, S.H. Increased brain dopamine and dopamine receptors in schizophrenia. Arch. Gen. Psychiatry 1982, 39, 991–997. [Google Scholar] [CrossRef]
- McCollum, L.A.; McCullumsmith, R.E.; Roberts, R.C. Tyrosine hydroxylase localization in the nucleus accumbens in schizophrenia. Brain Struct. Funct. 2016, 221, 4451–4458. [Google Scholar] [CrossRef]
- Pillai, A. Brain-derived neurotropic factor/TrkB signaling in the pathogenesis and novel pharmacotherapy of schizophrenia. NeuroSignals 2008, 16, 183–193. [Google Scholar] [CrossRef]
- Durany, N.; Thome, J. Neurotrophic factors and the pathophysiology of schizophrenic psychoses. Eur. Psychiatry 2004, 19, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Weickert, C.S.; Hyde, T.M.; Lipska, B.K.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2003, 8, 592–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Bergen, S.E.; Nguyen, Q.L.; Xu, B.; Monteggia, L.M.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 2005, 25, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iritani, S.; Niizato, K.; Nawa, H.; Ikeda, K.; Emson, P.C. Immunohistochemical study of brain-derived neurotrophic factor and its receptor, TrkB, in the hippocampal formation of schizophrenic brains. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2003, 27, 801–807. [Google Scholar] [CrossRef]
- Durany, N.; Michel, T.; Zöchling, R.; Boissl, K.W.; Cruz-Sánchez, F.F.; Riederer, P.; Thome, J. Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr. Res. 2001, 52, 79–86. [Google Scholar] [CrossRef]
- Fang, X.; Chen, Y.; Wang, Y.; Ren, J.; Zhang, C. Depressive symptoms in schizophrenia patients: A possible relationship between SIRT1 and BDNF. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2019, 95, 109673. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 2003, 60, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Castrén, E. Neurotrophic effects of antidepressant drugs. Curr. Opin. Pharmacol. 2004, 4, 58–64. [Google Scholar] [CrossRef]
- Pandey, G.N.; Ren, X.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Dwivedi, Y. Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int. J. Neuropsychopharmacol. 2008, 11, 1047–1061. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Chen, Z.Y. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol. Sin. 2011, 32, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Tebartz Van Elst, L.; Woermann, F.; Lemieux, L.; Trimble, M.R. Increased amygdala volumes in female and depressed humans. A quantitative magnetic resonance imaging study. Neurosci. Lett. 2000, 281, 103–106. [Google Scholar] [CrossRef]
- Frodl, T.; Meisenzahl, E.; Zetzsche, T.; Bottlender, R.; Born, C.; Groll, C.; Jäger, M.; Leinsinger, G.; Hahn, K.; Möller, H.J. Enlargement of the amygdala in patients with a first episode of major depression. Biol. Psychiatry 2002, 51, 708–714. [Google Scholar] [CrossRef]
- Tendilla-Beltrán, H.; Coatl-Cuaya, H.; Meneses-Prado, S.; Vázquez-Roque, R.A.; Brambila, E.; Tapia-Rodríguez, M.; Martín-Hernández, D.; Garcés-Ramírez, L.; Madrigal, J.L.M.; Leza, J.C.; et al. Neuroplasticity and inflammatory alterations in the nucleus accumbens are corrected after risperidone treatment in a schizophrenia-related developmental model in rats. Schizophr. Res. 2021, 235, 17–28. [Google Scholar] [CrossRef]
- Thylur, D.S.; Goldsmith, D.R. Brick by brick: Building a transdiagnostic understanding of inflammation in psychiatry. Harv. Rev. Psychiatry 2022, 30, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Sun, Y.; Wan, S.; Zhao, H.; Liu, R.; Li, X.; Wu, S.; Nedelska, Z.; Hort, J.; Qing, Z.; et al. Subregional structural alterations in hippocampus and nucleus accumbens correlate with the clinical impairment in patients with Alzheimer’s Disease clinical spectrum: Parallel combining volume and vertex-based approach. Front. Neurol. 2017, 8, 15. [Google Scholar] [CrossRef]
- Kawakami, I.; Hasegawa, M.; Arai, T.; Ikeda, K.; Oshima, K.; Niizato, K.; Aoki, N.; Omi, K.; Higashi, S.; Hosokawa, M.; et al. Tau accumulation in the nucleus accumbens in tangle-predominant dementia. Acta Neuropathol. Commun. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobili, A.; Latagliata, E.C.; Viscomi, M.T.; Cavallucci, V.; Cutuli, D.; Giacovazzo, G.; Krashia, P.; Rizzo, F.R.; Marino, R.; Federici, M.; et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Cordella, A.; Krashia, P.; Nobili, A.; Pignataro, A.; la Barbera, L.; Viscomi, M.T.; Valzania, A.; Keller, F.; Ammassari-Teule, M.; Mercuri, N.B.; et al. Dopamine loss alters the hippocampus-nucleus accumbens synaptic transmission in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2018, 116, 142–154. [Google Scholar] [CrossRef]
- Fernández-Pérez, E.J.; Gallegos, S.; Armijo-Weingart, L.; Araya, A.; Riffo-Lepe, N.O.; Cayuman, F.; Aguayo, L.G. Changes in neuronal excitability and synaptic transmission in nucleus accumbens in a transgenic Alzheimer’s disease mouse model. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef]
- Voineskos, A.N.; Lerch, J.P.; Felsky, D.; Shaikh, S.; Rajji, T.K.; Miranda, D.; Lobaugh, N.J.; Mulsant, B.H.; Pollock, B.G.; Kennedy, J.L. The brain-derived neurotrophic factor Val66Met polymorphism and prediction of neural risk for alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-S.; Yang, W.; Pan, Y.-F.; Min, J.; Zhang, Z.; Gao, H.-Z.; Qi, J.-S. Brain-derived neurotrophic factor prevents against amyloid beta protein-induced impairment of hippocampal in vivo long-term potentiation in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2012, 28, 425–429. [Google Scholar] [PubMed]
- Rohe, M.; Synowitz, M.; Glass, R.; Paul, S.M.; Nykjaer, A.; Willnow, T.E. Brain-derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. J. Neurosci. 2009, 29, 15472–15478. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Beiser, A.S.; Choi, S.H.; Preis, S.R.; Chen, T.C.; Vorgas, D.; Au, R.; Pikula, A.; Wolf, P.A.; DeStefano, A.L.; et al. Serum brain-derived neurotrophic factor and the risk for dementia: The framingham heart study. JAMA Neurol. 2014, 71, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Laske, C.; Stellos, K.; Hoffmann, N.; Stransky, E.; Straten, G.; Eschweiler, G.W.; Leyhe, T. Higher BDNF serum levels predict slower cognitive decline in Alzheimer’s disease patients. Int. J. Neuropsychopharmacol. 2011, 14, 399–404. [Google Scholar] [CrossRef]
- Guo, C.; Wen, D.; Zhang, Y.; Mustaklem, R.; Mustaklem, B.; Zhou, M.; Ma, T.; Ma, Y.-Y. Amyloid-β oligomers in the nucleus accumbens decrease motivation via insertion of calcium-permeable AMPA receptors. Mol. Psychiatry 2022, 1–12. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb. Exp. Pharmacol. 2015, 220, 223–250. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.P.; Feng, Z.G.; Yuan, B.L.; Yu, S.Z.; Li, Q.; Qu, H.Y.; Sun, M.J. Transduced PTD-BDNF fusion protein protects against beta amyloid peptide-induced learning and memory deficits in mice. Brain Res. 2008, 1191, 12–19. [Google Scholar] [CrossRef]
- Connor, B.; Young, D.; Yan, Q.; Faull, R.L.M.; Synek, B.; Dragunow, M. Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Mol. Brain Res. 1997, 49, 71–81. [Google Scholar] [CrossRef]
- Michalski, B.; Fahnestock, M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer’s disease. Mol. Brain Res. 2003, 111, 148–154. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Song, J.H.; Yu, J.T.; Tan, L. Brain-derived neurotrophic factor in alzheimer’s disease: Risk, mechanisms, and therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef]
- Beal, M.F.; Bird, E.D.; Langlais, P.J.; Martin, J.B. Somatostatin is increased in the nucleus accumbens in huntington’s disease. Neurology 1984, 34, 663–666. [Google Scholar] [CrossRef]
- Van den Bogaard, S.J.A.; Dumas, E.M.; Acharya, T.P.; Johnson, H.; Langbehn, D.R.; Scahill, R.I.; Tabrizi, S.J.; van Buchem, M.A.; van Der Grond, J.; Roos, R.A.C. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J. Neurol. 2011, 258, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, M.; Iritani, S.; Fujishiro, H.; Torii, Y.; Habuchi, C.; Sekiguchi, H.; Yoshida, M.; Ozaki, N. Clinicopathological differences between the motor onset and psychiatric onset of Huntington’s disease, focusing on the nucleus accumbens. Neuropathology 2019, 39, 331–341. [Google Scholar] [CrossRef]
- Beal, M.F.; Mazurek, M.F.; Ellison, D.W.; Swartz, K.J.; McGarvey, U.; Bird, E.D.; Martin, J.B. Somatostatin and neuropeptide Y concentrations in pathologically graded cases of huntington’s disease. Ann. Neurol. 1988, 23, 562–569. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kowall, N.W.; Beal, M.F.; Martin, J.B.; Bird, E.D.; Richardson, E.P. Morphologic and histochemical characteristics of a spared subset of striatal neurons in huntington’s disease. J. Neuropathol. Exp. Neurol. 1987, 46, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B.; Youngblood, W.W.; Manberg, P.J.; Prange, A.J.; Kizer, J.S. Regional brain concentrations of neuropeptides in Huntington’s chorea and schizophrenia. Science 1983, 221, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Palacios, J.M.; Rigo, M.; Chinaglia, G.; Probst, A. Reduced density of striatal somatostatin receptors in Huntington’s chorea. Brain Res. 1990, 522, 342–346. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kowall, N.W.; Richardson, E.P.; Bird, E.D.; Martin, J.B. Topography of enkephalin, substance P and acetylcholinesterase staining in Huntington’s disease striatum. Neurosci. Lett. 1986, 71, 283–288. [Google Scholar] [CrossRef]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 1987, 22, 386–389. [Google Scholar] [CrossRef]
- Ellison, D.W.; Beal, M.F.; Mazurek, M.F.; Malloy, J.R.; Bird, E.D.; Martin, J.B. Amino acid neurotransmitter abnormalities in huntington’s disease and the quinolinic acid animal model of Huntington’s disease. Brain 1987, 110, 1657–1673. [Google Scholar] [CrossRef] [PubMed]
- Hammond, P.; Brimijoin, S. Acetylcholinesterase in Huntington’s and Alzheimer’s diseases: Simultaneous enzyme assay and immunoassay of multiple brain regions. J. Neurochem. 1988, 50, 1111–1116. [Google Scholar] [CrossRef]
- Turjanski, N.; Weeks, R.; Dolan, R.; Harding, A.E.; Brooks, D.J. Striatal D1 and D2 receptor binding in patients with huntington’s disease and other choreas A PET study. Brain 1995, 118, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Ginovart, N.; Lundin, A.; Farde, L.; Halldin, C.; Bäckman, L.; Swahn, C.G.; Pauli, S.; Sedvall, G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain 1997, 120, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Couly, S.; Paucard, A.; Bonneaud, N.; Maurice, T.; Benigno, L.; Jourdan, C.; Cohen-Solal, C.; Vignes, M.; Maschat, F. Improvement of BDNF signalling by P42 peptide in Huntington’s disease. Hum. Mol. Genet. 2018, 27, 3012–3028. [Google Scholar] [CrossRef]
- Gharami, K.; Xie, Y.; An, J.J.; Tonegawa, S.; Xu, B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J. Neurochem. 2008, 105, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, V.; Giralt, A.; Masana, M.; Royes, A.; Espina, M.; Sieiro, E.; Alberch, J.; Castañé, A.; Girault, J.A.; Ginés, S. Cyclin-dependent kinase 5 dysfunction contributes to depressive-like behaviors in huntington’s disease by altering the DARPP-32 phosphorylation status in the nucleus accumbens. Biol. Psychiatry 2019, 86, 196–207. [Google Scholar] [CrossRef]
- Khan, A.R.; Hiebert, N.M.; Vo, A.; Wang, B.T.; Owen, A.M.; Seergobin, K.N.; MacDonald, P.A. Biomarkers of Parkinson’s disease: Striatal sub-regional structural morphometry and diffusion MRI. NeuroImage Clin. 2019, 21, 101597. [Google Scholar] [CrossRef]
- Mavridis, I.; Boviatsis, E.; Anagnostopoulou, S. The human nucleus accumbens suffers parkinsonism-related shrinkage: A novel finding. Surg. Radiol. Anat. 2011, 33, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Carriere, N.; Besson, P.; Dujardin, K.; Duhamel, A.; Defebvre, L.; Delmaire, C.; Devos, D. Apathy in Parkinson’s disease is associated with nucleus accumbens atrophy: A magnetic resonance imaging shape analysis. Mov. Disord. 2014, 29, 897–903. [Google Scholar] [CrossRef]
- Mavridis, I.N. Is nucleus accumbens atrophy correlated with cognitive symptoms of Parkinson’s disease? Brain 2015, 138, e319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinaz, S.; Kamel, S.; Aravala, S.S.; Sezgin, M.; Elfil, M.; Sinha, R. Distinct neural circuits are associated with subclinical neuropsychiatric symptoms in Parkinson’s disease. J. Neurol. Sci. 2021, 423. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, P.; Hapuarachchi, B.; Djamshidian, A.; Strand, K.; Lees, A.J.; de Silva, R.; Holton, J.L.; Warner, T.T. Lower nucleus accumbens a-synuclein load and D3 receptor levels in Parkinson’s disease with impulsive compulsive behaviours. Brain 2019, 142, 3580–3591. [Google Scholar] [CrossRef]
- White, F.J. Dopamine receptors get a boost. Nature 2001, 411, 35–38. [Google Scholar] [CrossRef]
- Bamford, N.S.; Wang, W. Corticostriatal plasticity in the nucleus accumbens core. J. Neurosci. Res. 2019, 97, 1559–1578. [Google Scholar] [CrossRef]
- Hammes, J.; Theis, H.; Giehl, K.; Hoenig, M.C.; Greuel, A.; Tittgemeyer, M.; Timmermann, L.; Fink, G.R.; Drzezga, A.; Eggers, C.; et al. Dopamine metabolism of the nucleus accumbens and fronto-striatal connectivity modulate impulse control. Brain 2019, 142, 733–743. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayassi-Jakowicka, M.; Lietzau, G.; Czuba, E.; Patrone, C.; Kowiański, P. More than Addiction—The Nucleus Accumbens Contribution to Development of Mental Disorders and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 2618. https://doi.org/10.3390/ijms23052618
Bayassi-Jakowicka M, Lietzau G, Czuba E, Patrone C, Kowiański P. More than Addiction—The Nucleus Accumbens Contribution to Development of Mental Disorders and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(5):2618. https://doi.org/10.3390/ijms23052618
Chicago/Turabian StyleBayassi-Jakowicka, Martyna, Grazyna Lietzau, Ewelina Czuba, Cesare Patrone, and Przemysław Kowiański. 2022. "More than Addiction—The Nucleus Accumbens Contribution to Development of Mental Disorders and Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 5: 2618. https://doi.org/10.3390/ijms23052618