
Virus Infection, Genetic Mutations, and Prion Infection in Prion Protein Conversion

Division of Molecular Neurobiology, The Institute for Enzyme Research (KOSOKEN), Tokushima University, 3-18-15 Kuramoto, Tokushima 770-8503, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(22), 12439; https://doi.org/10.3390/ijms222212439
Submission received: 28 October 2021
/
Revised: 14 November 2021
/
Accepted: 16 November 2021
/
Published: 18 November 2021
(This article belongs to the Special Issue Prions and Prion Diseases 2.0)
Abstract
:Conformational conversion of the cellular isoform of prion protein, PrPC, into the abnormally folded, amyloidogenic isoform, PrPSc, is an underlying pathogenic mechanism in prion diseases. The diseases manifest as sporadic, hereditary, and acquired disorders. Etiological mechanisms driving the conversion of PrPC into PrPSc are unknown in sporadic prion diseases, while prion infection and specific mutations in the PrP gene are known to cause the conversion of PrPC into PrPSc in acquired and hereditary prion diseases, respectively. We recently reported that a neurotropic strain of influenza A virus (IAV) induced the conversion of PrPC into PrPSc as well as formation of infectious prions in mouse neuroblastoma cells after infection, suggesting the causative role of the neuronal infection of IAV in sporadic prion diseases. Here, we discuss the conversion mechanism of PrPC into PrPSc in different types of prion diseases, by presenting our findings of the IAV infection-induced conversion of PrPC into PrPSc and by reviewing the so far reported transgenic animal models of hereditary prion diseases and the reverse genetic studies, which have revealed the structure-function relationship for PrPC to convert into PrPSc after prion infection.
1. Introduction
Prion diseases, or transmissible spongiform encephalopathies, are a group of fatal neurodegenerative disorders in humans and animals, caused by accumulation of proteinaceous infectious particles, termed “prions”, in the brain [1,2,3]. Characteristic histopathological features, such as spongiform vacuolation, neuronal cell death, and gliosis, are observed in the affected brains [1,2]. Prion diseases manifest as sporadic, hereditary, or acquired disorders. Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common prion disease in humans, accounting for 85–90% of total human cases [4,5,6]. The annual incidence of sCJD has been estimated to be 1–2 cases per a million people worldwide, although the reported incidence varies from country to country and is influenced by the method and extent of the study conducted [7]. 10–15% of human cases belong to hereditary prion diseases, such as familial CJD (fCJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI) [8,9]. They are causatively linked to specific mutations in the gene (Prnp) of prion protein (PrP) [8,9]. The remaining cases, accounting for less than 1%, are those of acquired prion diseases, which include iatrogenic CJD (iCJD), variant CJD (vCJD), and kuru. iCJD is a disease caused by human-to-human transmission of prions via medical treatments or procedures [10]. Dura mater, cornea, and growth hormone from the patients, and depth electroencephalogram electrodes and neurosurgical instruments used for the patients have been considered to transmit the disease [11]. vCJD is believed to be transmitted from bovine spongiform encephalopathy (BSE) via oral consumption of BSE prion-contaminated food [10]. As of July 2021, 229 cases of vCJD have been identified worldwide since 1996, with most of them in the U.K. (http://www.cjd.ed.ac.uk/sites/default/files/worldfigs.pdf, accessed on 21 July 2021). Kuru is a disease spread via ritualistic cannibalism among Fore people in Papua New Guinea [11].
BSE is an acquired animal prion disease in cattle spread through contaminated meat-and-bone meals since the mid-1980s [12]. Transmissible mink encephalopathy in minks, chronic wasting disease (CWD) in deer, and feline spongiform encephalopathy in cats also acquired prion diseases [13,14]. Scrapie is a sporadic animal prion disease in sheep [13]. Unlike the case of BSE, epidemiological studies suggest unlikelihood of the zoonotic transmissions of scrapie or CWD to humans [15,16,17,18,19].
Prions are widely believed to consist of the abnormally folded, amyloidogenic isoform of PrP, designated PrPSc, which is produced from the cellular isoform of PrP, PrPC, through its conformational conversion [1,2,3]. We and others have shown that mice devoid of PrPC (Prnp0/0) are resistant to prion infections, neither developing disease nor propagating prions in the brains after intracerebral inoculation with the prions [20,21,22,23], confirming that the conversion of PrPC into PrPSc eventually leading to accumulation of PrPSc in the brain is a key pathogenic event in prion diseases. In acquired prion diseases, prion infections drive the conversion of PrPC into PrPSc. Mutated PrPs in hereditary prion diseases may undergo conformational changes to form PrPSc [8,9]. We have recently reported that neurotrophic influenza A virus (IAV) caused the conversion of PrPC into PrPSc in cultured neuronal cells after infection [24], raising the intriguing possibility that IAV infection in neurons might be a cause of or be associated with sporadic prion diseases.
Here, we present our current findings of the IAV infection-induced conversion of PrPC into PrPSc. We also review the so far reported transgenic (Tg) mouse models of hereditary prion diseases, where mutated PrPs are spontaneously converted into PrPScs, and the reverse genetic studies using Prnp0/0 mice, which have revealed the important regions for PrPC to convert into PrPSc after prion infection.
2. PrP Conversion
2.1. PrPC and PrPSc
PrPC is a host-encoded glycoprotein tethered to the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor moiety and expressed most abundantly in the brain, particularly by neurons, and to lesser extents in other non-neuronal tissues, including heart, lung, pancreas, liver, spleen, testis, and kidney [25,26,27]. Many physiological functions have been suggested for PrPC, including oxidative stress mitigation, neuroprotection, and those associated with cell trafficking, cell adhesion, cell differentiation, cell signaling, and cell survival [28]. We have recently identified that PrPC could also function as a protective molecule against IAV infection, mitigating oxidative stress and epithelial cell apoptosis in IAV-infected lungs, and a regulator molecule in macrophage M1/M2 polarization, stimulating the polarization to M2 anti-inflammatory macrophages [29,30].
PrPC structurally consists of two domains; the flexible non-structural N-terminal domain including the so-called octapeptide repeat (OR) region in the middle, and the globular C-terminal domain with 2 short β-sheets and 3 α-helices [26,27]. Mouse PrPC is translated as a precursor protein comprised of 254 amino acids, with the 22 amino acids of an endoplasmic reticulum (ER) migration signal sequence at the N-terminus and the 23 amino acids of a GPI anchor addition signal sequence at the C-terminus [26,27]. Both signal sequences are removed in the ER as the nascently translated PrP enters the ER [26,27]. The C-terminal domain further undergoes posttranslational modifications, such as disulfide bond formation between cysteine residues at positions 178 and 213 and N-type sugar chain addition at asparagine residues of positions 180 and 196 [26,27].
PrPSc is specifically detectable in the tissues or cells infected with prions, including the central nervous system, particularly neurons, and follicular dendric cells of the lymphoreticular system [31]. In contrast to PrPC being detergent-soluble and sensitive to protease degradation, PrPSc is highly detergent-insoluble, easily assembling to form amyloid fibrils, termed prion rods, with a diameter of 10–20 nm and a length of 100–200 nm, and relatively resistant to protease degradation [32]. These biochemical differences of both molecules arise from their structural differences. PrPC is rich in α-helix contents while PrPSc abounds with β-sheet contents [33].
2.2. In Vitro PrP Conversion
Kocisko et al. succeeded for the first time in producing PrPSc-like proteinase K (PK)-resistant PrP from PrPC in vitro, by incubating PrPC from prion-uninfected cultured cells with PrPSc partially purified from 263K prion-infected hamster brains in denaturing-renaturing conditions [34]. The protein misfolding cyclic amplification (PMCA) technique was then developed to amplify PrPSc-like PrP in vitro by serially incubating PrPC with the diluted PMCA products under sonication, eventually enriching PrPSc-like PrP converted from PrPC and contrarily reducing original PrPSc added in the reactions to a negligible level [35]. Subsequently, PrPSc-like PrP generated by PMCA was shown to correlate to prion infectivity, strongly suggesting that PrPSc-like PrP is generated by PMCA is infectious [36]. Prion infectivity was also successfully generated from bacterial recombinant PrP without incubating with PrPSc. The β sheet-rich amyloid fibrils consisting of the N-terminally truncated recombinant mouse PrP were shown to be infectious [37]. PMCA with bacterial recombinant full-length mouse PrP together with the synthetic anionic phospholipid and RNA molecules was also shown to generate PrPSc-like PrP as well as having prion infectivity [38]. Dextran sulfate, mRNA molecules, and plasmid DNA were also reported to function as co-factors to produce infectious prions from bank vole PrP in PMCA, although bank vole PrP was convertible into infectious prions in PMCA without these cofactors [39]. These results confirm that PrPSc is an essential component for prions to be infectious, strongly supporting that prions consist of PrPSc alone. Consistent with this, prion infectivity has been shown to be sensitive to treatments with protein-denaturing agents, but resistant to nucleic acid-degrading treatments with nucleases or UV radiation [40]. No specific nucleic acids have been isolated in the highly purified prion fractions [41].
2.3. Seeded-Polymerization Mechanism in PrP Conversion
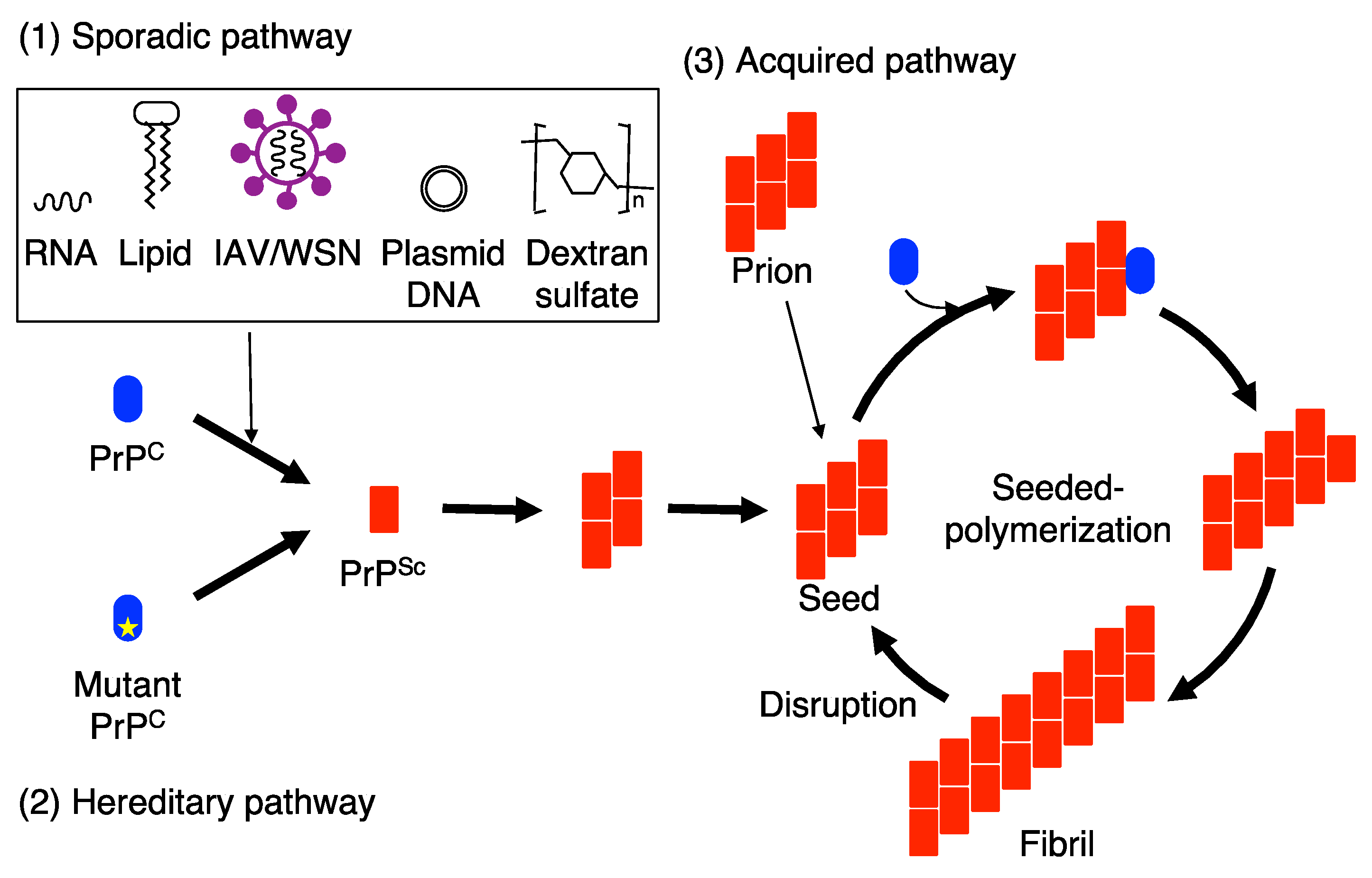
It has been suggested that a seeded-protein polymerization mechanism is an underlying mechanism for PrPSc propagation, or prion propagation [34,42]. Prion-infected brain fractions containing the aggregates of 14–28 PrPSc molecules were shown to be highly infectious compared to those containing large PrPSc fibrils or the aggregates of less than 5 PrPSc molecules [43], suggesting that prions are the multimeric aggregates of PrPSc. In the seeded-protein polymerization mechanism, the aggregates of PrPSc are considered to function as a seed or a scaffold for PrPC to undergo conformational conversion into PrPSc (Figure 1). PrPC is recruited onto the PrPSc seed and forced to convert into PrPSc on it through structural transition from α-helix to β-sheet [33], resulting in elongation of the PrPSc seeds, eventually leading to formation of the PrPSc fibrils (Figure 1). Prions, or PrPSc seeds, are postulated to then propagate by cleavage of the PrPSc fibrils into new multiple PrPSc seeds by unknown cellular mechanism (Figure 1) [34,35].
Structural models have been proposed for PrPSc fibrils. In a 4-rung β-solenoid model, a PrPSc molecule adopts a solenoid structure with 4 rungs, each rung consisting of 3 β-sheets and perpendicularly stacking with each other to form a PrPSc protofilament [44,45,46]. Two PrPSc protofilaments are then intertwined, forming a PrPSc fibril [44,45,46]. The PrPSc protofilaments might function as seeds, recruiting PrPC molecules to their upper and lower ß-solenoid rungs and inducing the formation of ß-solenoid rungs through the structural transition of α-helices to β-sheet, eventually converting the PrPC into PrPSc. In a parallel in-register intermolecular ß-sheet model, a PrPSc fibril is formed by stacking PrPSc molecules, each PrPSc comprising the entire cross-section of the fibril with many hairpins, through the intermolecular β-sheet interactions between them [47,48]. The fibrils function as seeds for PrPC to convert into PrPSc.
3. Virus Infections in PrP Conversion
3.1. Neurotropic IAV Infection in PrP Conversion
We have recently reported that infection of mouse neuroblastoma N2a cells overexpressing the exogenously transduced mouse PrPC, termed N2aC24 cells, with a neurotropic strain of influenza A/WSN/33 (H1N1) virus (hereafter referred to as IAV/WSN) induced spontaneous conformational changes in PrPC to form PrPSc-like PK-resistant PrP [24]. These results indicate that, like prion infection, IAV/WSN infection could induce the conformational conversion of PrPC into PrPSc. We also detected PrPSc-like PrP in parent N2a cells, which express endogenous mouse PrPC alone, after infection with IAV/WSN [24], indicating that overexpression of PrPC is not required for the IAV/WSN-induced conversion into PrPSc. We further showed that the anti-IAV agent oseltamivir and anti-IAV mouse antisera blocked IAV/WSN infection of N2aC24 cells and prevented formation of PrPSc-like PrP in the cells [24], not only ruling out the possibility of the contamination of laboratory prions in the cell culture, but also confirming that IAV/WSN infection is essential for the conversion of PrPC into PrPSc-like PrP in the cells. We finally demonstrated that wild-type (WT) mice intracerebrally inoculated with cell lysates from PrPSc-like PrP-producing N2aC24 cells developed prion disease, with abundant accumulation of PrPSc and spongiosis in their brains [24], indicating that, like authentic PrPSc produced by prion infection, PrPSc-like PrP produced in IAV/WSN-infected N2aC24 cells could form infectious prions. Taken together, these results suggest that IAV infection in neurons might be a cause of or be associated with sporadic prion diseases, including sCJD.
PrPSc-like PrP (hereafter simply referred to as PrPSc) was constitutively produced in IAV/WSN-infected N2aC24 cells even after IAV/WSN infection was cleared during long culture [24], indicating that persistent IAV/WSN infection is not necessary for the constitutive conversion PrPC into PrPSc in the cells. It is thus possible that IAV/WSN infection could induce the conversion of PrPC into PrPSc in a hit-and-run manner, and the nascent PrPSc could polymerize to form the PrPSc seeds, enabling the constitutive conversion of PrPC into PrPSc in the seeded-polymerization mechanism. IAVs are negative-stranded, segmented, enveloped RNA viruses [49]. It was reported that RNA molecules were able to convert human recombinant PrP into PK-resistant PrP in vitro [50]. Moreover, RNA molecules were shown to function as a cofactor, together with the synthetic anionic phospholipid, for the conversion of recombinant PrP into PrPSc in PMCA [38]. These results indicate that RNA molecules play an important role in the conversion of PrPC into PrPSc. IAV/WSN-derived RNA molecules might play the same role in the conversion of PrPC into PrPSc in N2aC24 cells. However, it has been reported that RNA molecules prepared from mammalian tissues but not from invertebrate species including bacteria, yeast, worms, and flies converted PrPC into PrPSc in PMCA [51]. For elucidation of the role of IAV/WSN-derived RNA molecules in the conversion of PrPC into PrPSc, it would be helpful to investigate if IAV/WSN-derived RNA molecules have an ability to convert PrPC into PrPSc in PMCA.
3.2. Other Virus Infections in Prion Infection
It was shown that mice co-infected with scrapie prions and mouse adenovirus accelerated prion disease compared to those infected with scrapie prions alone, suggesting that mouse adenovirus infection could enhance prion pathogenesis in mice [52]. Infection with the Cork strain of caprine arthritis encephalitis virus, a small-ruminant lentivirus, was also shown to increase PrPSc in scrapie prion-infected primary sheep microglia at levels 2-times higher than in control cells [53]. However, no data were available as to whether these virus infections could cause the de novo conversion of PrPC into PrPSc [53]. Mouse A9 fibroblasts infected with murine minute virus, a murine parvovirus, were reported to bind to exogenously added PrPSc molecules more abundantly and internalize them to lysosomal compartments, which are postulated to be one of the cellular sites for the conversion of PrPC into PrPSc, more efficiently than control cells [54]. Molony murine leukemia virus infection into 22L scrapie prion-infected NIH3T3 cells was reported to increase the release of PrPSc in culture medium by facilitating the incorporation of PrPSc into virus particles or exosomes [55]. It is thus possible that the increased levels of PrPSc in the prion-infected cells after infection with mouse adenovirus or caprine arthritis encephalitis virus might be due to the increased accessibility of PrPSc to PrPC through the enhanced binding of PrPSc molecules on the cells, their stimulated internalization to lysosomal compartments, and the increased release of PrPSc from prion-infected cells.
4. Tg Mouse Models of Hereditary Prion Diseases
4.1. Genetic Mutations in PrP Conversion
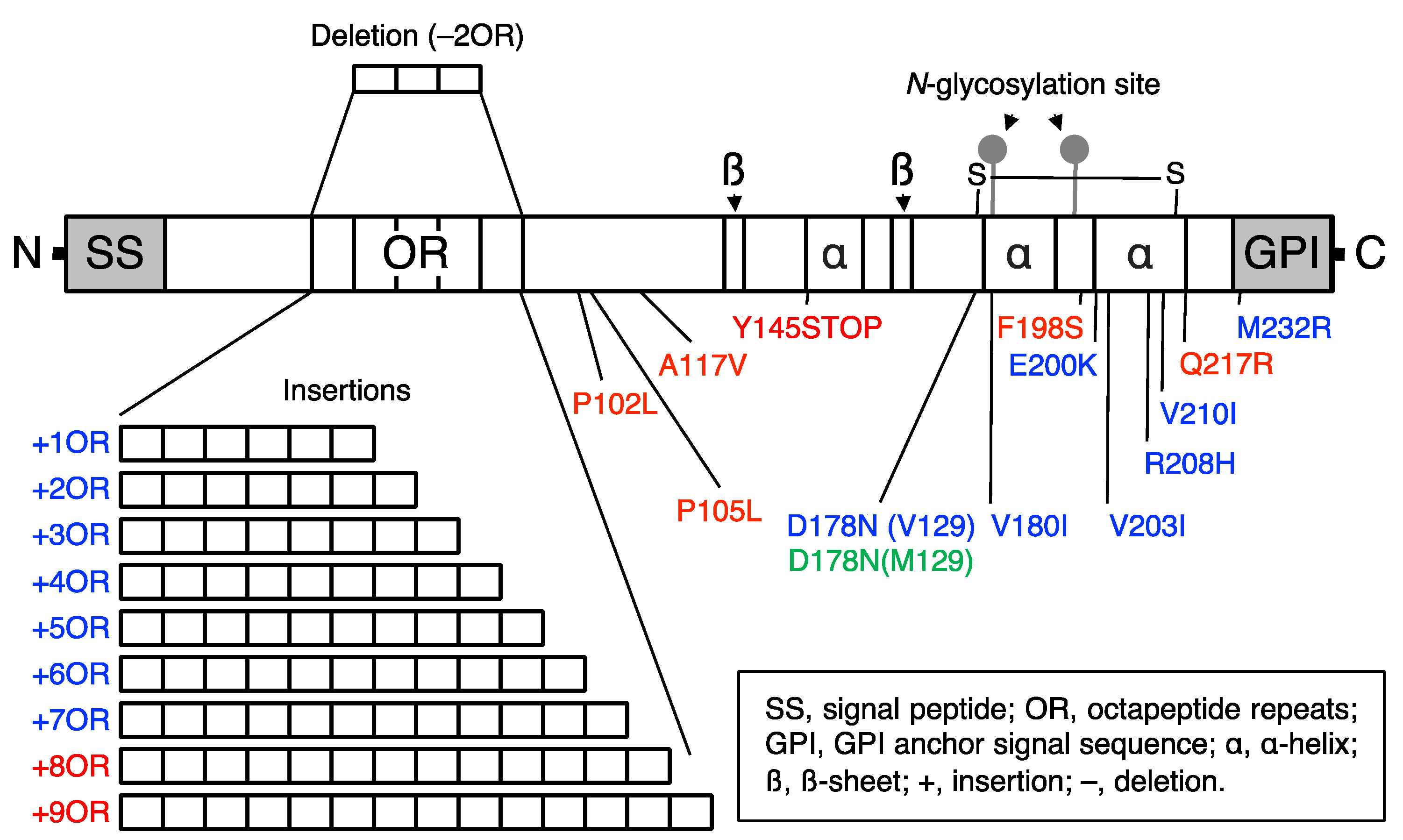
Many point mutations and insertions or a deletion of the OR sequence have been identified in hereditary human prion diseases, including GSS, fCJD, and FFI (Figure 2) [8,9]. To understand the pathogenic mechanism of hereditary prion diseases, many lines of Tg mice expressing the disease-associated mutant PrPs have been reported, uncovering biologically and structurally different forms of PrPSc, or neurotoxic and infectious PrPScs [56]. Some PrP mutants were shown to convert into neurotoxic forms but not to infectious forms while others converted to neurotoxic and infectious forms [56]. It is thus possible that PrPC might undergo conformational changes to convert into PrPSc, then polymerizing to form neurotoxic PrPSc oligomers and to neurotoxic and infectious PrPSc oligomers, or PrPSc fibrils.
4.2. Tg Mouse Models of GSS
The first reported Tg mouse model of hereditary prion diseases is Tg(PrP-P101L) mice, which express mouse PrP-P101L, the analogous mutation to human PrP-P102L in GSS (Table 1) [57]. They expressed the mutant PrP at levels at least 3 times higher than PrPC in WT mice and spontaneously developed prion disease-like disease at 150–600 days after birth, with the disease-characteristic pathologies of amyloid plaques, spongiform degeneration, and gliosis in their brains [57]. However, in contrast to authentic PrPSc, which is highly resistant to PK, PrPSc-P101L accumulated in the brains of ill Tg(PrP-P101L) mice was easily digested by a high dose of PK [57]. In addition, intracerebral inoculation with brain homogenates from ill Tg(PrP-P101L) mice transmitted the disease to 40% of Tg(PrP-P101L) mice, which express low levels of the mutant protein, and 10% of hamsters, but not to WT mice [58]. These results suggest that PrP-P101L could convert into neurotoxic and infectious PrPSc-P101L. However, the infectivity of PrPSc-P101L was dependent on recipient animals. Tg(PrP-A116V) mice, which express mouse PrP-A116V (the human homologue of PrP-A117V in GSS) at levels 4–6 times higher than PrPC in WT mice, also spontaneously developed neurological disease around 150 days after birth, with spongiosis and PrP amyloid plaques in their brains (Table 1) [59]. PrPSc-A117V in the brains of ill Tg(PrP-A116V) mice was partially detergent-insoluble and weakly resistant to PK [59]. No data are available as to whether PrPSc-A117V is infectious [59]. Tg(PG14)/Prnp0/0 mice are another GSS mouse model expressing a mutant PrP with 9 extra OR sequences in the OR region on the genetic background of Prnp0/0 (Table 1) [60,61]. These mice developed spontaneous cerebellar neurodegeneration, with granule cell death and very slight but substantial accumulation of PrPSc-PG14 in their brains [60,61]. PrPSc-PG14 showed weak resistance to PK and had no prion infectivity in animals [62]. These results indicate that PrP-PG14 could convert into neurotoxic PrPSc-PG14 but not into infectious PrPSc-PG14.
4.3. Tg Mouse Models of fCJD
Tg(Mo/Hu-PrP-E199K) mice, which express mouse-human chimeric PrP, or Mo/Hu-PrP, with the E199K mutation (the human homologue of E200K in fCJD) 2 times higher than PrPC in WT mice, were reported to develop neurological disease around 200 days of age, with hind-limb paralysis and kyphosis (Table 1) [63]. Mo/Hu-PrPSc-E199K in the brains of sick Tg(Mo/Hu-PrP-E199K) mice showed atypical PK-resistant bands on Western blotting [63]. Inoculation of brain homogenates from sick Tg(Mo/Hu-PrP-E199K) mice transmitted the disease into non-Tg mice with highly variable efficiency [63]. These results indicate that Mo/Hu-PrP-E199K could convert into neurotoxic and infectious Mo/Hu-PrPSc-E199K. However, interestingly, Tg mice overexpressing human PrP with the E200K mutation in the brains at levels 2 to 3 times higher than PrPC in human brains developed no neurological diseases (Table 1) [64]. In contrast, knock-in mice expressing mouse PrP-E199K with the 3F4 epitope, which derives from human PrP, exhibited abnormal behaviors, such as reduced performance on the rotarod test and decreased burrowing behavior, with spongiosis and punctuate deposition of PrPSc-E199K(3F4) in their brains (Table 1) [65]. PrPSc-E199K(3F4) was weakly resistant to PK [65]. Inoculation of brain homogenates from sick Tg[PrP-E199K(3F4)] mice transmitted the disease in Tg mice overexpressing Mo-PrP as well as in non-Tg mice [65]. It is thus possible that the human PrP-derived 3F4 sequence might affect mouse PrP to spontaneously convert into neurotoxic and infectious PrPSc.
Tg mice overexpressing Mo-PrP with the D177N mutation (the human homologue of D178N in fCJD) along with the polymorphic amino acid substitution of M128V (the human homologue of M129V) 2 times higher than PrPC in WT mice were also reported to develop neurological disease around 150 days of age, with ataxia, kyphosis, and foot clasping (Table 1) [66]. PrPSc-D177N(V128) was detergent-insoluble and mildly resistant to PK [66]. Inoculation of brain homogenates from sick Tg[PrP-D177N(V128)] mice failed to transmit the disease to non-Tg mice as well as Tg mice overexpressing mouse PrP and Tg mice expressing lower levels of PrP-D177N(V128) [67]. These results suggest that PrP-D177N(V128) could convert into neurotoxic PrPSc-D177N(V128) but not to infectious PrPSc-D177N(V128).
4.4. Tg Mouse Models of FFI
Tg(FFI)/Prnp0/0 mice, which express mouse PrP-D177N(M128) corresponding to the human homologue of PrP-D178N(M129) in FFI in their brains 2 times higher than PrPC in WT mice, were reported to develop fatal neurological disease with motor and cognitive deficits (Table 1) [67]. Circadian rhythms of sleep and motor activity were also disturbed in Tg(FFI)/Prnp0/0 mice, as observed in FFI patients [67]. Sick Tg(FFI)/Prnp0/0 mice accumulated weakly PK-resistant PrPSc-D177N(M128) in their brains [67]. Inoculation of brain homogenates from sick Tg(FFI)/Prnp0/0 mice failed to transmit the disease to WT mice and Tg mice overexpressing mouse PrP [67]. These results suggest that PrP-D177N(M128) could convert into neurotoxic PrPSc-D177N(M128) but not to infectious PrPSc-D177N(M128). However, knock-in mice expressing PrP-D178N(M129) with the 3F4 epitope sequence were shown to develop neurological disease with neuronal loss and gliosis in the thalamus and transmit the disease to Tg mice overexpressing mouse PrP after inoculation with their brain homogenates (Table 1) [68]. It is thus suggested that the 3F4 sequence could force PrP-D178N(M129) to undergo conformational changes to form neurotoxic PrPSc-D177N(M128) and then to neurotoxic and infectious PrPSc-D177N(M128).
5. Reverse Genetic Studies for Acquired Prion Diseases
5.1. The N-Terminal Polybasic Residues of PrPC in Prion Infection
Successful restoration of the susceptibility to prion infection in Prnp0/0 mice by transgenic expression of full-length mouse PrPC has enabled reverse genetic studies to identify the regions or amino acids in PrPC important for the conversion into PrPSc after prion infection [69]. Tg(PrP∆23–31)/Prnp0/0 mice, which express PrP with a deletion of the so-called N-terminal polybasic region consisting of residues 23–31 on the background of Prnp0/0, were shown to develop disease after longer incubation times with a slower accumulation of PrPSc∆23–31 in their brains after infection with RML scrapie prions, indicating that the polybasic region is important for PrPC to convert into PrPSc after prion infection (Table 2) [70]. We generated Tg(PrP3K3A)/Prnp0/0 mice, which express mouse PrPC with lysine residues at positions 23, 24 and 27 mutated to alanine residues in the polybasic region, and intracerebrally inoculated them with RML and 22L scrapie prions (Table 2) [71]. They were highly resistant to the prions, developing disease with longer incubation times [71], suggesting that lysine residues at 23, 24, and 27 are important for the polybasic region to support prion infection. Recombinant PrP∆23-31 was weakly interacted with PrPSc compared to full-length PrP [70]. The polybasic region might thus be involved in the intermolecular interaction with PrPC and PrPSc through the positively charged lysine residues, thereby supporting prion infection through driving the conversion of PrPC into PrPSc.
5.2. The Central Residues of PrPC in Prion Infection
Tg(PrP∆32–80)/Prnp0/0 mice, which express mouse PrP lacking residues 32–80 including most of the OR region, developed disease without prolonged incubation times after infection with RML prions and accumulated PrPSc∆32–80 in their brains (Table 2) [69]. However, Tg(PrP∆32–93)/Prnp0/0 mice expressing PrP with the deletion of residues 32–93 encompassing the entire OR region exhibited longer incubation times and reduced PrPSc∆32–93 in their brains after infection with RML prions (Table 2) [72]. Furthermore, Tg(PrP∆32–106)/Prnp0/0 mice expressing a mutant PrP with a deletion extended to residue 106 neither developed disease nor accumulated PrPSc∆32–106 in their brains even after intracerebral inoculation with RML prions (Table 2) [73]. These results suggest that the deletion of residues 91–106 not the OR region (residues 51–90) renders PrP∆32–106 unsusceptible to RML prions. Consistent with this, we demonstrated that, while Tg(PrP∆OR)/Prnp0/0 mice expressing PrP lacking the OR region alone were susceptible to RML and 22L prions (Table 2) [74], Tg(PrP∆91–106)/Prnp0/0 mice expressing PrP lacking residues 91–106 were resistant to RML and 22L prions, neither developing disease nor accumulating PrPSc∆91–106 in their brains (Table 2) [75]. Using RML prion-infected N2a cells, ScN2a, it was shown that residues 100–104 are important for PrPC to convert into PrPSc in the cells [76]. Residues 100–104 form a loop between the β-sheets in the PrPSc structural model [77,78,79]. It is thus possible that the loop region might play an important role in the conversion of PrPC to PrPSc after infection with RML and 22L prions. Intriguingly, in contrast to the case for RML and 22L prions, Tg(PrP∆91–106)/Prnp0/0 mice were susceptible to BSE prions and Tg(PrP∆OR)/Prnp0/0 mice had reduced their susceptibility to BSE prions (Table 2) [74,75]. These results indicate that, in contrast to RML and 22L prions, BSE prions might require the OR region partially but not residues 91–106 to induce the conversion of PrPC to PrPSc, suggesting that the OR region and residues 91–106 of PrPC are involved in prion infection in a prion strain-dependent manner.
6. Perspectives
The etiologies of sporadic prion diseases including sCJD remain unknown. Our findings that IAV/WSN infection induced the conversion of PrPC into PrPSc and infectious prions in N2a cells has raised the possibility that IAV infection in neurons might be an etiological mechanism of sporadic prion diseases [24]. Elucidation of (1) the mechanism of how IAV/WSN infection could induce the conversion of PrPC into PrPSc, (2) whether or not IAV/WSN infection in neurons could induce prion disease in animals, and (3) whether or not IAV infection in humans could be epidemiologically linked to sCJD are crucial for further understanding of the causative role of IAV infection in sporadic prion diseases. Also, addressing (1) the mechanism of the conversion of the hereditary prion disease-associated mutated PrPs into PrPSc, (2) the molecular nature of neurotoxic and infectious PrPSc molecules, and (3) the mechanism of how the N-terminal polybasic region and the central residues 91–106 are involved in the conversion of PrPC into PrPSc is important not only for further understanding of the pathogenic mechanisms of prion diseases, but also for the development of effective treatment and prevention measures for prion diseases.
Author Contributions
Conceptualization, H.H. and S.S.; writing—original draft preparation, H.H. and S.S.; Funding acquisition, H.H. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by JSPS KAKENHI Grant Number 21K07462, Takeda Science Foundation, Kobayashi Magobe Memorial Medical Foundation, and The Waksman Foundation of Japan to H.H. and JSPS KAKENHI Grant Number 19H03548 to S.S.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- DeArmond, S.J.; Prusiner, S.B. Etiology and pathogenesis of prion diseases. Am. J. Pathol. 1995, 146, 785–811. [Google Scholar] [PubMed]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Brandel, J.P.; Peckeu, L.; Haik, S. The French surveillance network of Creutzfeldt-Jakob disease. Epidemiological data in France and worldwide. Transfus. Clin. Biol. 2013, 20, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, U.; Krasnianski, A.; Meissner, B.; Varges, D.; Kallenberg, K.; Schulz-Schaeffer, W.J.; Steinhoff, B.J.; Grasbon-Frodl, E.M.; Kretzschmar, H.A.; Zerr, I. Creutzfeldt-Jakob disease in Germany: A prospective 12-year surveillance. Brain 2007, 130, 1350–1359. [Google Scholar] [CrossRef] [Green Version]
- Maddox, R.A.; Person, M.K.; Blevins, J.E.; Abrams, J.Y.; Appleby, B.S.; Schonberger, L.B.; Belay, E.D. Prion disease incidence in the United States: 2003–2015. Neurology 2020, 94, e153–e157. [Google Scholar] [CrossRef] [PubMed]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10. [Google Scholar] [CrossRef]
- Webb, T.E.; Poulter, M.; Beck, J.; Uphill, J.; Adamson, G.; Campbell, T.; Linehan, J.; Powell, C.; Brandner, S.; Pal, S.; et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain 2008, 131, 2632–2646. [Google Scholar] [CrossRef] [Green Version]
- Cracco, L.; Appleby, B.S.; Gambetti, P. Fatal familial insomnia and sporadic fatal insomnia. Handb. Clin. Neurol. 2018, 153, 271–299. [Google Scholar]
- Will, R.G.; Ironside, J.W.; Zeidler, M.; Cousens, S.N.; Estibeiro, K.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hofman, A.; Smith, P.G. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347, 921–925. [Google Scholar] [CrossRef]
- Will, R.G. Acquired prion disease: Iatrogenic CJD, variant CJD, kuru. Br. Med. Bull. 2003, 66, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, G.A.; Scott, A.C.; Johnson, C.T.; Gunning, R.F.; Hancock, R.D.; Jeffrey, M.; Dawson, M.; Bradley, R. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 1987, 121, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Orge, L.; Lima, C.; Machado, C.; Tavares, P.; Mendonca, P.; Carvalho, P.; Silva, J.; Pinto, M.L.; Bastos, E.; Pereira, J.C.; et al. Neuropathology of Animal Prion Diseases. Biomolecules 2021, 11, 466. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Young, S. Spongiform encephalopathy of Rocky Mountain elk. J. Wildl. Dis. 1982, 18, 465–471. [Google Scholar] [CrossRef]
- Wilson, R.; Plinston, C.; Hunter, N.; Casalone, C.; Corona, C.; Tagliavini, F.; Suardi, S.; Ruggerone, M.; Moda, F.; Graziano, S.; et al. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J. Gen. Virol. 2012, 93, 1624–1629. [Google Scholar] [CrossRef]
- Belay, E.D.; Maddox, R.A.; Williams, E.S.; Miller, M.W.; Gambetti, P.; Schonberger, L.B. Chronic wasting disease and potential transmission to humans. Emerg. Infect. Dis. 2004, 10, 977–984. [Google Scholar] [CrossRef] [Green Version]
- Belay, E.D.; Gambetti, P.; Schonberger, L.B.; Parchi, P.; Lyon, D.R.; Capellari, S.; McQuiston, J.H.; Bradley, K.; Dowdle, G.; Crutcher, J.M.; et al. Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch. Neurol. 2001, 58, 1673–1678. [Google Scholar] [CrossRef]
- Brown, P.; Cathala, F.; Raubertas, R.F.; Gajdusek, D.C.; Castaigne, P. The epidemiology of Creutzfeldt-Jakob disease: Conclusion of a 15-year investigation in France and review of the world literature. Neurology 1987, 37, 895–904. [Google Scholar] [CrossRef]
- Van Duijn, C.M.; Delasnerie-Laupretre, N.; Masullo, C.; Zerr, I.; de Silva, R.; Wientjens, D.P.; Brandel, J.P.; Weber, T.; Bonavita, V.; Zeidler, M.; et al. Case-control study of risk factors of Creutzfeldt-Jakob disease in Europe during 1993-95. European Union (EU) Collaborative Study Group of Creutzfeldt-Jakob disease (CJD). Lancet 1998, 351, 1081–1085. [Google Scholar] [CrossRef]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.; Serban, A.; Koehler, R.; Foster, D.; Torchia, M.; Burton, D.; Yang, S.L.; DeArmond, S.J. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. USA 1993, 90, 10608–10612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manson, J.C.; Clarke, A.R.; McBride, P.A.; McConnell, I.; Hope, J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994, 3, 331–340. [Google Scholar]
- Sakaguchi, S.; Katamine, S.; Shigematsu, K.; Nakatani, A.; Moriuchi, R.; Nishida, N.; Kurokawa, K.; Nakaoke, R.; Sato, H.; Jishage, K.; et al. Accumulation of proteinase K-resistant prion protein (PrP) is restricted by the expression level of normal PrP in mice inoculated with a mouse-adapted strain of the Creutzfeldt-Jakob disease agent. J. Virol. 1995, 69, 7586–7592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, H.; Chida, J.; Uchiyama, K.; Pasiana, A.D.; Takahashi, E.; Kido, H.; Sakaguchi, S. Neurotropic influenza A virus infection causes prion protein misfolding into infectious prions in neuroblastoma cells. Sci. Rep. 2021, 11, 10109. [Google Scholar] [CrossRef]
- Oesch, B.; Westaway, D.; Walchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [Green Version]
- Hackl, S.; Becker, C.F.W. Prion protein-Semisynthetic prion protein (PrP) variants with posttranslational modifications. J. Pept. Sci. 2019, 25, e3216. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef]
- Chida, J.; Hara, H.; Yano, M.; Uchiyama, K.; Das, N.R.; Takahashi, E.; Miyata, H.; Tomioka, Y.; Ito, T.; Kido, H.; et al. Prion protein protects mice from lethal infection with influenza A viruses. PLoS Pathog. 2018, 14, e1007049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chida, J.; Hara, H.; Uchiyama, K.; Takahashi, E.; Miyata, H.; Kosako, H.; Tomioka, Y.; Ito, T.; Horiuchi, H.; Matsuda, H.; et al. Prion protein signaling induces M2 macrophage polarization and protects from lethal influenza infection in mice. PLoS Pathog. 2020, 16, e1008823. [Google Scholar] [CrossRef]
- Prusiner, S.B. The prion diseases. Brain Pathol. 1998, 8, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; McKinley, M.P.; Bowman, K.A.; Bolton, D.C.; Bendheim, P.E.; Groth, D.F.; Glenner, G.G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 1983, 35, 349–358. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Castilla, J.; Saa, P.; Hetz, C.; Soto, C. In vitro generation of infectious scrapie prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Borges, N.; Di Bari, M.A.; Erana, H.; Sanchez-Martin, M.; Pirisinu, L.; Parra, B.; Elezgarai, S.R.; Vanni, I.; Lopez-Moreno, R.; Vaccari, G.; et al. Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol. 2018, 135, 179–199. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, H.; Bian, W.; McDonald, M.; Kendall, A.; Colby, D.W.; Bloch, L.; Ollesch, J.; Borovinskiy, A.L.; Cohen, F.E.; Prusiner, S.B.; et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Natl. Acad. Sci. USA 2009, 106, 16990–16995. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Fernandez, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016, 12, e1005835. [Google Scholar] [CrossRef] [PubMed]
- Spagnolli, G.; Rigoli, M.; Orioli, S.; Sevillano, A.M.; Faccioli, P.; Wille, H.; Biasini, E.; Requena, J.R. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019, 15, e1007864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskakov, I.V.; Caughey, B.; Requena, J.R.; Sevillano, A.M.; Surewicz, W.K.; Wille, H. The prion 2018 round tables (I): The structure of PrPSc. Prion 2019, 13, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groveman, B.R.; Dolan, M.A.; Taubner, L.M.; Kraus, A.; Wickner, R.B.; Caughey, B. Parallel in-register intermolecular beta-sheet architectures for prion-seeded prion protein (PrP) amyloids. J. Biol. Chem. 2014, 289, 24129–24142. [Google Scholar] [CrossRef] [Green Version]
- Ferhadian, D.; Contrant, M.; Printz-Schweigert, A.; Smyth, R.P.; Paillart, J.C.; Marquet, R. Structural and Functional Motifs in Influenza Virus RNAs. Front. Microbiol. 2018, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Adler, V.; Zeiler, B.; Kryukov, V.; Kascsak, R.; Rubenstein, R.; Grossman, A. Small, highly structured RNAs participate in the conversion of human recombinant PrPSen to PrPRes in vitro. J. Mol. Biol. 2003, 332, 47–57. [Google Scholar] [CrossRef]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Ehresmann, D.W.; Hogan, R.N. Acceleration of scrapie disease in mice by an adenovirus. Intervirology 1986, 25, 103–110. [Google Scholar] [CrossRef]
- Stanton, J.B.; Knowles, D.P.; O’Rourke, K.I.; Herrmann-Hoesing, L.M.; Mathison, B.A.; Baszler, T.V. Small-ruminant lentivirus enhances PrPSc accumulation in cultured sheep microglial cells. J. Virol. 2008, 82, 9839–9847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haviv, Y.; Avrahami, D.; Ovadia, H.; Ben-Hur, T.; Gabizon, R.; Sharon, R. Induced neuroprotection independently from PrPSc accumulation in a mouse model for prion disease treated with simvastatin. Arch. Neurol. 2008, 65, 762–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, P.; Alais, S.; Porto-Carreiro, I.; Lehmann, S.; Grassi, J.; Raposo, G.; Darlix, J.L. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J. 2006, 25, 2674–2685. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.C.; Prusiner, S.B. Experimental Models of Inherited PrP Prion Diseases. Cold Spring Harb. Perspect. Med. 2017, 7, a02715. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.K.; Scott, M.; Foster, D.; Groth, D.F.; DeArmond, S.J.; Prusiner, S.B. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 1990, 250, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.K.; Groth, D.; Scott, M.; Yang, S.L.; Serban, H.; Rapp, D.; Foster, D.; Torchia, M.; Dearmond, S.J.; Prusiner, S.B. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc. Natl. Acad. Sci. USA 1994, 91, 9126–9130. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Cook, J.; Rassbach, B.; Lemus, A.; DeArmond, S.J.; Mastrianni, J.A. A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP. J. Neurosci. 2009, 29, 10072–10080. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, R.; Piccardo, P.; Ghetti, B.; Harris, D.A. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998, 21, 1339–1351. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, R.; Drisaldi, B.; Quaglio, E.; Migheli, A.; Piccardo, P.; Ghetti, B.; Harris, D.A. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 5574–5579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasini, E.; Seegulam, M.E.; Patti, B.N.; Solforosi, L.; Medrano, A.Z.; Christensen, H.M.; Senatore, A.; Chiesa, R.; Williamson, R.A.; Harris, D.A. Non-infectious aggregates of the prion protein react with several PrPSc-directed antibodies. J. Neurochem. 2008, 105, 2190–2204. [Google Scholar] [CrossRef]
- Friedman-Levi, Y.; Meiner, Z.; Canello, T.; Frid, K.; Kovacs, G.G.; Budka, H.; Avrahami, D.; Gabizon, R. Fatal prion disease in a mouse model of genetic E200K Creutzfeldt-Jakob disease. PLoS Pathog. 2011, 7, e1002350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asante, E.A.; Gowland, I.; Grimshaw, A.; Linehan, J.M.; Smidak, M.; Houghton, R.; Osiguwa, O.; Tomlinson, A.; Joiner, S.; Brandner, S.; et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J. Gen. Virol. 2009, 90, 546–558. [Google Scholar] [CrossRef]
- Jackson, W.S.; Borkowski, A.W.; Watson, N.E.; King, O.D.; Faas, H.; Jasanoff, A.; Lindquist, S. Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 14759–14764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossena, S.; Imeri, L.; Mangieri, M.; Garofoli, A.; Ferrari, L.; Senatore, A.; Restelli, E.; Balducci, C.; Fiordaliso, F.; Salio, M.; et al. Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron 2008, 60, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouybayoune, I.; Mantovani, S.; Del Gallo, F.; Bertani, I.; Restelli, E.; Comerio, L.; Tapella, L.; Baracchi, F.; Fernandez-Borges, N.; Mangieri, M.; et al. Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS Pathog. 2015, 11, e1004796. [Google Scholar] [CrossRef]
- Jackson, W.S.; Borkowski, A.W.; Faas, H.; Steele, A.D.; King, O.D.; Watson, N.; Jasanoff, A.; Lindquist, S. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 2009, 63, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Rulicke, T.; Raeber, A.; Sailer, A.; Moser, M.; Oesch, B.; Brandner, S.; Aguzzi, A.; Weissmann, C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996, 15, 1255–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, J.A.; Unterberger, U.; Saa, P.; Massignan, T.; Fluharty, B.R.; Bowman, F.P.; Miller, M.B.; Supattapone, S.; Biasini, E.; Harris, D.A. The N-terminal, polybasic region of PrPC dictates the efficiency of prion propagation by binding to PrPSc. J. Neurosci. 2012, 32, 8817–8830. [Google Scholar] [CrossRef] [Green Version]
- Das, N.R.; Miyata, H.; Hara, H.; Chida, J.; Uchiyama, K.; Masujin, K.; Watanabe, H.; Kondoh, G.; Sakaguchi, S. The N-Terminal Polybasic Region of Prion Protein Is Crucial in Prion Pathogenesis Independently of the Octapeptide Repeat Region. Mol. Neurobiol. 2020, 57, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, E.; Shmerling, D.; Hegyi, I.; Raeber, A.J.; Fischer, M.; Cozzio, A.; von Mering, C.; Aguzzi, A.; Weissmann, C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 2000, 27, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Weissmann, C.; Flechsig, E. PrP knock-out and PrP transgenic mice in prion research. Br. Med. Bull. 2003, 66, 43–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, H.; Miyata, H.; Das, N.R.; Chida, J.; Yoshimochi, T.; Uchiyama, K.; Watanabe, H.; Kondoh, G.; Yokoyama, T.; Sakaguchi, S. Prion Protein Devoid of the Octapeptide Repeat Region Delays Bovine Spongiform Encephalopathy Pathogenesis in Mice. J. Virol. 2018, 92, e01368-17. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, K.; Miyata, H.; Yamaguchi, Y.; Imamura, M.; Okazaki, M.; Pasiana, A.D.; Chida, J.; Hara, H.; Atarashi, R.; Watanabe, H.; et al. Strain-Dependent Prion Infection in Mice Expressing Prion Protein with Deletion of Central Residues 91–106. Int. J. Mol. Sci. 2020, 21, 7260. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Okemoto-Nakamura, Y.; Shinkai-Ouchi, F.; Hanada, K.; Yamakawa, Y.; Hagiwara, K. Mouse prion protein (PrP) segment 100 to 104 regulates conversion of PrPC to PrPSc in prion-infected neuroblastoma cells. J. Virol. 2012, 86, 5626–5636. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, K.; Hara, H.; Hanada, K. Species-barrier phenomenon in prion transmissibility from a viewpoint of protein science. J. Biochem. 2013, 153, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, C.; Wille, H.; Prusiner, S.B.; Cohen, F.E. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc. Natl. Acad. Sci. USA 2004, 101, 8342–8347. [Google Scholar] [CrossRef] [Green Version]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551. [Google Scholar] [CrossRef]
Figure 1.
Seeded protein polymerization model for the conversion of PrPC into PrPSc in sporadic, hereditary, and acquired prion diseases. (1) IAV/WSN infection and interaction with RNA, lipid molecules plasmid DNA and dextran sulfate drives PrPC to undergo conformational changes to form PrPSc in sporadic prion diseases. (2) In hereditary prion diseases, mutated PrPs are prone to convert into PrPSc. The nascently generated PrPSc molecules in sporadic and hereditary prion diseases assemble to form small oligomeric PrPSc aggregates and then further polymerize to form PrPSc seeds, which are able to recruit and force PrPC to convert into PrPSc on them. (3) In acquired prion diseases, exogenously invading prions function as PrPSc seeds for the conversion of PrPC into PrPSc. As a result, the PrPSc oligomers are elongated to form fibrils in sporadic, hereditary, and acquired prion diseases. The fibrils are then disrupted into prions, or small PrPSc seeds, which then enter the conversion cycle of PrPC into PrPSc.
Figure 1.
Seeded protein polymerization model for the conversion of PrPC into PrPSc in sporadic, hereditary, and acquired prion diseases. (1) IAV/WSN infection and interaction with RNA, lipid molecules plasmid DNA and dextran sulfate drives PrPC to undergo conformational changes to form PrPSc in sporadic prion diseases. (2) In hereditary prion diseases, mutated PrPs are prone to convert into PrPSc. The nascently generated PrPSc molecules in sporadic and hereditary prion diseases assemble to form small oligomeric PrPSc aggregates and then further polymerize to form PrPSc seeds, which are able to recruit and force PrPC to convert into PrPSc on them. (3) In acquired prion diseases, exogenously invading prions function as PrPSc seeds for the conversion of PrPC into PrPSc. As a result, the PrPSc oligomers are elongated to form fibrils in sporadic, hereditary, and acquired prion diseases. The fibrils are then disrupted into prions, or small PrPSc seeds, which then enter the conversion cycle of PrPC into PrPSc.

Figure 2.
Schematic illustration of hereditary prion disease-associated mutations in PrP. GSS, fCJD, and FFI mutations are indicated in red, blue, and green, respectively.
Figure 2.
Schematic illustration of hereditary prion disease-associated mutations in PrP. GSS, fCJD, and FFI mutations are indicated in red, blue, and green, respectively.


{kind=link}
{kind=link}
Table 1.
Hereditary prion disease-associated mutations in PrP conversion.
| Hereditary Prion Diseases | Mutant PrPs | Conversion to Neurotoxic PrPSc/Infectious PrPSc | References |
|---|---|---|---|
| GSS | Mo-PrP-P101L | Yes/Yes | [57,58] |
| Mo-PrP-A116V | Yes/N.D. | [59] | |
| Mo-PrP-PG14 | Yes/No | [60,61,62] | |
| fCJD | Mo/Hu-PrP-E199K | Yes/Yes | [63] |
| Hu-PrP-E200K | No/No | [64] | |
| Mo-PrP-E199K(3F4) | Yes/Yes | [65] | |
| Mo-PrP-D177N(V128) | Yes/No | [66] | |
| FFI | Mo-PrP-D177N(M128) | Yes/No | [67] |
| Mo-PrP-D177N(M128)(3F4) | Yes/Yes | [68] |
Mo, mouse; Hu, human; N.D., not determined.
Table 2.
PrP deletional and point mutants in prion infections.
| PrPs | Susceptibility to Prions | References |
|---|---|---|
| PrP∆23–31 | Reduced to RML prions | [70] |
| PrP3K3A | Reduced to RML and 22L prions | [71] |
| PrP∆32–80 | Not reduced to RML prions | [72] |
| PrP∆32–93 | Reduced to RML prions | [73] |
| PrP∆32–106 | Resistant to RML prions | [73] |
| PrP∆OR | Not reduced to RML and 22L prions Reduced to BSE prions | [74] |
| PrP∆91–106 | Resistant to RML and 22L prions Reduced to BSE prions | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hara, H.; Sakaguchi, S. Virus Infection, Genetic Mutations, and Prion Infection in Prion Protein Conversion. Int. J. Mol. Sci. 2021, 22, 12439. https://doi.org/10.3390/ijms222212439
AMA Style
Hara H, Sakaguchi S. Virus Infection, Genetic Mutations, and Prion Infection in Prion Protein Conversion. International Journal of Molecular Sciences. 2021; 22(22):12439. https://doi.org/10.3390/ijms222212439
Chicago/Turabian StyleHara, Hideyuki, and Suehiro Sakaguchi. 2021. "Virus Infection, Genetic Mutations, and Prion Infection in Prion Protein Conversion" International Journal of Molecular Sciences 22, no. 22: 12439. https://doi.org/10.3390/ijms222212439
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.