
Novel Sulfonamide-Based Carbamates as Selective Inhibitors of BChE

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Evaluation of AChE- and BChE-Inhibiting Activity
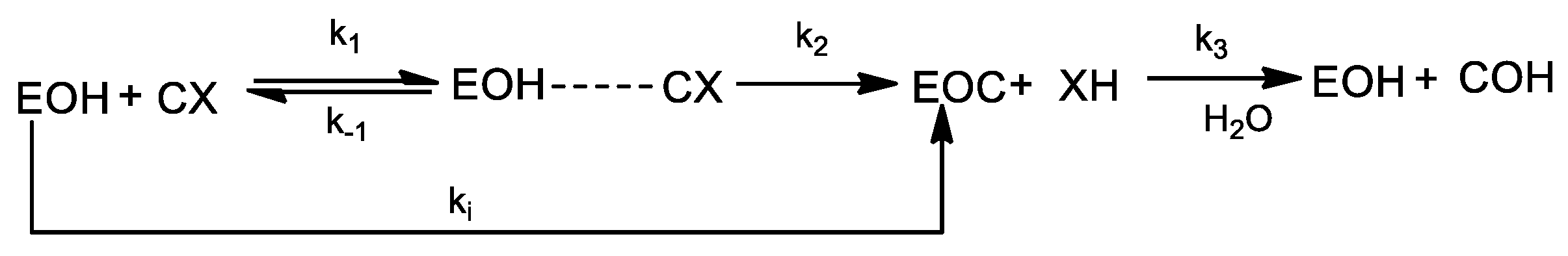
2.3. Evaluation of Carbamylation and Reactivation of BChE
2.4. Molecular Modeling Studies
QTAIM Calculations
2.5. Cytotoxicity Evaluation and ADME Predictions
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. General Experimental Procedure for the Synthesis of N-Cbz Alcohols 2a–c
3.2.2. General Experimental Procedure for the Synthesis of Mesylates 3a–c
3.2.3. General Experimental Procedure for the Synthesis of Thioacetates 4a–c
3.2.4. General Experimental Procedure for the Synthesis of the Investigated Sulfonates 5a–5n
3.3. AChE and BChE Inhibition Studies
3.4. Kinetic Studies
3.5. Molecular Modeling
3.5.1. Receptor Preparation and Docking Procedure
3.5.2. Molecular Dynamic Simulations
3.5.3. MM-GBSA Free Energy Decomposition
3.5.4. QTAIM Analysis
3.6. Cytotoxicity Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eikelboom, W.S.; Van den Berg, E.; Coesmans, M.; Singleton, E.H.; Papma, J.M. Neuropsychiatric and Cognitive Symptoms in Alzheimer’s Disease: A Study in Ad Biomarker Confirmed Patients across the Clinical Spectrum. Alzheimer’s Dement. 2019, 15, 567–568. [Google Scholar] [CrossRef]
- Lopez, O.L.; DeKosky, S.T. Clinical symptoms in Alzheimer’s disease. Handb. Clin. Neurol. 2008, 89, 207–216. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Milleri, D.S. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’S Assoc. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.; Palmer, A.; Snape, M.; Wilcock, G. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimer’s Dis. 2019, 6, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Blessed, G.; Tomlinson, B.E.; Roth, M. The Association between Quantitative Measures of Dementia and of Senile Change in the Cerebral Grey Matter of Elderly Subjects. Br. J. Psychiatry 1968, 114, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Tepper, M.; et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 2018, 99, 925–940. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lambb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarević-Pašti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A Review of Butyrylcholinesterase as a Therapeutic Target in the Treatment of Alzheimer’s Disease. Prim. Care Companion CNS Disord. 2013, 15. PCC.12r01412. [Google Scholar] [CrossRef]
- Darvesh, S. Butyrylcholinesterase as a Diagnostic and Therapeutic Target for Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Yu, Q.; Zhu, X.; Holloway, H.W.; Perry, T.A.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A New Therapeutic Target in Alzheimer’s Disease Treatment: Attention to Butyrylcholinesterase. Curr. Med Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, G.; Greig, N.H.; Khan, J.A.; Kamal, M.A. Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1432–1439. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Lange, M.; Bigl, V. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development—A study of molecular forms. Neurochem. Int. 1992, 21, 381–396. [Google Scholar] [CrossRef]
- Mesulam, M.M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Li, B.; Duysen, E.G.; Carlson, M.; Lockridge, O. The Butyrylcholinesterase Knockout Mouse as a Model for Human Butyrylcholinesterase Deficiency. J. Pharmacol. Exp. Ther. 2008, 324, 1146–1154. [Google Scholar] [CrossRef]
- Holmes, C.; Ballard, C.; Lehmann, D.; Smith, A.D.; Beaumont, H.; Day, I.N.; Khan, M.N.; Lovestone, S.; McCulley, M.; Morris, C.M.; et al. Rate of Progression of Cognitive Decline in Alzheimer’s Disease: Effect of Butyrylcholinesterase K Gene Variation. J. Neurol. Neurosur. Psychiatry 2005, 76, 640–643. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.; Kiewert, C.; Duysen, E.G.; Lockridge, O.; Greig, N.H.; Klein, J. Excessive Hippocampal Acetylcholine Levels in Acetylcholinesterase-Deficient Mice Are Moderated by Butyrylcholinesterase Activity. J. Neurochem. 2007, 100, 1421–1429. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective Butyrylcholinesterase Inhibition Elevates Brain Acetylcholine, Augments Learning and Lowers Alzheimer B-Amyloid Peptide in Rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa-Hibi, Y.; Alkam, T.; Nitta, A.; Matsuyama, A.; Mizoguchi, H.; Suzuki, K.; Moussaoui, S.; Yu, Q.S.; Greig, N.H.; Nagai, T.; et al. Butyrylcholinesterase Inhibitors Ameliorate Cognitive Dysfunction Induced by Amyloid-B Peptide in Mice. Behav. Brain Res. 2011, 225, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, N.H.; Yu, Q.; Brossi, A.; Soncrant, T.T.; Hausman, M. Highly Selective Butyrylcholinesterase Inhibitors for the Treatment and Diagnosis of Alzheimer’s Disease and Dementias. U.S. Patent 20020094999, 18 July 2002. [Google Scholar]
- Giacobini, E. Cholinesterase Inhibitors: New Roles and Therapeutic Alternatives. J. Ital. Pharmacol. Soc. 2004, 50, 433–440. [Google Scholar]
- McGleenon, B.M.; Dynan, K.B.; Passmore, A.P. Acetylcholinesterase Inhibitors in Alzheimer’s Disease. Br. J. Clin. Pharmacol. 1999, 48, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Ballard, C.; Greig, N.; Guillozet-Bongaarts, A.; Enz, A.; Darvesh, S. Cholinesterases: Roles in the Brain During Health and Disease. Curr. Alzheimer Res. 2005, 2, 307–318. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Dighe, S.N.; Tippana, M.; van Akker, S.; Collet, T.A. Structure-based scaffold repurposing toward the discovery of novel cholinesterase inhibitors. ACS Omega 2020, 5, 30971–30979. [Google Scholar] [CrossRef]
- Lane, R.M.; Potkin, S.G.A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef]
- Alcantara, V.M.; Chautard-Freire-Maia, E.A.; Scartezini, M.; Cerci, M.S.J.; Braun-Prado, K.; Picheth, G. Butyrylcholinesterase Activity and Risk Factors for Coronary Artery Disease. Scand. J. Clin. Lab. Investig. 2002, 62, 399–404. [Google Scholar] [CrossRef]
- Stojanov, M.; Stefanovič, A.; Džingalaševič, G.; Mandič-Radič, S.; Prostran, M. Butyrylcholinesterase Activity in Young Men and Women: Association with Cardiovascular Risk Factors. Clin. Biochem. 2011, 44, 623–626. [Google Scholar] [CrossRef]
- Iwasaki, T.; Yoneda, M.; Nakajima, A.; Terauchi, Y. Serum Butyrylcholinesterase is Strongly Associated with Adiposity, the Serum Lipid Profile and Insulin Resistance. Intern. Med. 2007, 46, 1633–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.K.; Hayashi, T.; Maeda, I.; Koh, H.; Harita, N.; Uehara, S.; Onishi, Y.; Oue, K.; Nakamura, Y.; Endo, G.; et al. Serum Butyrylcholinesterase and the Risk of Future Type 2 Diabetes: The Kansai Healthcare Study. Clin. Endocrinol. 2014, 80, 362–367. [Google Scholar] [CrossRef]
- Li, B.; Duysen, E.G.; Lockridge, O. The Butyrylcholinesterase Knockout Mouse is Obese on a High-Fat Diet. Chem.-Biol. Interact. 2008, 175, 88–91. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Brus, B.; Košak, U.; Turk, S.; Pišlar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.P.; Gobec, S.J. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef]
- Sawatzky, E.; Wehle, S.; Kling, B.; Wendrich, J.; Bringmann, G.; Christoph, A.; Sotriffer, C.A.; Heilmann, J.; Decker, M. Discovery of highly selective and nanomolar carbamate-based butyrylcholinesterase inhibitors by rational investigation into their inhibition mode. J. Med. Chem. 2016, 59, 2067–2082. [Google Scholar] [CrossRef]
- Kumar, A.; Pintus, F.; Di Petrillo, A.; Medda, A.R.; Caria, P.; Matos, M.J.; Vina, D.; Pieroni, E.; Delogu, F.; Era, B.; et al. Novel 2-pheynlbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci. Rep. 2018, 8, 4424. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Tu, Y.; Li, Z.; Li, Y. Highly selective carbamate-based butyrylcholinesterase inhibitors derived from a naturally occurring pyranoisoflavone. Bioorg. Chem. 2019, 88, 102949. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Stiller, C.; Endres, E.; Scheiner, M.; Gunesch, S.; Sotriffer, C.; Maurice, T.; Decker, M. Highly selective butyrylcholinesterase inhibitors with tunable duration of action by chemical modification of transferable carbamate units exhibit pronounced neuroprotective effect in an Alzheimer’s disease mouse model. J. Med. Chem. 2019, 62, 9116–9140. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Kozakiewicz, D.; Gajcy, K.; Strub, D.J.; Swietlicka, A.; Stepankova, S.; Imramovsky, A.; Polanski, J.; Smolinski, A.; et al. Novel benzene-based carbamates for AChE/BChE inhibition: Synthesis and ligand/structure-oriented SAR study. Int. J. Mol. Sci. 2019, 20, 1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielczewska, U.; Jorda, R.; Gonzalez, G.; Morzycki, J.W.; Ajani, H.; Svrckova, K.; Stepankova, S.; Wojtkielewicz, A. The synthesis and cholinesterase inhibitory activities of solasodine analogues with seven-membered F ring. J. Steroid Biochem. Mol. Biol. 2021, 205, 105776. [Google Scholar] [CrossRef]
- Jann, M.W.; Shirley, K.L.; Small, G.W. Clinical Pharmacokinetics and Pharmacodynamics of Cholinesterase Inhibitors. Clin. Pharmacokinet. 2002, 41, 719–739. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, M.; Cavrini, V.; Andrisano, V. Characterization of reversible and pseudo-irreversible acetylcholinesterase inhibitors by means of an immobilized enzyme reactor. J. Chromatogr. A 2007, 1144, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.B.; Hatch, M.A.; Ginsburg, S. Carbamylation of Acetylcholinesterase. J. Biol. Chem. 1960, 235, 2312–2315. [Google Scholar] [CrossRef]
- Wilson, I.B.; Harrison, M.A.; Ginsburg, S. Carbamyl derivatives of acetylcholinesterase. J. Biol. Chem. 1961, 236, 1498–1500. [Google Scholar] [CrossRef]
- Xie, Q.; Zheng, Z.; Shao, B.; Fu, W.; Xia, Z.; Li, W.; Sun, J.; Zheng, W.; Zhang, W.; Sheng, W.; et al. Pharmacophore-based design and discovery of (−)-meptazinol carbamates as dual modulators of cholinesterase and amyloidogenesis. J. Enzym. Inhib. Med. Chem. 2017, 32, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Kazaz, C.; Levent, S.; Gulcin, I. Synthesis, structure elucidation, and in vitro pharmacological evaluation of novel polyfluoro substituted pyrazoline type sulfonamides as multi-target agents for inhibition of acetylcholinesterase and carbonic anhydrase I and II enzymes. Bioorg. Chem. 2020, 96, 103627. [Google Scholar] [CrossRef]
- Bag, S.; Tulsan, R.; Sood, A.; Cho, H.; Redjeb, W.; Zhou, H.; LeVine, B.; Török, M. Sulfonamides as multifunctional agents for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015, 25, 626–630. [Google Scholar] [CrossRef]
- Košak, U.; Brus, B.; Knez, D.; Šink, R.; Žakelj, S.; Trontelj, J.; Pišlar, A.; Šlenc, J.; Gobec, M.; Živin, M.; et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016, 6, 39495. [Google Scholar] [CrossRef] [Green Version]
- Imramovsky, A.; Stepankova, S.; Vanco, J.; Pauk, K.; Monreal-Ferriz, J.; Vinsova, J.; Jampilek, J. Acetylcholinesterase-inhibiting activity of salicylanilide N-alkylcarbamates and their molecular docking. Molecules 2012, 17, 10142–10158. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pejchal, V.; Stepankova, S.; Vorcakova, K.; Jampilek, J.; Vanco, J.; Simunek, P.; Kralovec, K.; Bruckova, L.; Mandikova, J.; et al. Synthesis and in vitro evaluation of new derivatives of 2-substituted-6-fluorobenzo[d]thiazoles as cholinesterase inhibitors. Bioorg. Med. Chem. 2013, 21, 1735–1748. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Rodriguez, M.; Llinares, M.; Doulut, S.; Heitz, A.; Martinez, J. A facile synthesis of chiral of chiral N-protected β-amino alcohols. Tet. Lett. 1991, 32, 923–926. [Google Scholar] [CrossRef]
- Dubiella, C.; Cui, H.; Gersch, M.; Brouwer, A.J.; Sieber, S.A.; Kruger, A.; Liskamp, R.M.J.; Groll. M. Selective inhibition of the immunoproteasome by ligand-induced crosslinking of the active site. Angew. Chem. Int. Ed. Engl. 2014, 53, 11969–11973. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- DrugBank—Rivastigmine. Available online: https://go.drugbank.com/drugs/DB00989 (accessed on 19 June 2021).
- Ariel, N.; Ordentlich, A.; Barak, D.; Bino, T.; Velan, B.; Shafferman, A. The ‘aromatic patch’ of three proximal residues in the human acetylcholinesterase active centre allows for versatile interaction modes with inhibitors. Biochem. J. 1998, 335, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Rojas, S.; Parravicini, O.; Vettorazzi, M.; Tosso, R.; Garro, A.; Gutierrez, L.; Andujar, S.; Enriz, R. Combined MD/QTAIM techniques to evaluate ligand-receptor interactions. Scope and limitations. Eur. J. Med. Chem. 2020, 208, 112792. [Google Scholar] [CrossRef]
- Campos, L.E.; Garibotto, F.; Angelina, E.; Kos, J.; Gonec, T.; Marvanova, P.; Vettorazzi, M.; Oravec, M.; Jendrzejewska, I.; Jampilek, J.; et al. Hydroxynaphthalenecarboxamides and Substituted Piperazinylpropandiols, Two New Series of BRAF Inhibitors. A theoretical and experimental study. Bioorg. Chem. 2020, 103, 104145. [Google Scholar] [CrossRef] [PubMed]
- Vettorazzi, M.; Insuasty, D.; Lima, S.; Gutierrez, L.; Nogueras, M.; Marchal, A.; Abonia, R.; Andujar, S.; Spiegel, S.; Cobo, J.; et al. Design of new quinolin-2-one-pyrimidine hybrids as sphingosine kinases inhibitors. Bioorg. Chem. 2020, 94, 103414. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Angelina, E.; Lima, S.; Gonec, T.; Otevrel, J.; Marvanova, P.; Padrtova, T.; Mokry, P.; Bobal, P.; Acosta, L.M.; et al. An integrative study to identify novel scaffolds for sphingosine kinase 1 inhibitors. Eur. J. Med. Chem. 2017, 139, 461–481. [Google Scholar] [CrossRef] [PubMed]
- Tosso, R.D.; Andujar, S.A.; Gutierrez, L.; Angelina, E.; Rodriguez, R.; Nogueras, M.; Baldoni, H.; Suvire, F.D.; Cobo, J.; Enriz, R.D. Molecular modeling study of dihydrofolate reductase inhibitors. Molecular dynamics simulations, quantum mechanical calculations, and experimental corroboration. J. Chem. Inf. Model. 2013, 53, 2018–2032. [Google Scholar] [CrossRef]
- Padrtova, T.; Marvanova, P.; Odehnalova, K.; Kubinova, R.; Parravicini, O.; Garro, A.; Enriz, R.; Humpa, O.; Oravec, M.; Mokry, P. Synthesis, analysis, cholinesterase-inhibiting activity and molecular modelling studies of 3-(dialkylamino)-2-hydroxypropyl 4-[(alkoxy-carbonyl)amino]benzoates and their quaternary ammonium salts. Molecules 2017, 22, 2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parravicini, O.; Angelina, E.; Spinelli, R.; Garibotto, F.; Siano, A.S.; Vila, L.; Cabedo, N.; Cortes, D.; Enriz, R.D. Design, synthesis, biological evaluation and molecular modelling of substituted pyrrolo[2,1-a]isoquinolinone derivatives: Discovery of potent inhibitors of AChE and BChE. New J. Chem. 2021, in press. [Google Scholar] [CrossRef]
- DrugBank—Galantamine. Available online: https://go.drugbank.com/drugs/DB00674 (accessed on 19 June 2021).
- Ortiz, J.E.; Garro, A.; Pigni, N.B.; Agüero, M.B.; Roitman, G.; Slanis, A.; Enriz, R.D.; Feresin, G.E.; Bastida, J.; Tapia, A. Cholinesterase-inhibitory effect and in silico analysis of alkaloids from bulbs of hieronymiella species. Phytomedicine 2018, 39, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, J.E.; Pigni, N.B.; Andujar, S.A.; Roitman, G.; Suvire, F.D.; Enriz, R.D.; Tapia, A.; Bastida, J.; Feresin, G.E. Alkaloids from hippeastrum argentinum and their cholinesterase-inhibitory activities: An in vitro and in silico study. J. Nat. Prod. 2016, 79, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, H.M.; Kryger, G.; Lewis, T.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with (−)-galanthamine at 2.3 Å resolution. FEBS Lett. 1999, 463, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Viglianco, F.; Zaragoza-Puchol, D.; Parravicini, O.; Garro, A.; Enriz, R.D.; Feresin, G.E.; Kurina-Sanz, M.; Orden, A.A. Synthesis, biological evaluation and molecular modeling studies of substituted N-benzyl-2-phenylethanamines as cholinesterase inhibitors. New J. Chem. 2020, 44, 9466–9476. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Gibney, G.; Camp, S.; Dionne, M.; MacPhee-Quigley, K.; Taylor, P. Mutagenesis of essential functional residues in acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1990, 87, 7546–7550. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, A.S.; Bhanumathy, C.D. Noncholinergic functions of cholinesterases. FASEB J. 1993, 7, 1354–1358. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modeling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L. Drug-Like Properties: Concepts. Structure Design and Methods: From ADME to Toxicity Optimization; Academic Press: San Diego, CA, USA, 2008. [Google Scholar]
- Wermuth, C.; Aldous, D.; Raboisson, P.; Rognan, D. The Practice of Medicinal Chemistry, 4th ed.; Academic Press: San Diego, CA, USA, 2015. [Google Scholar]
- Fukunishi, Y.; Nakamura, H. Definition of drug-likeness for compound affinity. J. Chem. Inf. Model. 2011, 51, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J. Potential of agricultural fungicides for antifungal drug discovery. Expert Opin. Drug Dis. 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Kwok, K.C.; Wang, Y.; Bao, H. An improved method to determine SH and –S–S– group content in soymilk protein. Food Chem. 2004, 88, 317–320. [Google Scholar] [CrossRef]
- Carletti, E.; Aubrek, N.; Gillon, E.; Loiodice, M.; Nicolet, Y.; Fontecilla-Camps, J.-C.; Masson, P.; Thiermann, H.; Nachon, F.; Worek, F. Structure–activity analysis of aging and reactivation of human butyrylcholinesterase inhibited by analogues of tabun. Biochem. J. 2009, 421, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krátký, M.; Štěpánková, Š.; Vorčáková, K.; Vinšová, J. Salicylanilide diethyl phosphates as cholinesterases inhibitors. Bioorg. Chem. 2015, 58, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.; Belew, R.; Goodsell, D.; Olson, A. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.; Dror, R.; Shaw, D. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.; Caldwell, J.; Kollman, P.; Case, D. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Izaguirre, J.; Catarello, D.; Wozniak, J.; Skeel, R. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Kozik, V.; Pindjakova, D.; Jankech, T.; Smolinski, A.; Stepankova, S.; Hosek, J.; Oravec, M.; Jampilek, J.; Bak, A. Synthesis and Hybrid SAR Property Modeling of Novel Cholinesterase Inhibitors. Int. J. Mol. Sci. 2021, 22, 3444. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Structure | IC50 (µM) | SI * | |
|---|---|---|---|---|
| AChE | BChE | |||
| 5a |  | 90.56 ± 5.70 | 100.25 ± 4.26 | 0.90 |
| 5b |  | 76.95 ± 1.52 | 86.12 ± 1.91 | 0.89 |
| 5c |  | 91.81 ± 4.89 | 8.52 ± 0.32 | 10.78 |
| 5d |  | 55.64 ± 1.38 | 16.54 ± 0.10 | 3.36 |
| 5e |  | 108.46 ± 2.39 | 58.94 ± 1.88 | 1.84 |
| 5f |  | 114.73 ± 6.22 | 15.29 ± 0.38 | 7.50 |
| 5g |  | 78.83 ± 0.63 | 12.61 ± 0.31 | 6.25 |
| 5h |  | 61.39 ± 0.47 | 11.54 ± 0.28 | 5.32 |
| 5i |  | 61.65 ± 1.47 | 112.21 ± 3.41 | 0.55 |
| 5j |  | 67.47 ± 0.37 | 6.57 ± 0.12 | 10.27 |
| 5k |  | 150.29 ± 3.82 | 4.33 ± 0.07 | 34.71 |
| 5l |  | 92.91 ± 2.47 | 55.64 ± 1.38 | 1.67 |
| 5m |  | 131.85 ± 0.42 | 19.99 ± 0.17 | 5.70 |
| 5n |  | 95.14 ± 2.77 | 13.51 ± 0.51 | 7.04 |
| RIV | – | 56.10 ± 1.41 | 38.40 ± 1.97 | 1.46 |
 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | R | MW | logP | HBD | HBA | RB | TPSA | HJS (cm/s) | ka (min−1) | logKaHSA | %PPB | logPS | logBB |
| 5c |  | 404.52 | 3.69 | 2 | 6 | 11 | 92.88 | 7.22 × 10−4 | 0.050 | 5.18 | 90.52 | −1.3 | 0.49 |
| 5f |  | 418.55 | 4.04 | 2 | 6 | 12 | 92.88 | 7.13 × 10−4 | 0.049 | 5.22 | 91.70 | −1.3 | 0.63 |
| 5j |  | 438.97 | 4.68 | 2 | 6 | 11 | 92.88 | 7.15 × 10−4 | 0.0449 | 5.34 | 94.64 | −1.2 | 0.74 |
| 5k |  | 434.55 | 3.95 | 2 | 7 | 12 | 102.11 | 7.09 × 10−4 | 0.049 | 5.20 | 89.60 | −1.3 | 0.68 |
| 5n |  | 459.39 | 5.17 | 2 | 6 | 10 | 92.88 | 7.17 × 10−4 | 0.049 | 5.68 | 96.90 | −1.8 | 0.42 |
| rivastigmine | 250.34 | 2.29 | 0 | 4 | 5 | 32.78 | 6.89 × 10−4 | 0.047 | 3.57 | 33.55 | −1.9 | 0.51 | |
| galantamine | 287.35 | 1.55 | 1 | 4 | 1 | 41.93 | 4.37 × 10−4 | 0.030 | 3.38 | 19.99 | −2.2 | 0.28 | |
| Ro5 | <500 | <5 | <5 | <10 | – | – | – | – | – | – | – | – | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magar, P.; Parravicini, O.; Štěpánková, Š.; Svrčková, K.; Garro, A.D.; Jendrzejewska, I.; Pauk, K.; Hošek, J.; Jampílek, J.; Enriz, R.D.; et al. Novel Sulfonamide-Based Carbamates as Selective Inhibitors of BChE. Int. J. Mol. Sci. 2021, 22, 9447. https://doi.org/10.3390/ijms22179447
Magar P, Parravicini O, Štěpánková Š, Svrčková K, Garro AD, Jendrzejewska I, Pauk K, Hošek J, Jampílek J, Enriz RD, et al. Novel Sulfonamide-Based Carbamates as Selective Inhibitors of BChE. International Journal of Molecular Sciences. 2021; 22(17):9447. https://doi.org/10.3390/ijms22179447
Chicago/Turabian StyleMagar, Pratibha, Oscar Parravicini, Šárka Štěpánková, Katarina Svrčková, Adriana D. Garro, Izabela Jendrzejewska, Karel Pauk, Jan Hošek, Josef Jampílek, Ricardo D. Enriz, and et al. 2021. "Novel Sulfonamide-Based Carbamates as Selective Inhibitors of BChE" International Journal of Molecular Sciences 22, no. 17: 9447. https://doi.org/10.3390/ijms22179447