
Modified Low-Temperature Extraction Method for Isolation of Bletilla striata Polysaccharide as Antioxidant for the Prevention of Alzheimer’s Disease


Abstract
:1. Introduction
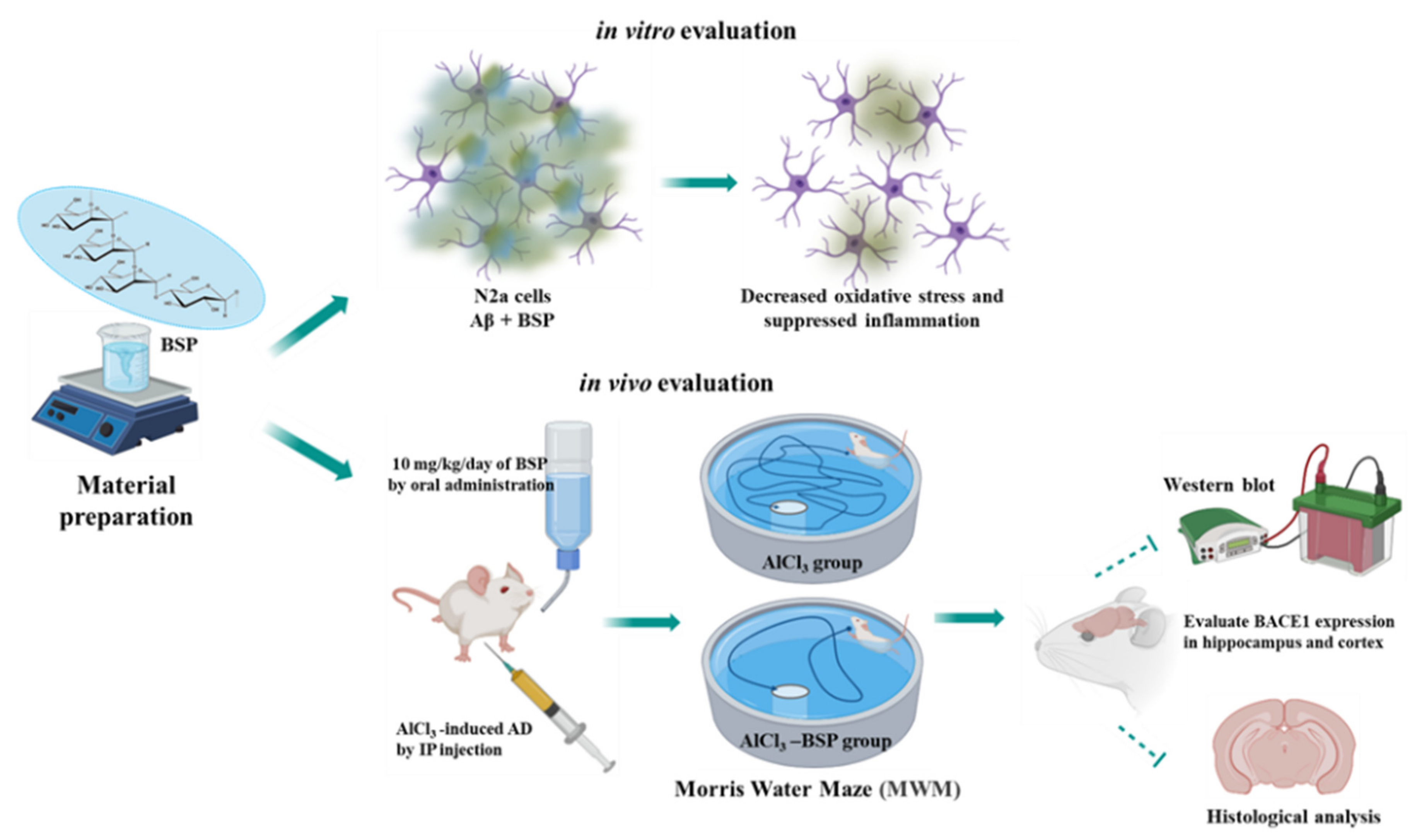
2. Results
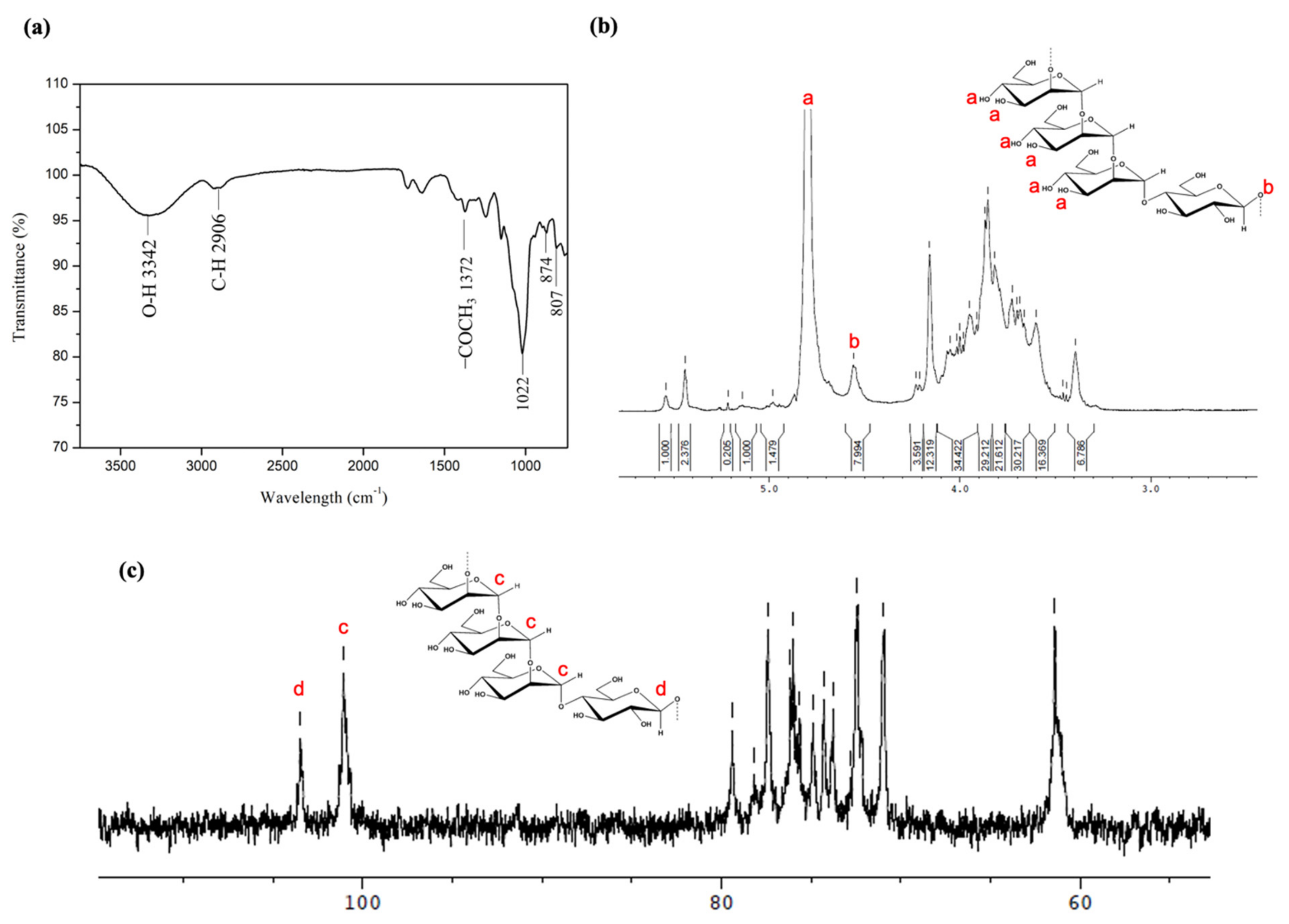
2.1. Identification of Molecular Structure and Functional Group
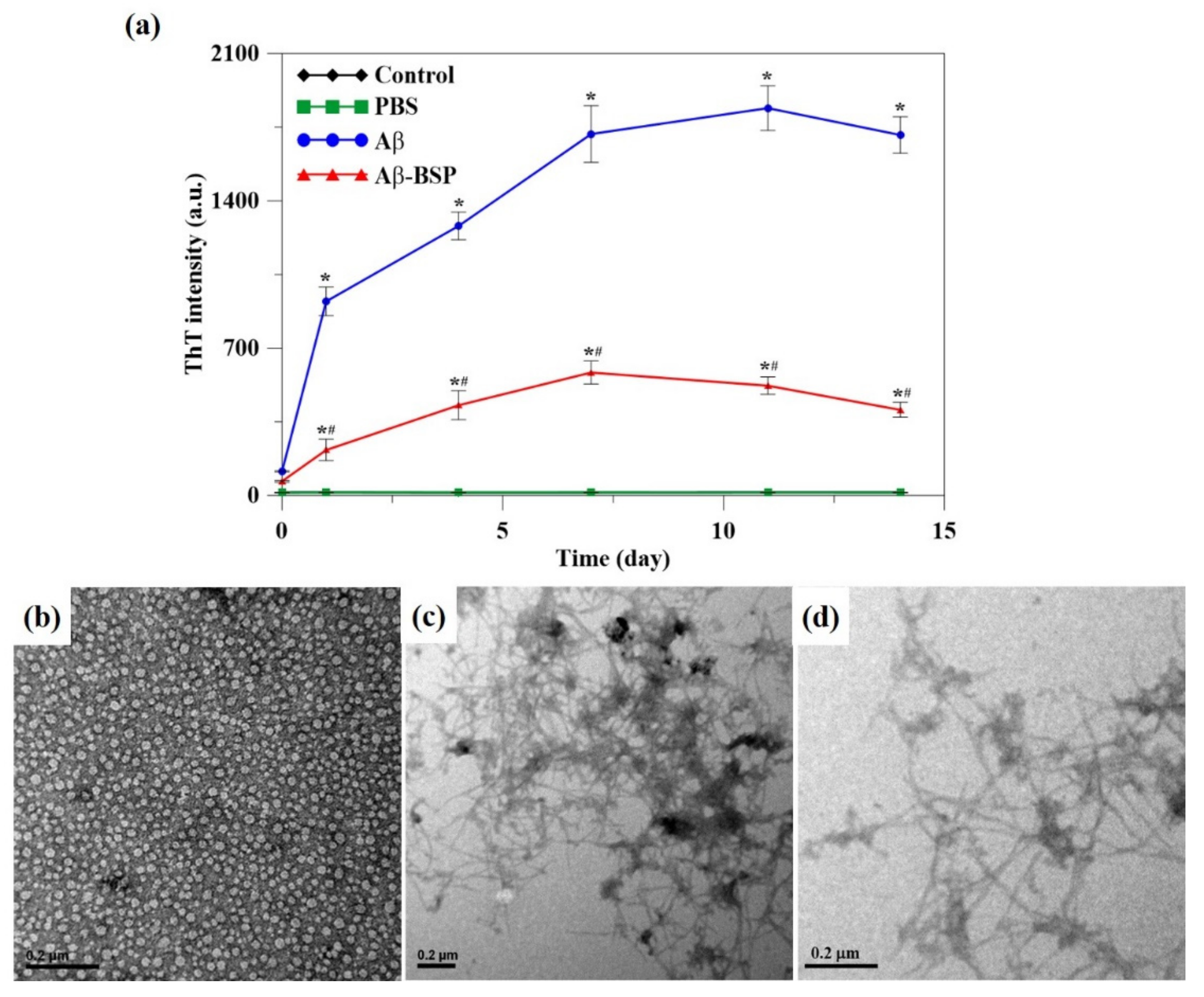
2.2. BSP Inhibit Aβ Fibril Formation
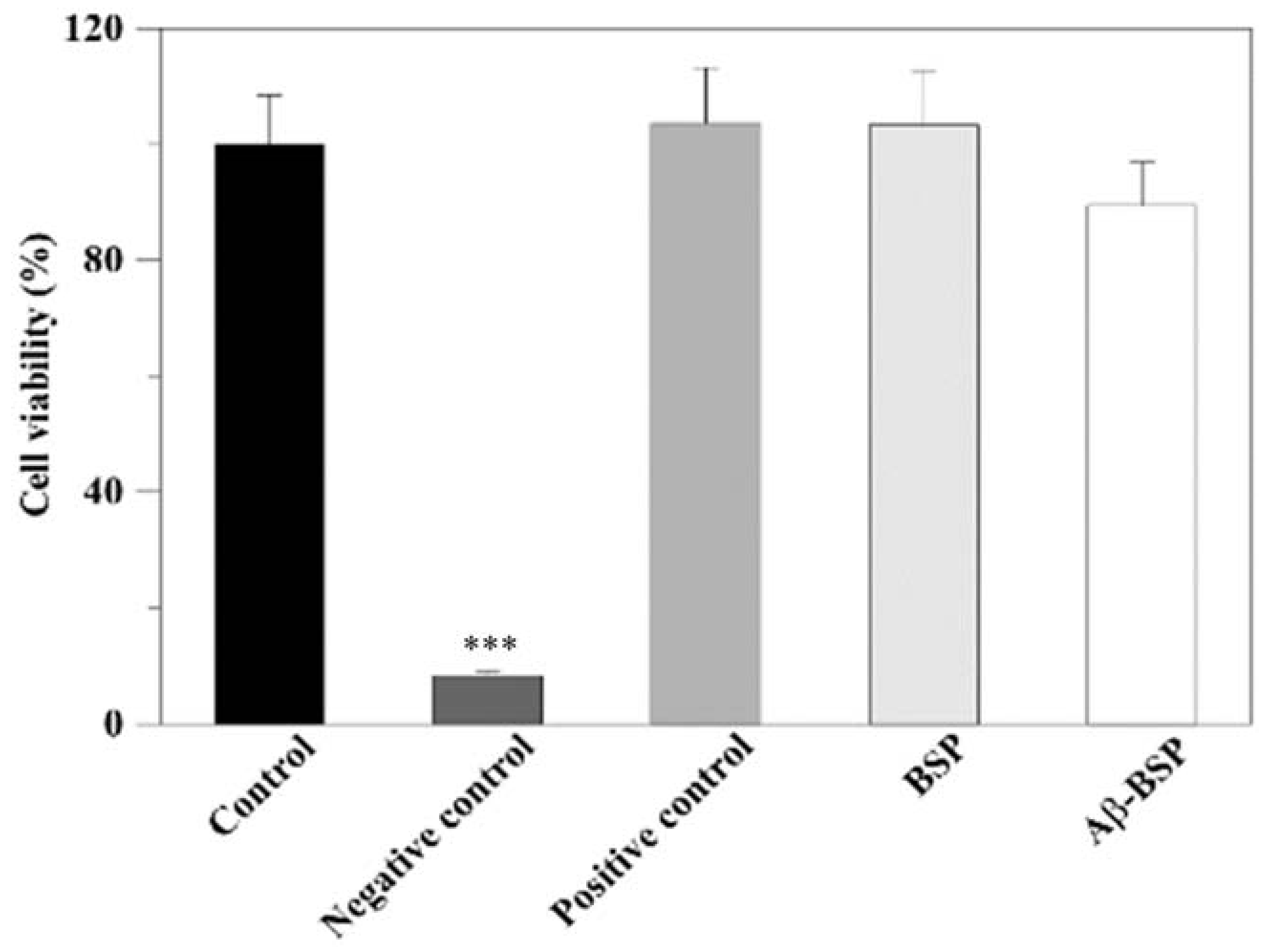
2.3. Effects of BSP on Aβ-Induced Cytotoxicity and Cell Viability
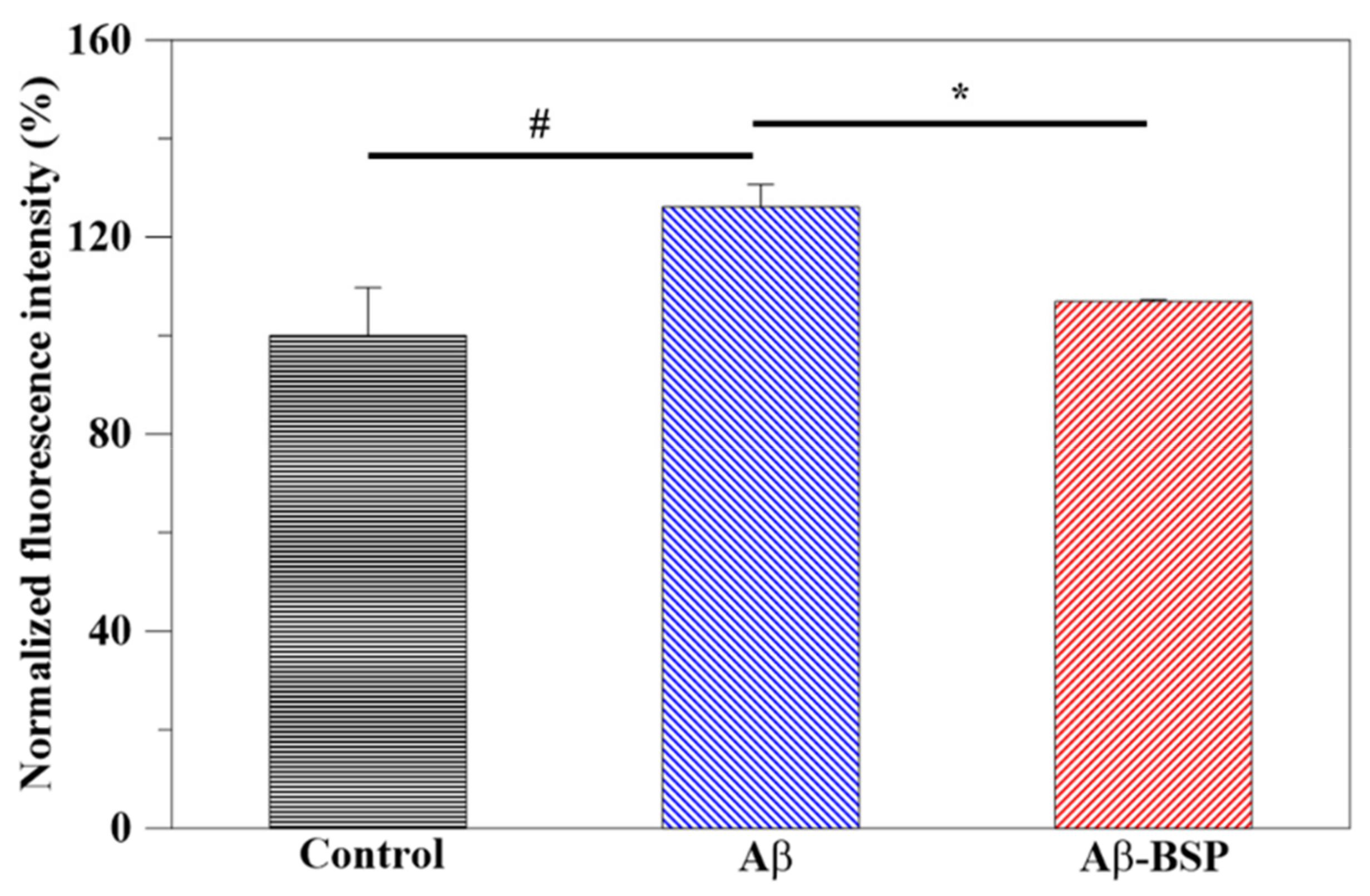
2.4. Antioxidant Activity of BSP
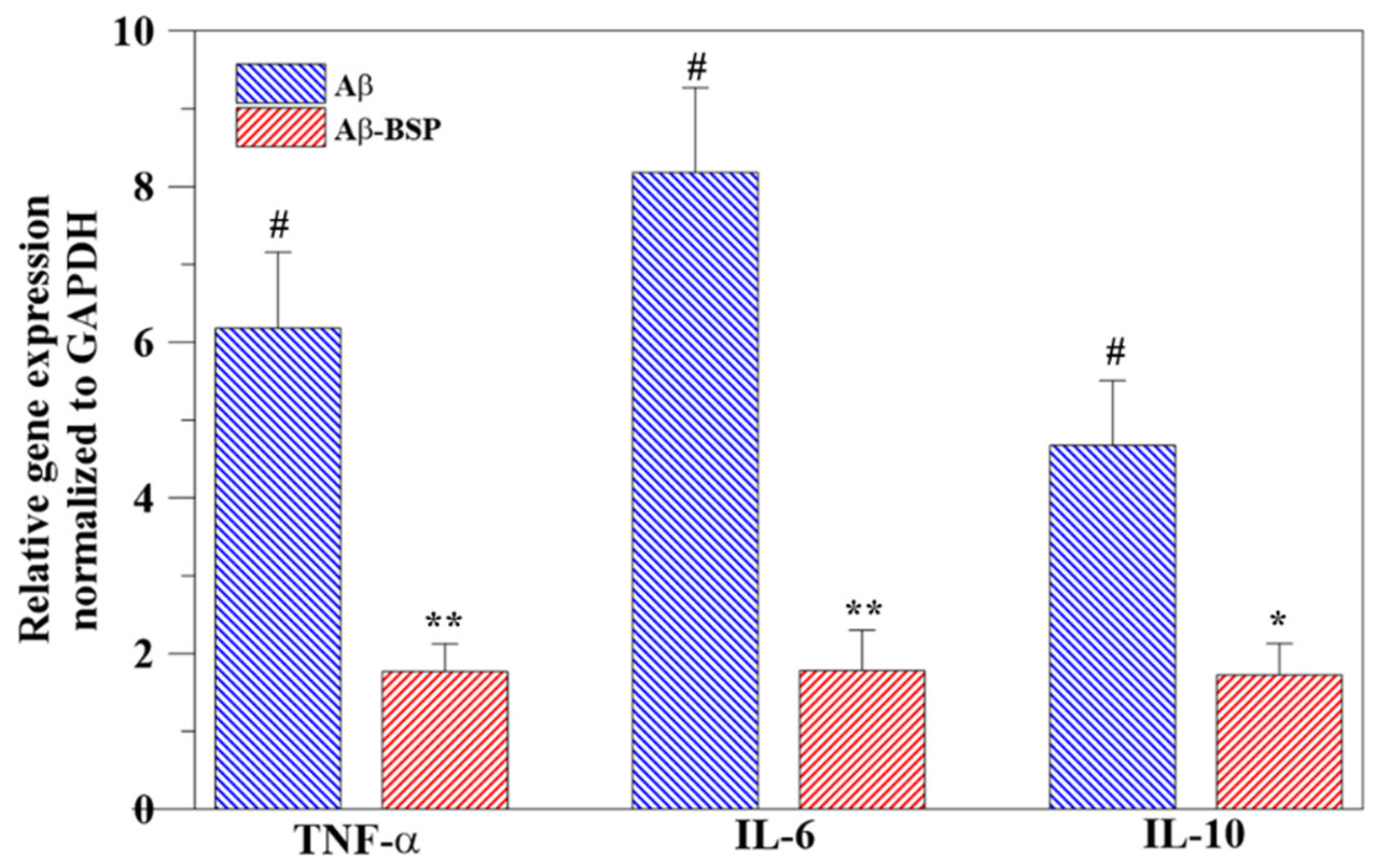
2.5. Anti-Inflammatory Activity of BSP
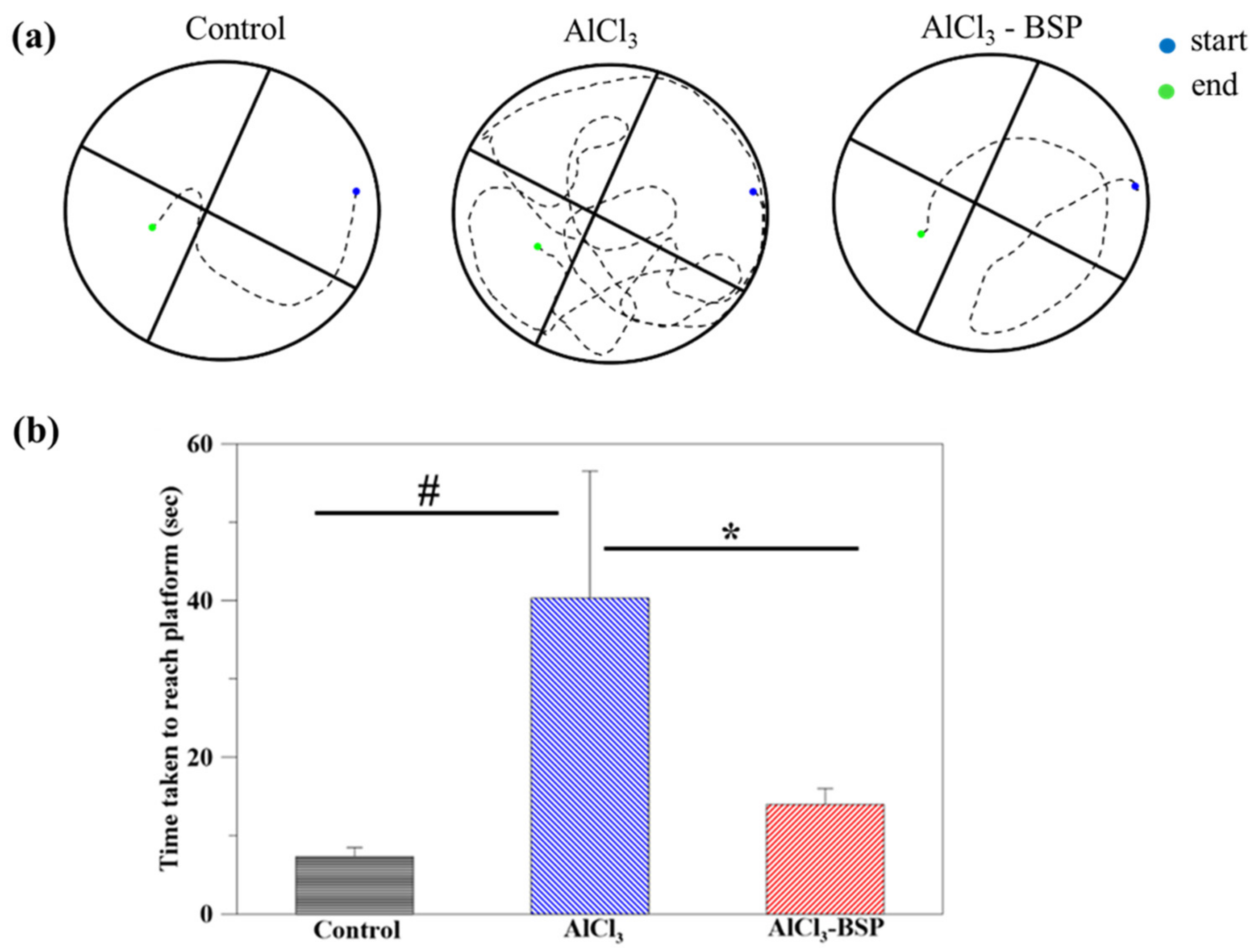
2.6. Morris Water Maze Test
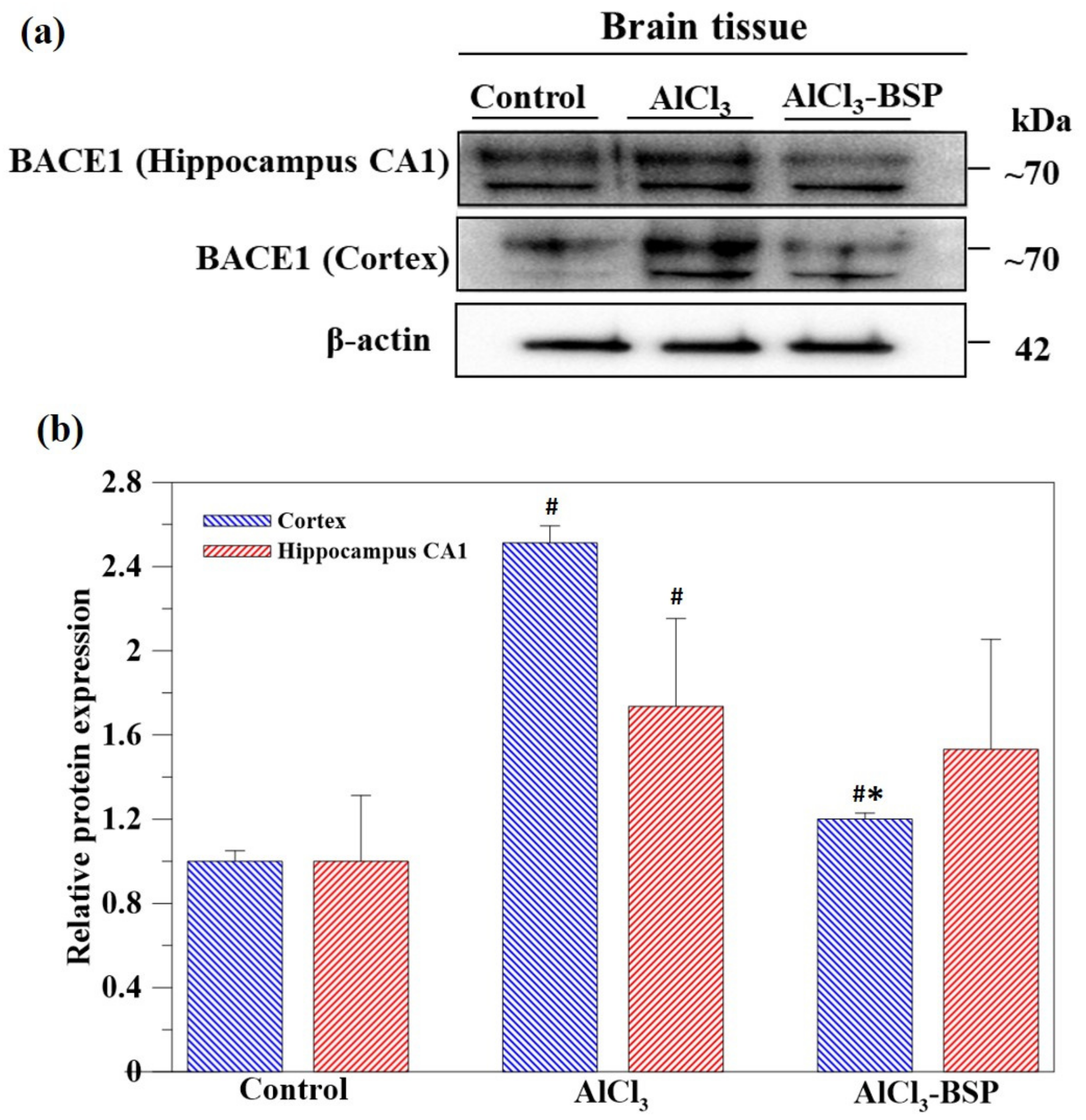
2.7. BSP Treatment Downregulated the BACE1 Treatment
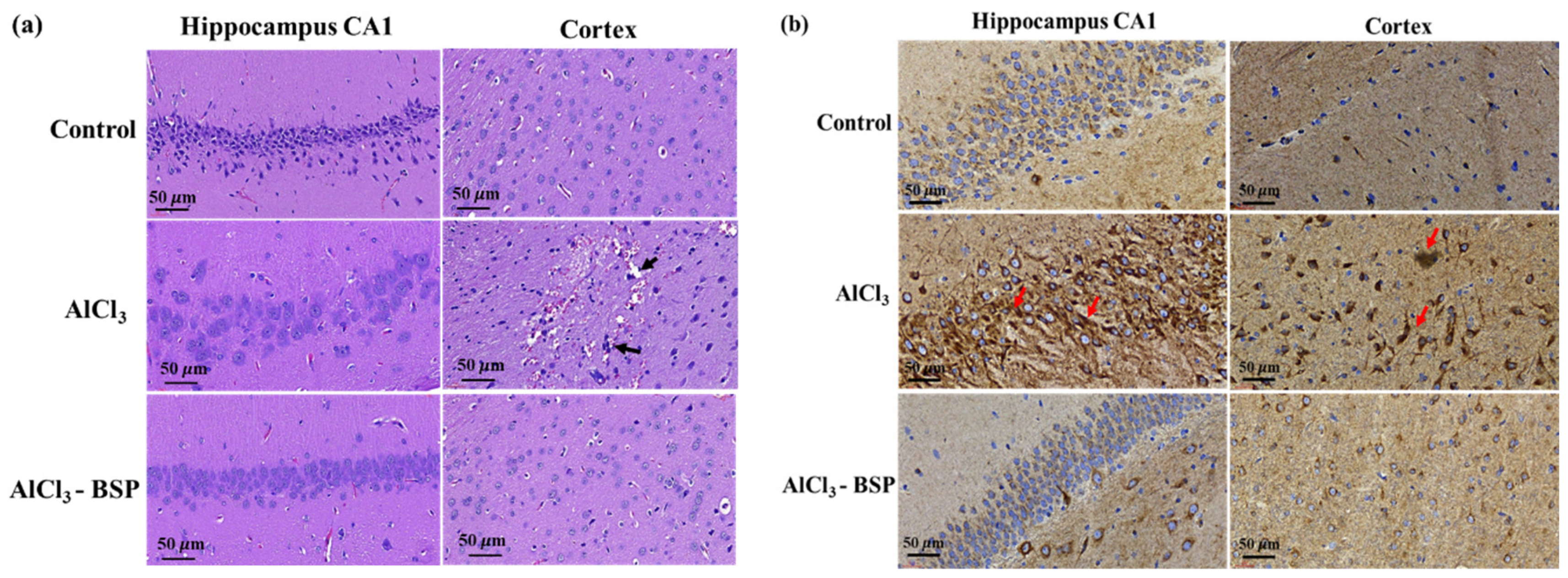
2.8. BSP Treatment Reduced Morphological Changes in the Neurons of the AD Rat Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. BSP Extraction and Purification
4.3. The Characterization of Extracted BSP by Fourier-Transform Infrared (FTIR) Spectroscopy and Nuclear Magnetic Resonance (NMR)
4.4. Preparation of Aβ Fibrils
4.5. Measurements of Thioflavin T Fluorescence
4.6. Transmission Electron Microscopy (TEM)
4.7. Evaluation of Cell Viability
4.8. Evaluation of Cytotoxicity
4.9. Determination of Cellular ROS Generation
4.10. Gene Expression Analysis
4.11. AlCl3-Induced AD Rat Model to Develop Alzheimer’s Disease (AD)
4.12. Morris Water Maze Test
4.13. Blood Analysis
4.14. Western Blotting
4.15. Histological Analysis and Immunohistochemical (IHC) Staining
4.16. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 101–108. [Google Scholar] [PubMed]
- 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Abate, G.; Vezzoli, M.; Polito, L.; Guaita, A.; Albani, D.; Marizzoni, M.; Garrafa, E.; Marengoni, A.; Forloni, G.; Frisoni, G.B.; et al. A Conformation Variant of p53 Combined with Machine Learning Identifies Alzheimer Disease in Preclinical and Prodromal Stages. J. Pers. Med. 2020, 11, 14. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Long-Smith, C.; O’Riordan, K.J.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota-Gut-Brain Axis: New Therapeutic Opportunities. Annu. Rev. Pharm. Toxicol. 2020, 60, 477–502. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sun, J.; Luo, Y.; Xue, W.; Diao, H.; Dong, L.; Chen, J.; Zhang, J. A polysaccharide isolated from the medicinal herb Bletilla striata induces endothelial cells proliferation and vascular endothelial growth factor expression in vitro. Biotechnol. Lett. 2006, 28, 539–543. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, D.; Chen, S.; Wang, Y.; Jiang, H.; Yin, H. A new glucomannan from Bletilla striata: Structural and anti-fibrosis effects. Fitoterapia 2014, 92, 72–78. [Google Scholar] [CrossRef]
- Yue, L.; Wang, W.; Wang, Y.; Du, T.; Shen, W.; Tang, H.; Wang, Y.; Yin, H. Bletilla striata polysaccharide inhibits angiotensin II-induced ROS and inflammation via NOX4 and TLR2 pathways. Int. J. Biol. Macromol. 2016, 89, 376–388. [Google Scholar] [CrossRef]
- Prema, A.; Thenmozhi, A.J.; Manivasagam, T.; Essa, M.M.; Akbar, M.D.; Akbar, M. Fenugreek Seed Powder Nullified Aluminium Chloride Induced Memory Loss, Biochemical Changes, Abeta Burden and Apoptosis via Regulating Akt/GSK3beta Signaling Pathway. PLoS ONE 2016, 11, e0165955. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Niu, F.; Sun, Z.; Cao, W.; Zhang, X.; Guan, D.; Lv, Z.; Zhang, B.; Xu, Y. Altered expression of Abeta metabolism-associated molecules from D-galactose/AlCl(3) induced mouse brain. Mech Ageing Dev. 2009, 130, 248–252. [Google Scholar] [CrossRef]
- Frid, P.; Anisimov, S.V.; Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 2007, 53, 135–160. [Google Scholar] [CrossRef]
- Brown, M.R.; Radford, S.E.; Hewitt, E.W. Modulation of β-amyloid fibril formation in Alzheimer’s disease by microglia and infection. Front. Mol. Neurosci. 2020, 13, 228. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Amyloid-β-induced reactive oxygen species production and priming are differentially regulated by ion channels in microglia. J. Cell. Physiol. 2011, 226, 3295–3302. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharm. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, S.L.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimer’s Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, M.; Li, C.; Jiang, X.; Su, Y.; Zhang, Y. Benefits of Vitamins in the Treatment of Parkinson’s Disease. Oxid. Med. Cell Longev. 2019, 2019, 9426867. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wilde, M.C.; Vellas, B.; Girault, E.; Yavuz, A.C.; Sijben, J.W. Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimer’s Dement. (N. Y.) 2017, 3, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, A.B.; Usman, M.; Ali, F.; Satti, S.A. Vitamin Supplementation as an Adjuvant Treatment for Alzheimer’s Disease. J. Clin. Diagn. Res. 2016, 10, Oe7–Oe11. [Google Scholar] [CrossRef]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C.S.; Grp, C.C.S. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements—The Cache County Study. Arch. Neurol. Chic. 2004, 61, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Littlejohns, T.J.; Henley, W.E.; Lang, I.A.; Annweiler, C.; Beauchet, O.; Chaves, P.H.M.; Fried, L.; Kestenbaum, B.R.; Kuller, L.H.; Langa, K.M.; et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology 2014, 83, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Khemka, V.K.; Ganguly, A.; Roy, D.; Ganguly, U.; Chakrabarti, S. Vitamin D and Alzheimer’s Disease: Neurocognition to Therapeutics. Int. J. Alzheimer’s Dis. 2015, 2015, 192747. [Google Scholar] [CrossRef] [Green Version]
- Diesel, B.; Radermacher, J.; Bureik, M.; Bernhardt, R.; Seifert, M.; Reichrath, J.; Fischer, U.; Meese, E. Vitamin D(3) metabolism in human glioblastoma multiforme: Functionality of CYP27B1 splice variants, metabolism of calcidiol, and effect of calcitriol. Clin. Cancer. Res. 2005, 11, 5370–5380. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F. Vitamin D and brain health: The need for vitamin D supplementation and sensible sun exposure. J. Intern. Med. 2015, 277, 90–93. [Google Scholar] [CrossRef]
- Neveu, I.; Naveilhan, P.; Menaa, C.; Wion, D.; Brachet, P.; Garabedian, M. Synthesis of 1,25-dihydroxyvitamin D3 by rat brain macrophages in vitro. J. Neurosci. Res. 1994, 38, 214–220. [Google Scholar] [CrossRef]
- Brown, J.; Bianco, J.I.; McGrath, J.J.; Eyles, D.W. 1,25-dihydroxyvitamin D3 induces nerve growth factor, promotes neurite outgrowth and inhibits mitosis in embryonic rat hippocampal neurons. Neurosci. Lett. 2003, 343, 139–143. [Google Scholar] [CrossRef]
- Orme, R.P.; Bhangal, M.S.; Fricker, R.A. Calcitriol imparts neuroprotection in vitro to midbrain dopaminergic neurons by upregulating GDNF expression. PLoS ONE 2013, 8, e62040. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The Neurotrophins and Their Role in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Calissano, P.; Matrone, C.; Amadoro, G. Nerve Growth Factor as a Paradigm of Neurotrophins Related to Alzheimer’s Disease. Dev. Neurobiol. 2010, 70, 372–383. [Google Scholar] [CrossRef]
- Veenstra, T.D.; Fahnestock, M.; Kumar, R. An AP-1 site in the nerve growth factor promoter is essential for 1,25-dihydroxyvitamin D-3-mediated nerve growth factor expression in osteoblasts. Biochemistry 1998, 37, 5988–5994. [Google Scholar] [CrossRef]
- La Fata, G.; Weber, P.; Mohajeri, M.H. Effects of Vitamin E on Cognitive Performance during Ageing and in Alzheimer’s Disease. Nutrients 2014, 6, 5453–5472. [Google Scholar] [CrossRef] [Green Version]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin E and Alzheimer’s disease: What do we know so far? Clin. Interv. Aging 2019, 14, 1303–1317. [Google Scholar] [CrossRef] [Green Version]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Role of Vitamin E in the Treatment of Alzheimer’s Disease: Evidence from Animal Models. Int. J. Mol. Sci. 2017, 18, 2504. [Google Scholar] [CrossRef] [Green Version]
- Baroni, L.; Bonetto, C.; Rizzo, G.; Bertola, C.; Caberlotto, L.; Bazzerla, G. Association Between Cognitive Impairment and Vitamin B12, Folate, and Homocysteine Status in Elderly Adults: A Retrospective Study. J. Alzheimer’s Dis. 2019, 70, 441–451. [Google Scholar] [CrossRef]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef] [Green Version]
- Mielech, A.; Puscion-Jakubik, A.; Markiewicz-Zukowska, R.; Socha, K. Vitamins in Alzheimer’s Disease-Review of the Latest Reports. Nutrients 2020, 12, 3458. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, P.; Steinbusch, H.W.M.; Vamanu, E.; Ashraf, G.; Singh, M.P. The Role of Vitamins in Neurodegenerative Disease: An Update. Biomedicines 2021, 9, 1284. [Google Scholar] [CrossRef]
- Gauthier, E.; Fortier, I.; Courchesne, F.; Pepin, P.; Mortimer, J.; Gauvreau, D. Aluminum forms in drinking water and risk of Alzheimer’s disease. Environ. Res. 2000, 84, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Walton, J. An aluminum-based rat model for Alzheimer’s disease exhibits oxidative damage, inhibition of PP2A activity, hyperphosphorylated tau, and granulovacuolar degeneration. J. Inorg. Biochem. 2007, 101, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Rossner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression--implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-L.; Lin, Y.-Y.; Sadhasivam, S.; Kuan, C.-Y.; Chi, C.-y.; Dong, G.-C.; Lin, F.-H. Efficacy of Bletilla striata polysaccharide on hydrogen peroxide-induced apoptosis of osteoarthritic chondrocytes. J. Polym. Res. 2018, 25, 49. [Google Scholar] [CrossRef]
- Stine, W.B.; Jungbauer, L.; Yu, C.; LaDu, M.J. Preparing synthetic Aβ in different aggregation states. In Alzheimer’s Disease and Frontotemporal Dementia; Springer: New York, NY, USA, 2010; pp. 13–32. [Google Scholar]
- Klunk, W.E.; Jacob, R.F.; Mason, R.P. Quantifying amyloid β-peptide (Aβ) aggregation using the Congo Red-Aβ (CR–Aβ) spectrophotometric assay. Anal. Biochem. 1999, 266, 66–76. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Lavysh, A.V.; Maskevich, A.A.; Kuznetsova, I.M.; Turoverov, K.K. Thioflavin T fluoresces as excimer in highly concentrated aqueous solutions and as monomer being incorporated in amyloid fibrils. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Goldsbury, C.; Baxa, U.; Simon, M.N.; Steven, A.C.; Engel, A.; Wall, J.S.; Aebi, U.; Müller, S.A. Amyloid structure and assembly: Insights from scanning transmission electron microscopy. J. Struct. Biol. 2011, 173, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wallin, R.F.; Arscott, E. A practical guide to ISO 10993-5: Cytotoxicity. Med. Device Diagn. Ind. 1998, 20, 96–98. [Google Scholar]
- Sena-Lopes, Â.; Remião, M.H.; Alves, M.S.D.; da Rocha Fonseca, B.; Seixas, F.K.; Collares, T.; Borsuk, S. Cell viability analysis of Toxocara cati larvae with LIVE/DEAD® Viability/Cytotoxicity kit. Exp. Parasitol. 2020, 212, 107871. [Google Scholar] [CrossRef]
- Velagapudi, R.; Olajide, O. Neuroprotective Effects of Thymoquinone in Aβ1–42-induced Toxicity in SK-N-SH Neuronal Cells. FASEB J. 2016, 30, lb509. [Google Scholar]
- Fox, D.A. Cytokine blockade as a new strategy to treat rheumatoid arthritis: Inhibition of tumor necrosis factor. Arch. Intern. Med. 2000, 160, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Aal, R.A.; Assi, A.-A.A.; Kostandy, B.B. Rivastigmine reverses aluminum-induced behavioral changes in rats. Eur. J. Pharmacol. 2011, 659, 169–176. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef] [Green Version]
- Thippeswamy, A.H.; Rafiq, M.; Viswantha, G.L.; Kavya, K.J.; Anturlikar, S.D.; Patki, P.S. Evaluation of Bacopa monniera for its synergistic activity with rivastigmine in reversing aluminum-induced memory loss and learning deficit in rats. J. Acupunct. Meridian Stud. 2013, 6, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848. [Google Scholar] [CrossRef] [Green Version]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Singh, N.A.; Bhardwaj, V.; Ravi, C.; Ramesh, N.; Mandal, A.K.A.; Khan, Z.A. EGCG Nanoparticles Attenuate Aluminum Chloride Induced Neurobehavioral Deficits, Beta Amyloid and Tau Pathology in a Rat Model of Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 244. [Google Scholar] [CrossRef]
- Baird, M.F.; Graham, S.M.; Baker, J.S.; Bickerstaff, G.F. Creatine-kinase-and exercise-related muscle damage implications for muscle performance and recovery. J. Nutr. Metab. 2012, 2012, 960363. [Google Scholar] [CrossRef] [Green Version]
- Carroll, M.F.; Schade, D.S. A practical approach to hypercalcemia. Am. Fam. Physician 2003, 67, 1959–1966. [Google Scholar]
- Zhiyou, C.; Yong, Y.; Shanquan, S.; Jun, Z.; Liangguo, H.; Ling, Y.; Jieying, L. Upregulation of BACE1 and β-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer’s disease. Neurochem. Res. 2009, 34, 1226–1235. [Google Scholar] [CrossRef]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and eosin staining of tissue and cell sections. Cold Spring Harb. Protoc. 2008, 2008, prot4986. [Google Scholar] [CrossRef]
- Youmans, K.L.; Tai, L.M.; Kanekiyo, T.; Stine Jr, W.B.; Michon, S.-C.; Nwabuisi-Heath, E.; Manelli, A.M.; Fu, Y.; Riordan, S.; Eimer, W.A. Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol. Neurodegener. 2012, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.Y.; Chen, S.H.; Chen, C.H.; Chou, P.Y.; Yang, C.C.; Lin, F.H. Polysaccharide Extracted from Bletilla striata Promotes Proliferation and Migration of Human Tenocytes. Polymers 2020, 12, 2567. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| TNF-α-Forward | 5′-CATCTTCTCAAAATTCGAGTACAA-3′ |
| TNF-α-Reverse | 5′-TGGGAGTAGACAAGGTACAACCC-3′ |
| IL-6-Forward | 5′-GGAGCCCACCAAGAACGATAGTCA-3′ |
| IL-6-Reverse | 5′-GAAGTAGGGAAGGCCGTGGTT-3′ |
| IL-10-Forward | 5′-TAAGGCTGGCCACACTTGAG-3′ |
| IL-10-Reverse | 5′-GTTTTCAGGGATGAAGCGGC-3′ |
| GAPDH-Forward | 5′-TGCTGAGTATGTCGTGGAGTCT-3′ |
| GAPDH-Reverse | 5′-AATGGGAGTTGCTGTTGAAGTC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-W.; Fang, C.-H.; Liang, Y.-J.; Liao, H.-H.; Lin, F.-H. Modified Low-Temperature Extraction Method for Isolation of Bletilla striata Polysaccharide as Antioxidant for the Prevention of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12760. https://doi.org/10.3390/ijms222312760
Lin Y-W, Fang C-H, Liang Y-J, Liao H-H, Lin F-H. Modified Low-Temperature Extraction Method for Isolation of Bletilla striata Polysaccharide as Antioxidant for the Prevention of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(23):12760. https://doi.org/10.3390/ijms222312760
Chicago/Turabian StyleLin, Yi-Wen, Chih-Hsiang Fang, Ya-Jyun Liang, Hong-Hsiang Liao, and Feng-Huei Lin. 2021. "Modified Low-Temperature Extraction Method for Isolation of Bletilla striata Polysaccharide as Antioxidant for the Prevention of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 23: 12760. https://doi.org/10.3390/ijms222312760