
Role of the Transcription Factor FOSL1 in Organ Development and Tumorigenesis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. FOSL1 Binding to the DNA and Its Interactions with Other Transcription Factors
1.2. Tissue-Specific Expression of FOSL1 Gene
2. Stabilization and Degradation of FOSL1 in the Cell
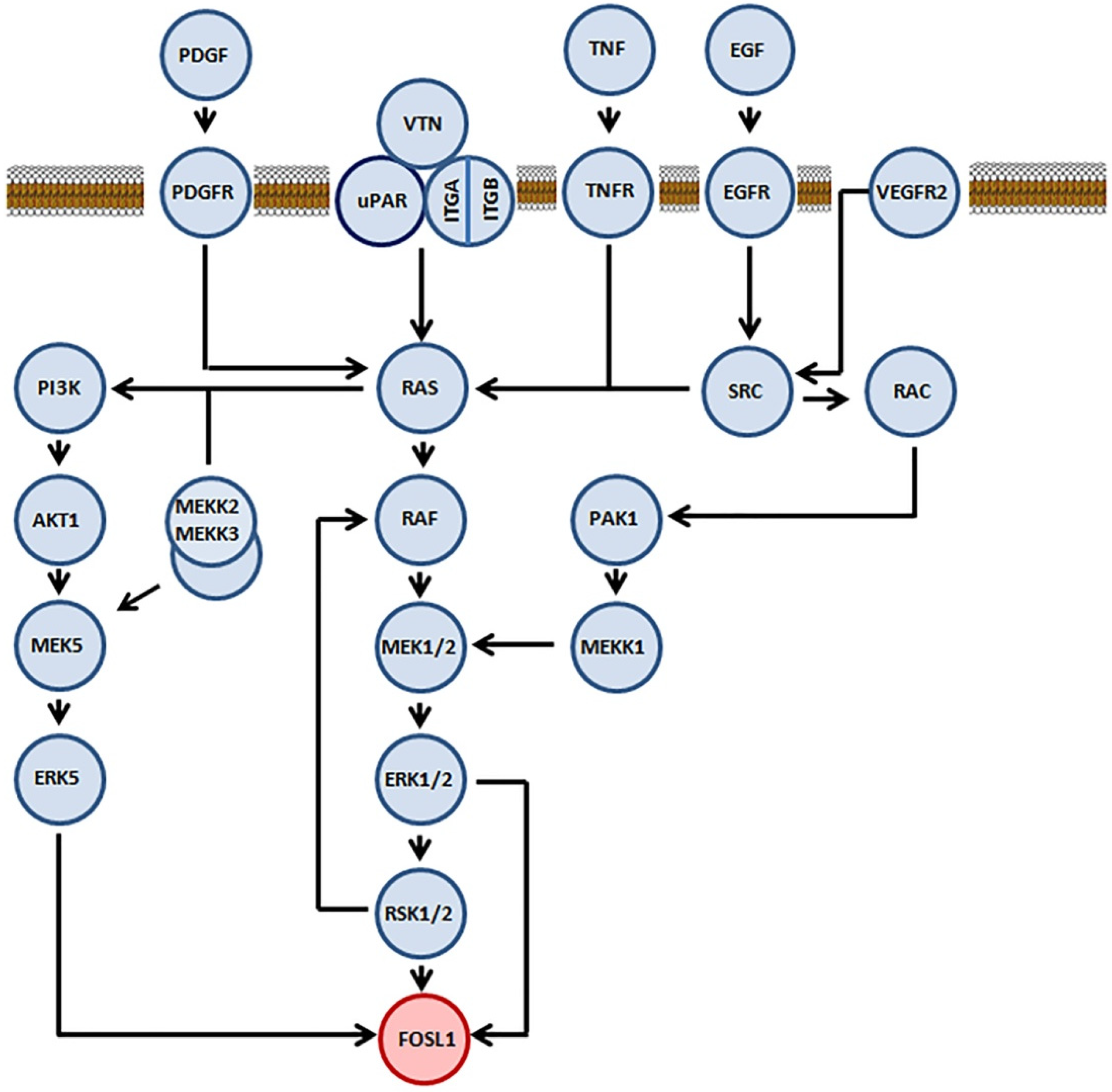
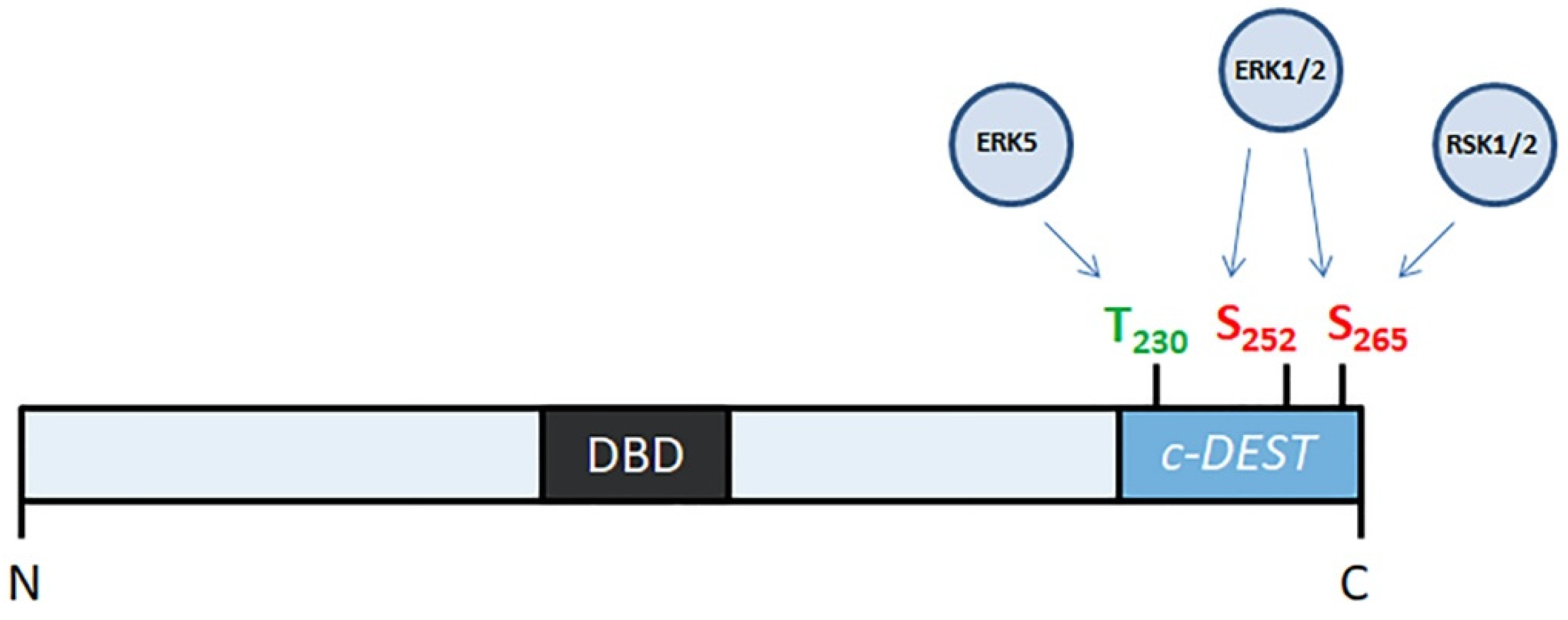
2.1. Role of Mitogen-Activated Protein Kinases in Stabilization of FOSL1
2.2. FOSL1 Phosphorylation by PKCθ
2.3. Degradation of FOSL1 in Proteasomes
3. Role of FOSL1 in Regulation of Gene Expression
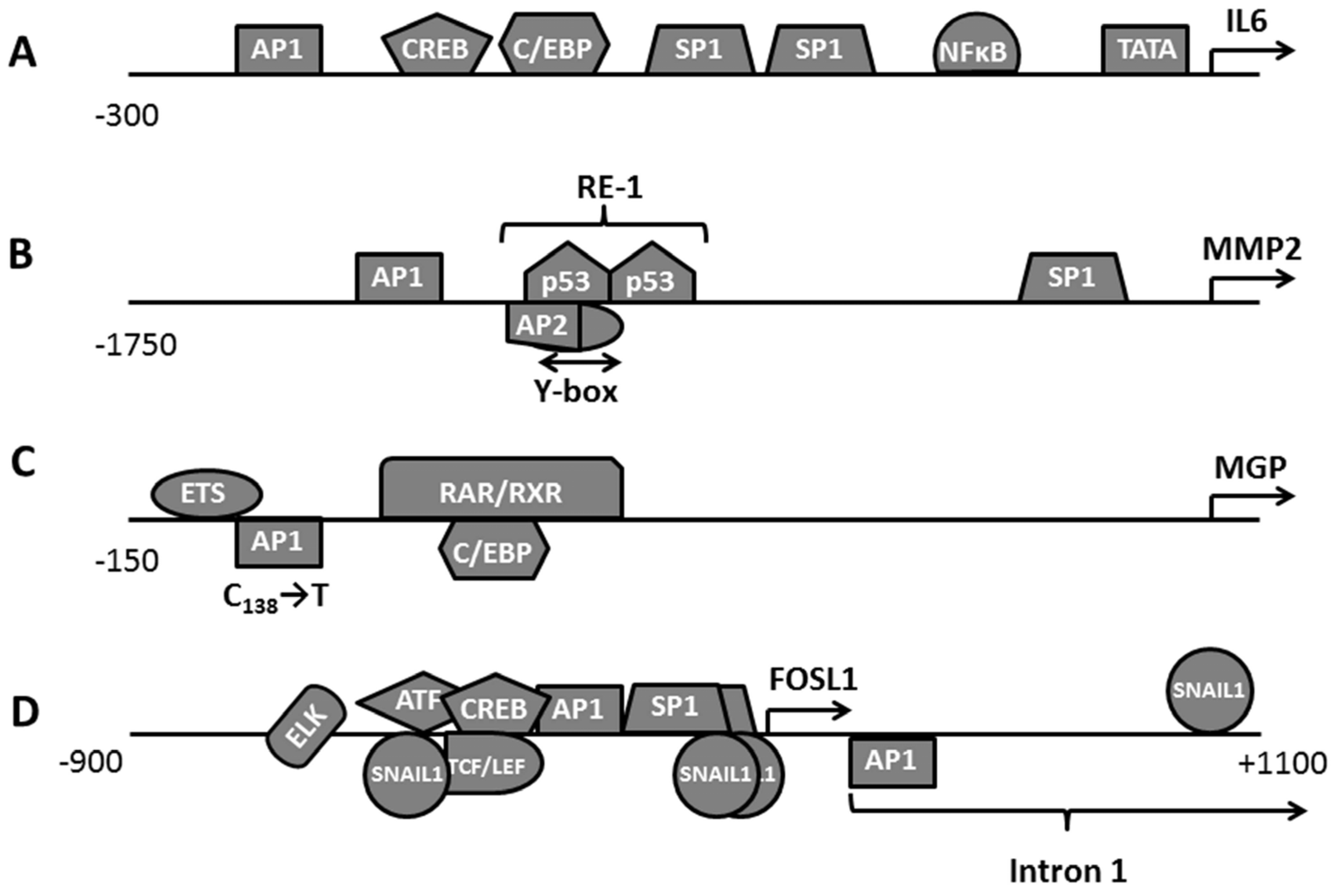
3.1. Assessment of FOSL1 Binding Sites in the Genome
3.2. Clustering FOSL1 with Other Transcription Factors and Participation in the Transcription Factors Network
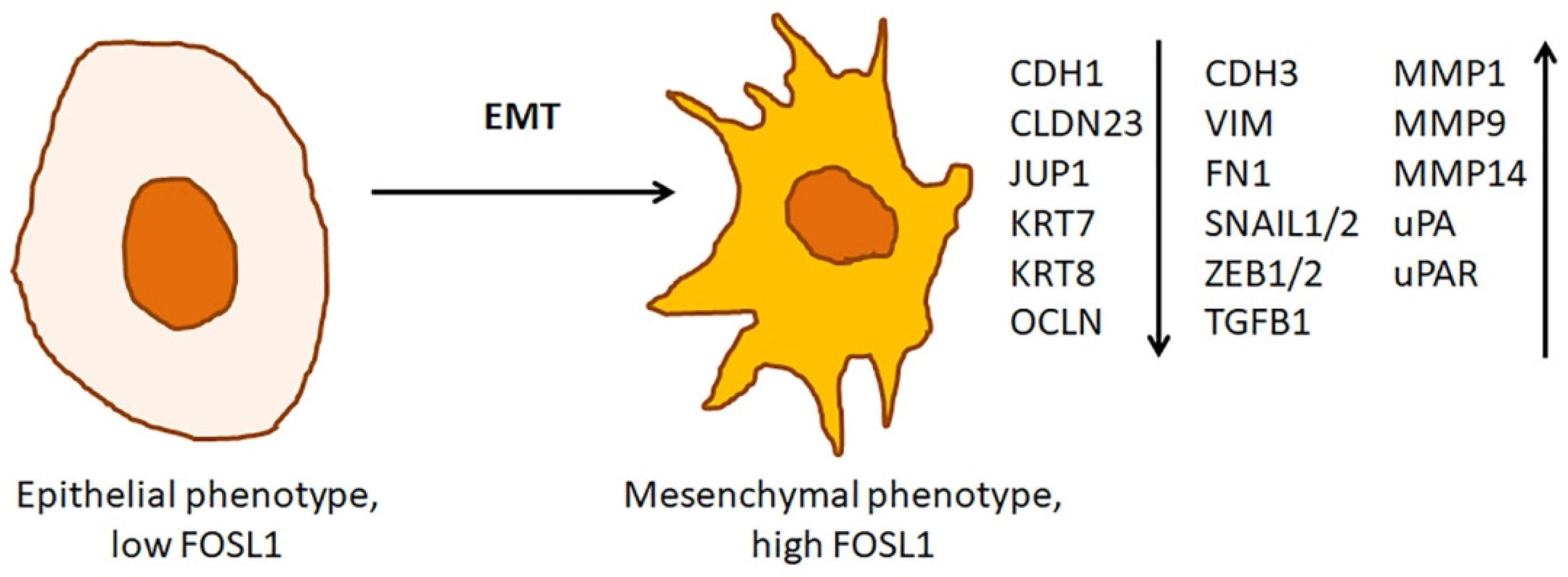
3.3. The Role of FOSL1 in Epithelial-Mesenchymal Transition
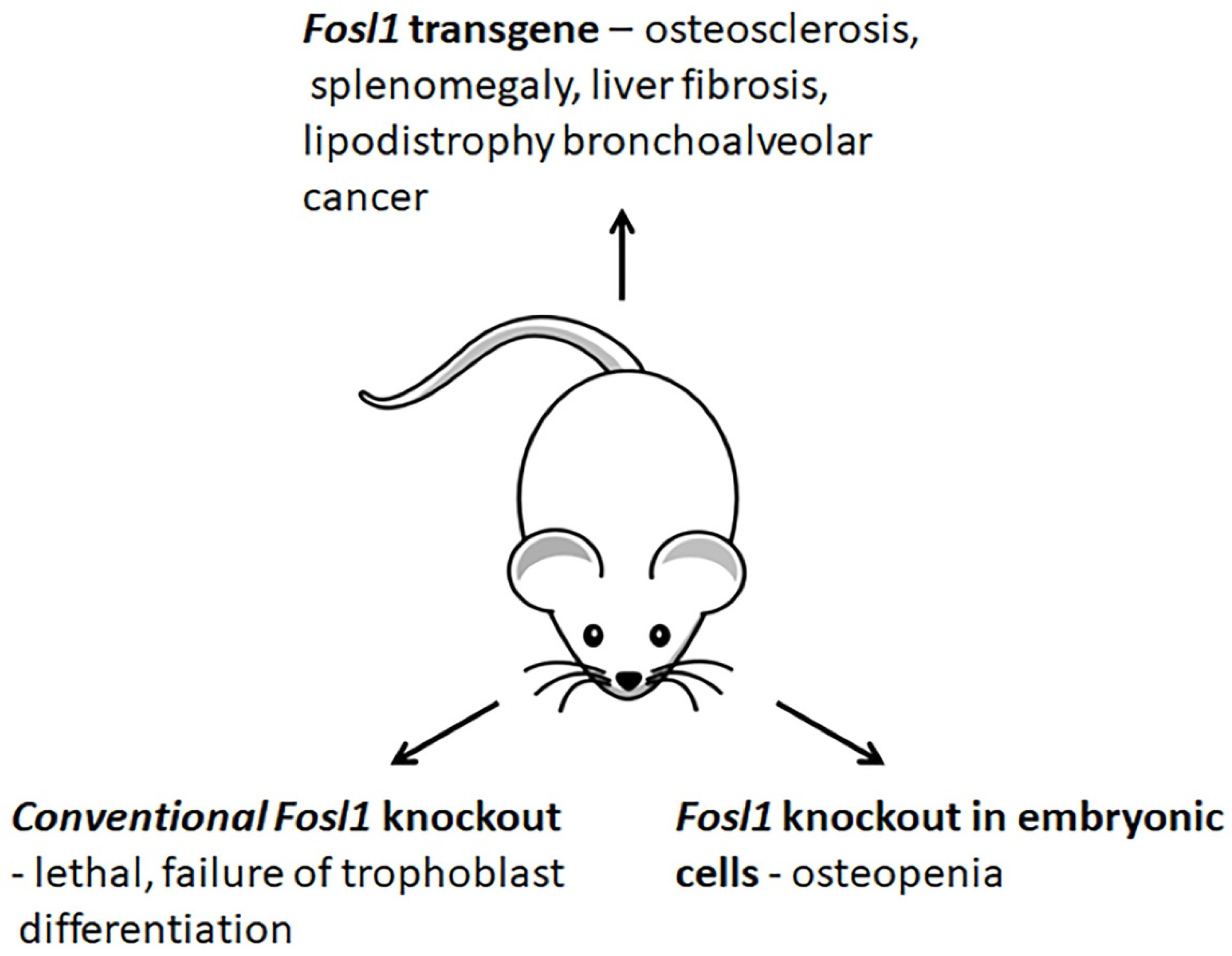
3.4. Changes in Phenotypes of Fosl1-Deficient and -Overexpressing Mice
4. Role of FOSL1 in Tumorigenesis
4.1. FOSL1 Expression Pattern in Solid Tumors
4.2. FOSL1 as a Prognostic Tool
4.3. FOSL1 as a Molecular Target for Anticancer Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACACA | acetyl CoA carboxylase 1 |
| ADIPOQ | adiponectin |
| ALP | alkaline phosphatase |
| AP1 | activator protein 1 |
| BGN | biglycan |
| BGLAP | osteocalcin encoding gene |
| CCND1 | cyclin D1 |
| CDH1 | cadherin E |
| ChIP | chromatin immunoprecipitation |
| CRE | cyclic AMP responsive element |
| CREB | cAMP response element-binding protein |
| DCN | decorin |
| ECM | extracellular matrix |
| EMT | epithelial-mesenchymal transformation |
| ER | estrogen receptor |
| FN1 | fibronectin |
| FAK | focal adhesion kinase |
| FASN | fatty acid synthase |
| FOSL1 | FOSL1 encoding gene |
| LEP | leptin |
| LPL | lipoprotein lipase |
| LPS | lipopolysaccharides |
| MCP | macrophage chemoattractant protein |
| MEK | mitogen-activated or extracellular signal-regulated protein kinase |
| MET | mesenchymal-epithelial transformation |
| MGP | matrix Gla protein |
| MMP | metalloproteinase |
| PKC | protein kinase C |
| PNPLA2 | patatin-like phospholipase domain-containing 2, lipolytic enzyme |
| PXN | paxillin |
| RETN | resistin |
| SCD1 | stearoyl CoA desaturase |
| SNP | single nucleotide polymorphism |
| TNBC | triple negative breast carcinomas |
| TRE | TPA responsive element |
| VIM | vimentin |
References
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta 2019, 1872, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Pognonec, P.; E Boulukos, K.; Aperlo, C.; Fujimoto, M.; Ariga, H.; Nomoto, A.; Kato, H. Cross-family interaction between the bHLHZip USF and bZip Fra1 proteins results in down-regulation of AP1 activity. Oncogene 1997, 14, 2091–2098. [Google Scholar] [CrossRef] [Green Version]
- Samoylenko, A.; Dimova, E.Y.; Horbach, T.; Teplyuk, N.; Immenschuh, S.; Kietzmann, T. Opposite Expression of the Antioxidant Heme Oxygenase-1 in Primary Cells and Tumor Cells: Regulation by Interaction of USF-2 and Fra-1. Antioxid. Redox Signal. 2008, 10, 1163–1174. [Google Scholar] [CrossRef]
- Evellin, S.; Galvagni, F.; Zippo, A.; Neri, F.; Orlandini, M.; Incarnato, D.; Dettori, D.; Neubauer, S.; Kessler, H.; Wagner, E.F.; et al. FOSL1 Controls the Assembly of Endothelial Cells into Capillary Tubes by Direct Repression of αv and β3 Integrin Transcription. Mol. Cell. Biol. 2013, 33, 1198–1209. [Google Scholar] [CrossRef] [Green Version]
- Burch, P.M.; Yuan, Z.; Loonen, A.; Heintz, N.H. An Extracellular Signal-Regulated Kinase 1- and 2-Dependent Program of Chromatin Trafficking of c-Fos and Fra-1 Is Required for Cyclin D1 Expression during Cell Cycle Reentry. Mol. Cell. Biol. 2004, 24, 4696–4709. [Google Scholar] [CrossRef] [Green Version]
- Motrich, R.D.; Castro, G.M.; Caputto, B.L. Old Players with a Newly Defined Function: Fra-1 and c-Fos Support Growth of Human Malignant Breast Tumors by Activating Membrane Biogenesis at the Cytoplasm. PLoS ONE 2013, 8, e53211. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Song, S.; Zhang, D.; Zhang, Y.; Chen, L.; Qian, L.; Shi, M.; Zhao, H.; Jiang, Z.; Guo, N. An association of a simultaneous nuclear and cytoplasmic localization of Fra-1 with breast malignancy. BMC Cancer 2006, 6, 298. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, M.; Wang, Z.; Jochum, W.; Fetka, I.; Elliott, C.; Wagner, E.F. Placental vascularisation requires the AP-1 component fra1. Development 2000, 127, 4937–4948. [Google Scholar] [CrossRef]
- Eferl, R.; Hoebertz, A.; Schilling, A.F.; Rath, M.; Karreth, F.; Kenner, L.; Amling, M.; Wagner, E.F. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 2004, 23, 2789–2799. [Google Scholar] [CrossRef]
- Persico, A.M.; Schindler, C.W.; O’Hara, B.F.; Brannock, M.T.; Uhl, G.R. Brain transcription factor expression: Effects of acute and chronic amphetamine and injection stress. Mol. Brain Res. 1993, 20, 91–100. [Google Scholar] [CrossRef]
- Hannan, R.D.; Stennard, F.A.; West, A.K. Expression of c-fos and Related Genes in the Rat Heart in Response to Norepinephrine. J. Mol. Cell. Cardiol. 1993, 25, 1137–1148. [Google Scholar] [CrossRef]
- Park, K.; Chung, M.; Kim, S. Inhibition of myogenesis by okadaic acid, an inhibitor of protein phosphatases, 1 and 2A, correlates with the induction of AP1. J. Biol. Chem. 1992, 267, 10810–10815. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Grigoriadis, A.E.; Wagner, E.F. Stable murine chondrogenic cell lines derived from c-fos-induced cartilage tumors. J. Bone Miner. Res. 2009, 8, 839–847. [Google Scholar] [CrossRef]
- Adiseshaiah, P.; Papaiahgari, S.R.; Vuong, H.; Kalvakolanu, D.V.; Reddy, S.P. Multiple cis-Elements Mediate the Transcriptional Activation of Human fra-1 by 12-O-Tetradecanoylphorbol-13-acetate in Bronchial Epithelial Cells. J. Biol. Chem. 2003, 278, 47423–47433. [Google Scholar] [CrossRef] [Green Version]
- Sobolev, V.V.; Zolotarenko, A.D.; Soboleva, A.G.; E Sautin, M.; A Il’Ina, S.; Sarkisova, M.K.; Golukhova, E.Z.; Elkin, A.M.; A Bruskin, S.; Abdeev, R.M. Expression of the FOSL1 gene in psoriasis and atherosclerosis. Russ. J. Genet. 2010, 46, 104–110. [Google Scholar] [CrossRef]
- Boise, L.H.; Petryniak, B.; Mao, X.; June, C.H.; Wang, C.Y.; Lindsten, T.; Bravo, R.; Kovary, K.; Leiden, J.M.; Thompson, C.B. The NFAT-1 DNA binding complex in activated T cells contains Fra-1 and JunB. Mol. Cell. Biol. 1993, 13, 1911–1919. [Google Scholar]
- Braselmann, S.; Graninger, P.; Busslinger, M. A selective transcriptional induction system for mammalian cells based on Gal4-estrogen receptor fusion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 1657–1661. [Google Scholar] [CrossRef] [Green Version]
- Stephens, J.M.; Butts, M.; Stone, R.; Pekala, P.H.; Bernlohr, D.A. Regulation of transcription factor mRNA accumulation during 3T3-L1 preadipocyte differentiation by antagonists of adipogenesis. Mol. Cell. Biochem. 1993, 123, 63–71. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Tulchinsky, E. FRA-1 as a driver of tumour heterogeneity: A nexus between oncogenes and embryonic signalling pathways in cancer. Oncogene 2014, 34, 4421–4428. [Google Scholar] [CrossRef] [Green Version]
- Terasawa, K.; Okazaki, K.; Nishida, E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells 2003, 8, 263–273. [Google Scholar] [CrossRef]
- Tower, G.B.; Coon, C.I.; Belguise, K.; Chalbos, D.; Brinckerhoff, C.E. Fra-1 targets the AP-1 site/2G single nucleotide polymorphism (ETS site) in the MMP-1 promoter. Eur. J. Biochem. 2003, 270, 4216–4225. [Google Scholar] [CrossRef] [Green Version]
- Gruda, M.C.; Kovary, K.; Metz, R.; Bravo, R. Regulation of Fra-1 and Fra-2 phosphorylation differs during the cell cycle of fibroblasts and phosphorylation in vitro by MAP kinase affects DNA binding activity. Oncogene 1994, 9, 2537–2547. [Google Scholar]
- Doehn, U.; Hauge, C.; Frank, S.R.; Jensen, C.J.; Duda, K.; Nielsen, J.V.; Cohen, M.S.; Johansen, J.V.; Winther, B.R.; Lund, L.R.; et al. RSK Is a Principal Effector of the RAS-ERK Pathway for Eliciting a Coordinate Promotile/Invasive Gene Program and Phenotype in Epithelial Cells. Mol. Cell 2009, 35, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Vial, E.; Marshall, C.J. Elevated ERK-MAP kinase activity protects the FOS family member FRA-1 against proteasomal degradation in colon carcinoma cells. J. Cell Sci. 2003, 116, 4957–4963. [Google Scholar] [CrossRef] [Green Version]
- Hollestelle, A.; Elstrodt, F.; Nagel, J.H.; Kallemeijn, W.W.; Schutte, M. Phosphatidylinositol-3-OH Kinase or RAS Pathway Mutations in Human Breast Cancer Cell Lines. Mol. Cancer Res. 2007, 5, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Belguise, K.; Milord, S.; Galtier, F.; Moquet-Torcy, G.; Piechaczyk, M.; Chalbos, D. The PKCθ pathway participates in the aberrant accumulation of Fra-1 protein in invasive ER-negative breast cancer cells. Oncogene 2012, 31, 4889–4897. [Google Scholar] [CrossRef] [Green Version]
- Delpire, E.; Gagnon, K.B.E. Genome-wide analysis of SPAK/OSR1 binding motifs. Physiol. Genom. 2007, 28, 223–231. [Google Scholar] [CrossRef]
- Gomard, T.; Jariel-Encontre, I.; Basbous, J.; Bossis, G.; Mocquet-Torcy, G.; Piechaczyk, M. Fos family protein degradation by the proteasome. Biochem. Soc. Trans. 2008, 36, 858–863. [Google Scholar] [CrossRef]
- Zhang, M.; Pickart, C.M.; Coffino, P. Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J. 2003, 22, 1488–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakay, J.L.; Diesch, J.; Gilan, O.; Yip, Y.-Y.; Sayan, E.; Kolch, W.; Mariadason, J.M.; Hannan, R.D.; Tulchinsky, E.; Dhillon, A.S. A 19S proteasomal subunit cooperates with an ERK MAPK-regulated degron to regulate accumulation of Fra-1 in tumour cells. Oncogene 2011, 31, 1817–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basbous, J.; Chalbos, D.; Hipskind, R.; Jariel-Encontre, I.; Piechaczyk, M. Ubiquitin-Independent Proteasomal Degradation of Fra-1 Is Antagonized by Erk1/2 Pathway-Mediated Phosphorylation of a Unique C-Terminal Destabilizer. Mol. Cell. Biol. 2007, 27, 3936–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, L.O.; MacKeigan, J.; Blenis, J. A Network of Immediate Early Gene Products Propagates Subtle Differences in Mitogen-Activated Protein Kinase Signal Amplitude and Duration. Mol. Cell. Biol. 2004, 24, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Stelniec-Klotz, I.; Legewie, S.; Tchernitsa, O.; Witzel, F.; Klinger, B.; Sers, C.; Herzel, H.; Blüthgen, N.; Schäfer, R. Reverse engineering a hierarchical regulatory network downstream of oncogenic KRAS. Mol. Syst. Biol. 2012, 8, 601. [Google Scholar] [CrossRef]
- Diesch, J.; Sanij, E.; Gilan, O.; Love, C.; Tran, H.; Fleming, N.I.; Ellul, J.; Amalia, M.; Haviv, I.; Pearson, R.; et al. Widespread FRA1-Dependent Control of Mesenchymal Transdifferentiation Programs in Colorectal Cancer Cells. PLoS ONE 2014, 9, e88950. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Manzano, G.; Lovett, D.H.; Kim, H.T. Role of AP-1 and RE-1 binding sites in matrix metalloproteinase-2 transcriptional regulation in skeletal muscle atrophy. Biochem. Biophys. Res. Commun. 2010, 396, 219–223. [Google Scholar] [CrossRef]
- Xiao, W.; Hodge, D.R.; Wang, L.; Yang, X.; Zhang, X.; Farrar, W.L. NF-kappaB activates IL-6 expression through cooperation with c-Jun and IL6-AP1 site, But is independent of its IL6-NFkappaB regulatory site in autocrine human multiple myeloma cells. Cancer Biol. Ther. 2004, 3, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Hershko, D.D.; Robb, B.W.; Luo, G.; Hasselgren, P.-O. Multiple transcription factors regulating the IL-6 gene are activated by cAMP in cultured Caco-2 cells. Am. J. Physiol. Integr. Comp. Physiol. 2002, 283, R1140–R1148. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Hodge, D.R.; Wang, L.; Yang, X.; Zhang, X.; Farrar, W.L. Co-operative functions between nuclear factors NF?B and CCAT/enhancer-binding protein-? (C/EBP-?) regulate the IL-6 promoter in autocrine human prostate cancer cells. Prostate 2004, 61, 354–370. [Google Scholar] [CrossRef]
- Farzaneh-Far, A.; Davies, J.D.; Braam, L.A.; Spronk, H.M.; Proudfoot, D.; Chan, S.-W.; O’Shaughnessy, K.M.; Weissberg, P.L.; Vermeer, C.; Shanahan, C. A Polymorphism of the Human Matrix γ-Carboxyglutamic Acid Protein Promoter Alters Binding of an Activating Protein-1 Complex and Is Associated with Altered Transcription and Serum Levels. J. Biol. Chem. 2001, 276, 32466–32473. [Google Scholar] [CrossRef] [Green Version]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guío-Carrión, A.; Hasenfuss, S.C.; Eger, A.; Müller, M.; Beug, H.; Wagner, E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2014, 22, 336–350. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Qiao, Y.; Jonsson, P.; Wang, J.; Xu, L.; Rouhi, P.; Sinha, I.; Cao, Y.; Williams, C.; Dahlman-Wright, K. Genome-wide Profiling of AP-1–Regulated Transcription Provides Insights into the Invasiveness of Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 3983–3994. [Google Scholar] [CrossRef] [Green Version]
- Sobolev, V.V.; Zolotorenko, A.D.; Soboleva, A.G.; Elkin, A.M.; Il’Ina, S.A.; Serov, D.N.; Potekaev, N.N.; Tkachenko, S.B.; Minnibaev, M.T.; Piruzyan, A.L. Effects of Expression of Transcriptional Factor AP-1 FOSL1 Gene on Psoriatic Process. Bull. Exp. Biol. Med. 2011, 150, 632–634. [Google Scholar] [CrossRef]
- Wang, W.-M.; Wu, S.-Y.; Lee, A.-Y.; Chiang, C.-M. Binding Site Specificity and Factor Redundancy in Activator Protein-1-driven Human Papillomavirus Chromatin-dependent Transcription. J. Biol. Chem. 2011, 286, 40974–40986. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, W.; De Bosscher, K.; Boone, E.; Plaisance, S.; Haegeman, G. The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J. Biol. Chem. 1999, 274, 32091–32098. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Pan, H.; Li, J.; Zhong, Q.; Chen, X.; Dry, S.M.; Wang, C.-Y. Epigenetic Activation of AP1 Promotes Squamous Cell Carcinoma Metastasis. Sci. Signal. 2013, 6, ra28. [Google Scholar] [CrossRef] [Green Version]
- Crish, J.F.; Eckert, R.L. Synergistic Activation of Human Involucrin Gene Expression by Fra-1 and p300—Evidence for the Presence of a Multiprotein Complex. J. Investig. Dermatol. 2008, 128, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Vincenti, M.P. The matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) genes. Transcriptional and posttranscriptional regulation, signal transduction and cell-type-specific expression. Methods Mol. Biol. 2001, 151, 121–148. [Google Scholar]
- Mertens, P.R.; Alfonso-Jaume, M.A.; Steinmann, K.; Lovett, D.H. A Synergistic Interaction of Transcription Factors AP2 and YB-1 Regulates Gelatinase A Enhancer-dependent Transcription. J. Biol. Chem. 1998, 273, 32957–32965. [Google Scholar] [CrossRef] [Green Version]
- Mertens, P.R.; Steinmann, K.; Alfonso-Jaume, M.A.; En-Nia, A.; Sun, Y.; Lovett, D.H. Combinatorial Interactions of p53, Activating Protein-2, and YB-1 with a Single Enhancer Element Regulate Gelatinase A Expression in Neoplastic Cells. J. Biol. Chem. 2002, 277, 24875–24882. [Google Scholar] [CrossRef] [Green Version]
- Bergman, M.R.; Cheng, S.; Honbo, N.; Piacentini, L.; Karliner, J.S.; Lovett, D.H. A functional activating protein 1 (AP-1) site regulates matrix metalloproteinase 2 (MMP-2) transcription by cardiac cells through interactions with JunB-Fra1 and JunB-FosB heterodimers. Biochem. J. 2003, 369, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Farzaneh-Far, A.; Weissberg, P.; Proudfoot, D.; Shanahan, C. Transcriptional regulation of matrix gla protein. Z. Kardiol. 2001, 90 (Suppl. 3), 38–42. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Zhang, Y.; Yu, W.; Zhang, C.; Gong, L.; Shao, L.; Lu, J.; Gao, Y.; Chen, X.; et al. Common Genetic Variants of MGP Are Associated With Calcification on the Arterial Wall but Not With Calcification Present in the Atherosclerotic Plaques. Circ. Cardiovasc. Genet. 2013, 6, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Roustazadeh, A.; Amirfarhangi, A.; Kazemi, B. Matrix Gla protein (MGP) promoter polymorphic variants and its serum level in stenosis of coronary artery. Mol. Biol. Rep. 2014, 41, 1779–1786. [Google Scholar] [CrossRef]
- Lu, X.; Gao, B.; Liu, Z.; Tian, X.; Mao, X.; Emmanuel, N.; Zhu, Q.; Xiao, C. A polymorphism of matrix Gla protein gene is associated with kidney stone in the Chinese Han population. Gene 2012, 511, 127–130. [Google Scholar] [CrossRef]
- Misra, D.; Booth, S.L.; Crosier, M.D.; Ordovas, J.M.; Felson, D.T.; Neogi, T. Matrix Gla Protein Polymorphism, But Not Concentrations, Is Associated with Radiographic Hand Osteoarthritis. J. Rheumatol. 2011, 38, 1960–1965. [Google Scholar] [CrossRef] [Green Version]
- Zippo, A.; Serafini, R.; Rocchigiani, M.; Pennacchini, S.; Krepelova, A.; Oliviero, S. Histone Crosstalk between H3S10ph and H4K16ac Generates a Histone Code that Mediates Transcription Elongation. Cell 2009, 138, 1122–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchernitsa, O.I.; Sers, C.; Zuber, J.; Hinzmann, B.; Grips, M.; Schramme, A.; Lund, P.; Schwendel, A.; Rosenthal, A.; Schäfer, R.; et al. Transcriptional basis of KRAS oncogene-mediated cellular transformation in ovarian epithelial cells. Oncogene 2004, 23, 4536–4555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlobec, I.; Lugli, A. Epithelial mesenchymal transition and tumor budding in aggressive colorectal cancer: Tumor budding as oncotarget. Oncotarget 2010, 1, 651–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T. To differentiate or not — routes towards metastasis. Nat. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Lee, B.-K.; Uprety, N.; Jang, Y.J.; Tucker, S.K.; Rhee, C.; LeBlanc, L.; Beck, S.; Kim, J. Fosl1 overexpression directly activates trophoblast-specific gene expression programs in embryonic stem cells. Stem Cell Res. 2018, 26, 95–102. [Google Scholar] [CrossRef]
- Vaz, M.; Reddy, N.M.; Rajasekaran, S.; Reddy, S.P. Genetic Disruption of Fra-1 Decreases Susceptibility to Endotoxin-Induced Acute Lung Injury and Mortality in Mice. Am. J. Respir. Cell Mol. Biol. 2012, 46, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Takada, Y.; Gresh, L.; Bozec, A.; Ikeda, E.; Kamiya, K.; Watanabe, M.; Kobayashi, K.; Asano, K.; Toyama, Y.; Wagner, E.F.; et al. Interstitial lung disease induced by gefitinib and Toll-like receptor ligands is mediated by Fra-1. Oncogene 2011, 30, 3821–3832. [Google Scholar] [CrossRef] [Green Version]
- Elangovan, I.M.; Vaz, M.; Tamatam, C.R.; Potteti, H.R.; Reddy, N.M.; Reddy, S.P. FOSL1 Promotes Kras-induced Lung Cancer through Amphiregulin and Cell Survival Gene Regulation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 625–635. [Google Scholar] [CrossRef]
- Jochum, W.; David, J.-P.; Elliott, C.; Wutz, A.; Plenk, H.; Matsuo, K.; Wagner, E.F. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med. 2000, 6, 980–984. [Google Scholar] [CrossRef]
- Kireva, T.; Erhardt, A.; Tiegs, G.; Tilg, H.; Denk, H.; Haybaeck, J.; Aigner, E.; Moschen, A.; Distler, J.H.; Schett, G.; et al. Transcription factor Fra-1 induces cholangitis and liver fibrosis. Hepatology 2011, 53, 1287–1297. [Google Scholar] [CrossRef]
- Luther, J.; Driessler, F.; Megges, M.; Hess, A.; Herbort, B.; Mandic, V.; Zaiss, M.; Reichardt, A.; Zech, C.; Tuckermann, J.P.; et al. Elevated Fra-1 expression causes severe lipodystrophy. J. Cell Sci. 2011, 124, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Takada, Y.; Maruyama, K.; Shimoda, K.; Arai, Y.; Nango, N.; Kosaki, N.; Takaishi, H.; Toyama, Y.; Matsuo, K. Fra-1/AP-1 Impairs Inflammatory Responses and Chondrogenesis in Fracture Healing. J. Bone Miner. Res. 2009, 24, 2056–2065. [Google Scholar] [CrossRef]
- Hasenfuss, S.; Bakiri, L.; Thomsen, M.; Williams, E.; Auwerx, J.; Wagner, E.F. Regulation of Steatohepatitis and PPARγ Signaling by Distinct AP-1 Dimers. Cell Metab. 2014, 19, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Shim, J.S. Targeting Epithelial–Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecues 2016, 21, 965. [Google Scholar] [CrossRef] [Green Version]
- Abdulla, N.; Vincent, C.T.; Kaur, M. Mechanistic Insights Delineating the Role of Cholesterol in Epithelial Mesenchymal Transition and Drug Resistance in Cancer. Front. Cell Dev. Biol. 2021, 9, 728325. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Nowak, E.; Bednarek, I. Aspects of the Epigenetic Regulation of EMT Related to Cancer Metastasis. Cells 2021, 10, 3435. [Google Scholar] [CrossRef]
- Chiappetta, G.; Tallini, G.; De Biasio, M.C.; Pentimalli, F.; de Nigris, F.; Losito, S.; Fedele, M.; Battista, S.; Verde, P.; Santoro, M.; et al. FRA-1 expression in hyperplastic and neoplastic thyroid diseases. Clin. Cancer Res. 2000, 6, 4300–4306. [Google Scholar] [CrossRef] [Green Version]
- Usui, A.; Hoshino, I.; Akutsu, Y.; Sakata, H.; Nishimori, T.; Murakami, K.; Kano, M.; Shuto, K.; Matsubara, H. The molecular role of Fra-1 and its prognostic significance in human esophageal squamous cell carcinoma. Cancer 2011, 118, 3387–3396. [Google Scholar] [CrossRef]
- E Sayan, A.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2011, 31, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- Chiappetta, G.; Ferraro, A.; Botti, G.; Monaco, M.; Pasquinelli, R.; Vuttariello, E.; Arnaldi, L.; Di Bonito, M.; D’Aiuto, G.; Pierantoni, G.M.; et al. FRA-1 protein overexpression is a feature of hyperplastic and neoplastic breast disorders. BMC Cancer 2007, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhao, Y.-P.; Zhang, Y.-Q. Low expression ofFOSL1is associated with favorable prognosis and sensitivity to radiation/pharmaceutical therapy in lower grade glioma. Neurol. Res. 2020, 42, 522–527. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, H.-Y.; Zhong, L.-P.; Wei, K.-J.; Yang, X.; Li, J.; Shen, G.-F.; Zhang, Z. Fos-related activator-1 is overexpressed in oral squamous cell carcinoma and associated with tumor lymph node metastasis. J. Oral Pathol. Med. 2010, 39, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Li, J.; Li, Y.; Ma, Z.; Yu, Y.; Wang, C.-Y. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Logullo, A.F.; Stiepcich, M.M.Á.; Osório, C.A.B.d.T.; Nonogaki, S.; Pasini, F.S.; Rocha, R.M.; Soares, F.A.; Brentani, M.M. Role of Fos-related antigen 1 in the progression and prognosis of ductal breast carcinoma. Histopathology 2011, 58, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, H.; Mizuta, N.; Sakaguchi, K.; Fujiwara, I.; Mizuta, M.; Furukawa, C.; Chang, Y.-C.; Magae, J. Aberrant Expression of Fra-1 in Estrogen Receptor-negative Breast Cancers and Suppression of their Propagation In Vivo by Ascochlorin, an Antibiotic that Inhibits Cellular Activator Protein-1 Activity. J. Antibiot. 2007, 60, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. Interleukin-13 Receptor 2, EphA2, and Fos-Related Antigen 1 as Molecular Denominators of High-Grade Astrocytomas and Specific Targets for Combinatorial Therapy. Clin. Cancer Res. 2008, 14, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Oh, J.H.; Kim, N.H.; Choi, K.M.; Kim, S.J.; Baik, S.H.; Choi, D.S.; Lee, E.S. Fra-1 Expression in Malignant and Benign Thyroid Tumor. Korean J. Intern. Med. 2001, 16, 93–97. [Google Scholar] [CrossRef]
- Marques, C.; Unterkircher, T.; Kroon, P.; Oldrini, B.; Izzo, A.; Dramaretska, Y.; Ferrarese, R.; Kling, E.; Schnell, O.; Nelander, S.; et al. NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1. eLife 2021, 10. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M. Fos-related antigen 1 (Fra-1) pairing with and transactivation of JunB in GBM cells. Cancer Biol. Ther. 2011, 11, 254–262. [Google Scholar] [CrossRef] [Green Version]
- Talotta, F.; Casalino, L.; Verde, P. The nuclear oncoprotein Fra-1: A transcription factor knocking on therapeutic applications’ door. Oncogene 2020, 39, 4491–4506. [Google Scholar] [CrossRef]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Kandelman, J.D.; Waitzberg, A.F.L.; Szejnfeld, J.; Smith, R.L. Expression of claudin, paxillin and FRA-1 in non-nodular breast lesions in association with microcalcifications. Sao Paulo Med. J. 2013, 131, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Kharman-Biz, A.; Gao, H.; Ghiasvand, R.; Zhao, C.; Zendehdel, K.; Dahlman-Wright, K. Expression of activator protein-1 (AP-1) family members in breast cancer. BMC Cancer 2013, 13, 441. [Google Scholar] [CrossRef] [Green Version]
- Serino, L.T.R.; Jucoski, T.S.; Bath de Morais, S.; Fernandes, C.C.C.; de Lima, R.S.; Urban, C.A.; Cavalli, L.R.; Cavalli, I.J.; Ribeiro, E.M.D.S.F. Association of FOSL1 copy number alteration and triple negative breast tumors. Genet. Mol. Biol. 2019, 42, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Reisfeld, R.A. The Tumor Microenvironment: A Target for Combination therapy of Breast Cancer. Crit. Rev. Oncog. 2013, 18, 115–133. [Google Scholar] [CrossRef]
- O’Leary, K.A.; Rugowski, D.E.; Sullivan, R.; Schuler, L.A. Prolactin cooperates with loss of p53 to promote claudin-low mammary carcinomas. Oncogene 2013, 33, 3075–3082. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.C.; Lam, K.Y.; Law, S.; Wong, J.; Srivastava, G. Identification of differentially expressed genes in esophageal squamous cell carcinoma (ESCC) by cDNA expression array: Overexpression of Fra-1, Neogenin, Id-1, and CDC25B genes in ESCC. Clin. Cancer Res. 2001, 7, 2213–2221. [Google Scholar]
- Desmet, C.J.; Gallenne, T.; Prieur, A.; Reyal, F.; Visser, N.L.; Wittner, B.S.; Smit, M.A.; Geiger, T.R.; Laoukili, J.; Iskit, S.; et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5139–5144. [Google Scholar] [CrossRef] [Green Version]
- Britten, C.D. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013, 71, 1395–1409. [Google Scholar] [CrossRef]
- Zeiser, R. Trametinib. In Small Molecules in Oncology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 201, pp. 241–248. [Google Scholar]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Gibney, G.T.; Sondak, V.K.; Smalley, K.S.M. Beyond BRAF: Where next for melanoma therapy? Br. J. Cancer 2015, 112, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Gregory, P.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G. A Double-Negative Feedback Loop between ZEB1-SIP1 and the microRNA-200 Family Regulates Epithelial-Mesenchymal Transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Kim, H.S.; Li, X.-Y.; Lee, I.; Choi, H.-S.; Kang, S.E.; Cha, S.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial–mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Siemens, H.; Jackstadt, R.; Hünten, S.; Kaller, M.; Menssen, A.; Götz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.-H.; Gibbons, D.L.; Chakravarti, D.; Creighton, C.J.; Rizvi, Z.H.; Adams, H.P.; Pertsemlidis, A.; Gregory, P.; Wright, J.A.; Goodall, G.; et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J. Clin. Investig. 2012, 122, 3170–3183. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Li, Y.; Gao, J.; Zhang, T.; Li, S.; Luo, A.; Chen, H.; Ding, F.; Wang, X.; Liu, Z. MicroRNA-34 suppresses breast cancer invasion and metastasis by directly targeting Fra-1. Oncogene 2012, 32, 4294–4303. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, G.; Lv, L.; Ren, Y.-F.; Zhang, X.-J.; Xue, Y.-F.; Li, G.; Lu, X.; Sun, Z.; Tang, K.-F. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis 2011, 33, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, H.; Shiozaki, A.; Bai, X.-H.; Iitaka, D.; Kim, H.; Yang, B.B.; Keshavjee, S.; Liu, M. XB130, a New Adaptor Protein, Regulates Expression of Tumor Suppressive MicroRNAs in Cancer Cells. PLoS ONE 2013, 8, e59057. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Wang, C.; Liu, X.; Mu, W.; Chen, Z.; Yu, D.; Wang, A.; Dai, Y.; Zhou, X. Molecular Characterization of the MicroRNA-138-Fos-like Antigen 1 (FOSL1) Regulatory Module in Squamous Cell Carcinoma. J. Biol. Chem. 2011, 286, 40104–40109. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-K. Circulating microRNAs and long non-coding RNAs in gastric cancer diagnosis: An update and review. World J. Gastroenterol. 2015, 21, 9863–9886. [Google Scholar] [CrossRef]
- Kamath, V. Cancer vaccines: An unkept promise? Drug Discov. Today 2021, 26, 1347–1352. [Google Scholar] [CrossRef]
- Poczobutt, J.M.; De, S.; Yadav, V.; Nguyen, T.T.; Li, H.; Sippel, T.R.; Weiser-Evans, M.C.M.; Nemenoff, R.A. Expression Profiling of Macrophages Reveals Multiple Populations with Distinct Biological Roles in an Immunocompetent Orthotopic Model of Lung Cancer. J. Immunol. 2016, 196, 2847–2859. [Google Scholar] [CrossRef]
- Mandelli, G.E.; Missale, F.; Bresciani, D.; Gatta, L.B.; Scapini, P.; Caveggion, E.; Roca, E.; Bugatti, M.; Monti, M.; Cristinelli, L.; et al. Tumor Infiltrating Neutrophils Are Enriched in Basal-Type Urothelial Bladder Cancer. Cells 2020, 9, 291. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobolev, V.V.; Khashukoeva, A.Z.; Evina, O.E.; Geppe, N.A.; Chebysheva, S.N.; Korsunskaya, I.M.; Tchepourina, E.; Mezentsev, A. Role of the Transcription Factor FOSL1 in Organ Development and Tumorigenesis. Int. J. Mol. Sci. 2022, 23, 1521. https://doi.org/10.3390/ijms23031521
Sobolev VV, Khashukoeva AZ, Evina OE, Geppe NA, Chebysheva SN, Korsunskaya IM, Tchepourina E, Mezentsev A. Role of the Transcription Factor FOSL1 in Organ Development and Tumorigenesis. International Journal of Molecular Sciences. 2022; 23(3):1521. https://doi.org/10.3390/ijms23031521
Chicago/Turabian StyleSobolev, Vladimir V., Asiat Z. Khashukoeva, Olga E. Evina, Natalia A. Geppe, Svetlana N. Chebysheva, Irina M. Korsunskaya, Ekaterina Tchepourina, and Alexandre Mezentsev. 2022. "Role of the Transcription Factor FOSL1 in Organ Development and Tumorigenesis" International Journal of Molecular Sciences 23, no. 3: 1521. https://doi.org/10.3390/ijms23031521