
Identification of Rf Genes in Hexaploid Wheat (Triticumaestivum L.) by RNA-Seq and Paralog Analyses

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
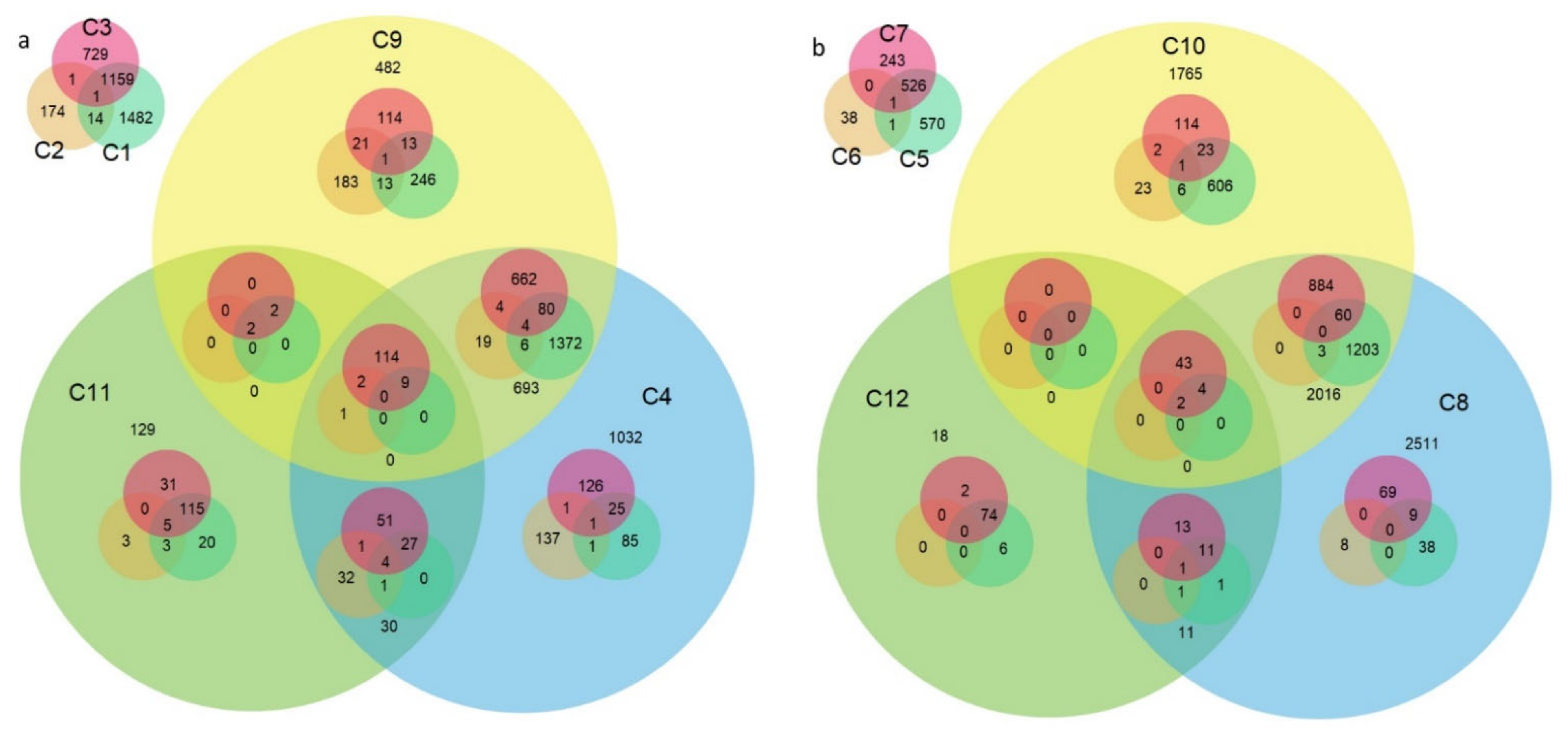
2.1. RNA-Seq Analysis
2.1.1. General Characteristics of the RNA-Seq Data
2.1.2. Targeted Analysis of RNA-Seq Results
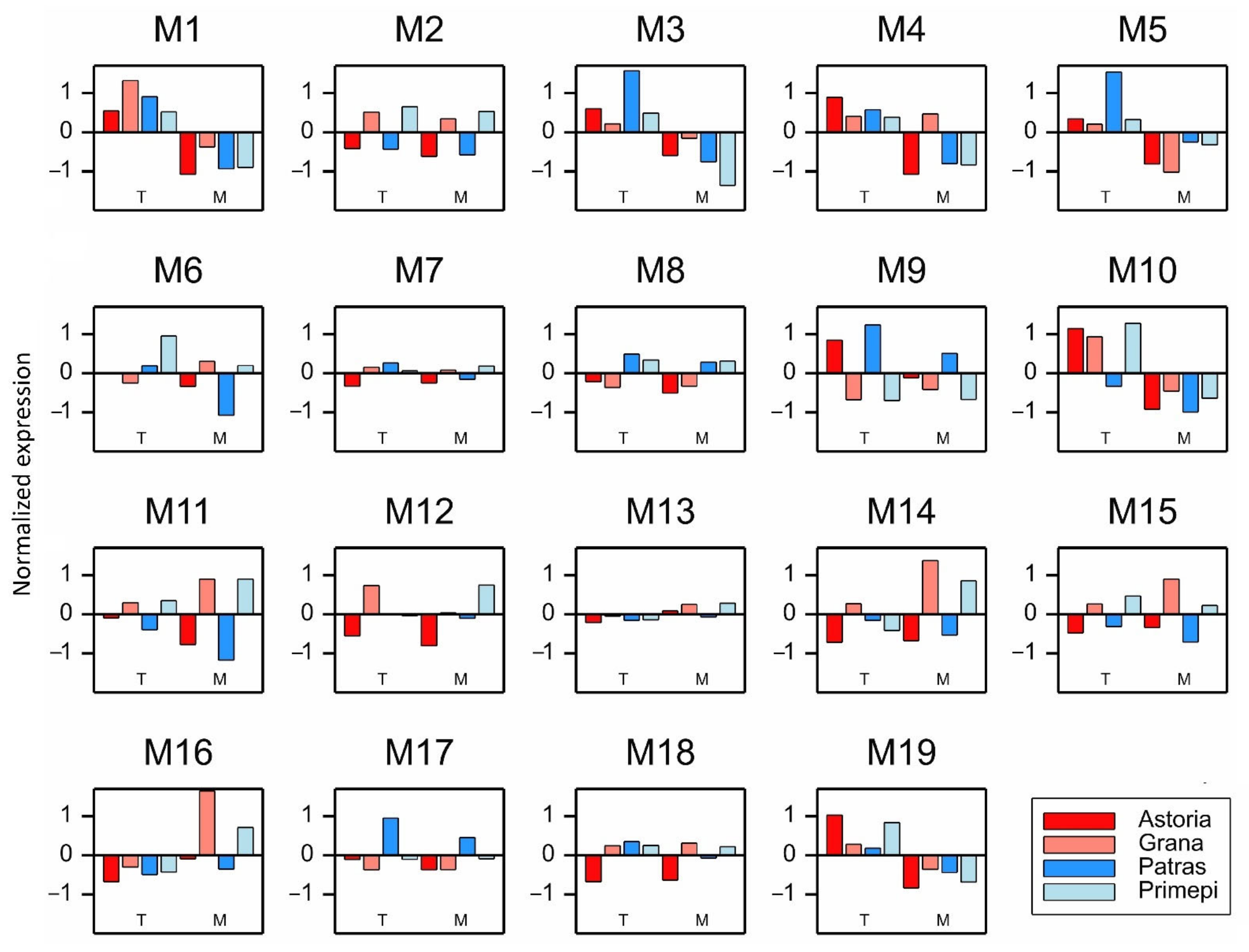
2.2. Analysis of Paralogous Genes
2.3. In Silico Analysis of the Promoters of Selected DEGs and PAGs
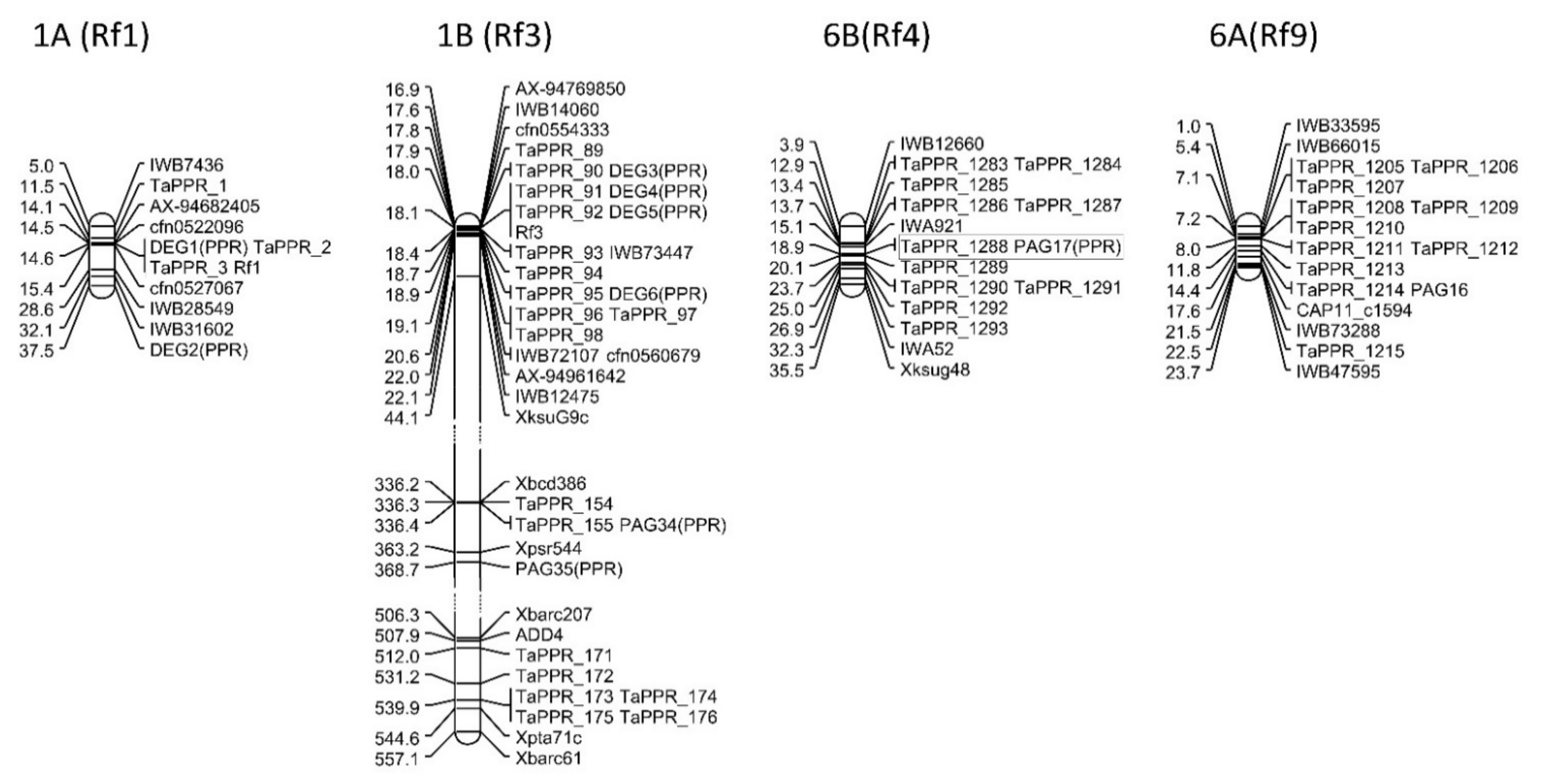
2.4. Mapping DEGs and PAGs in Rf Gene Regions
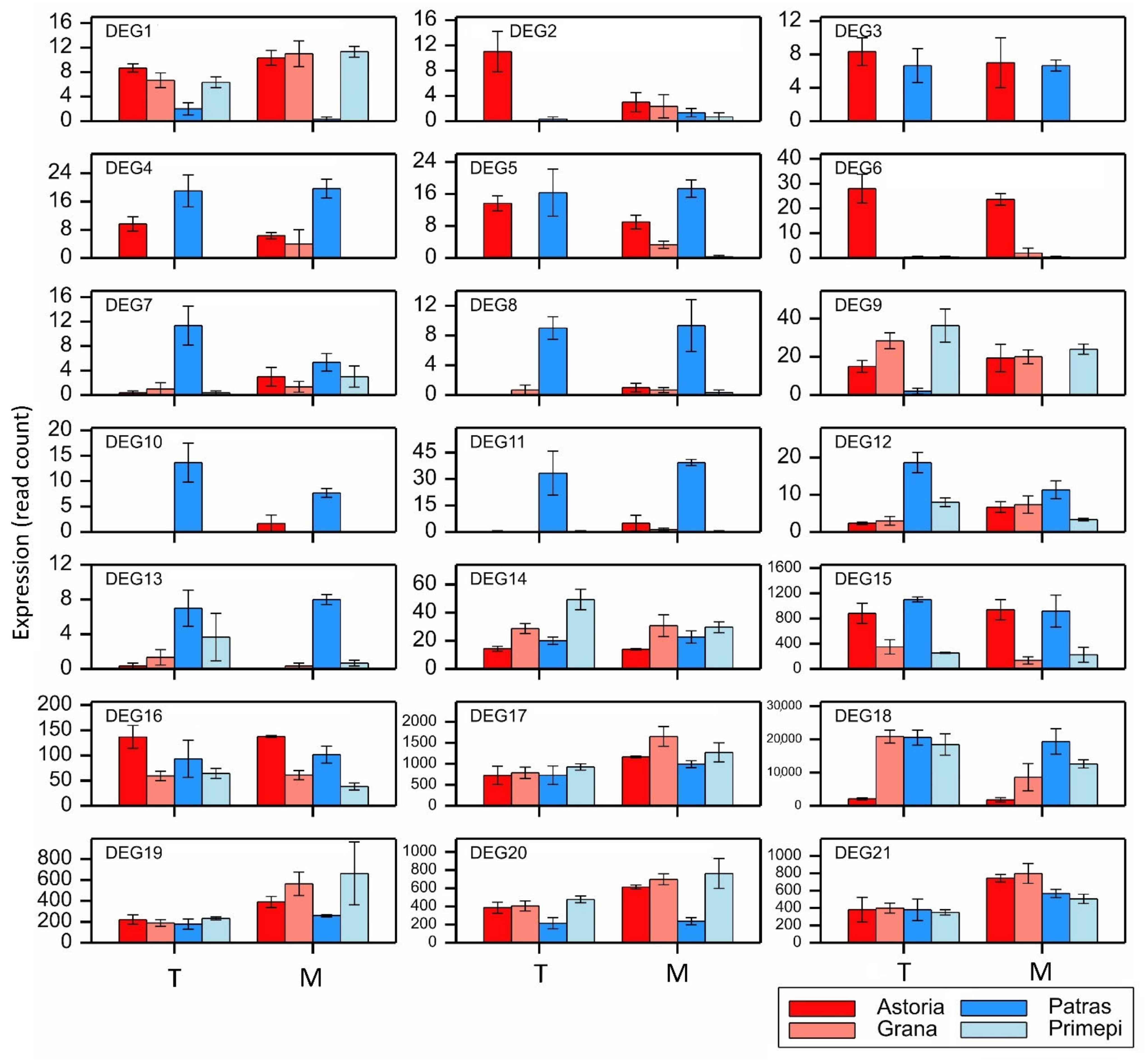
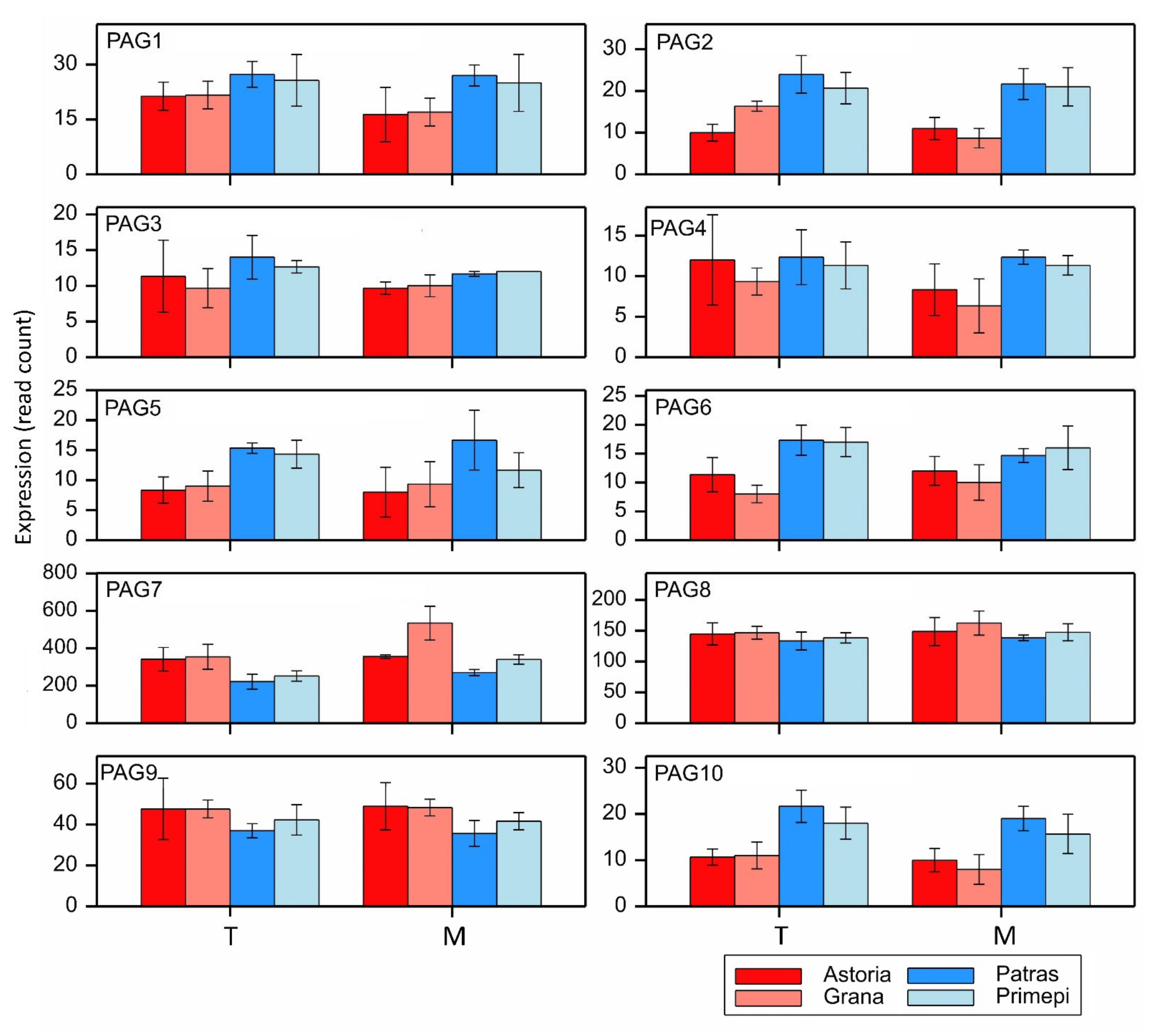
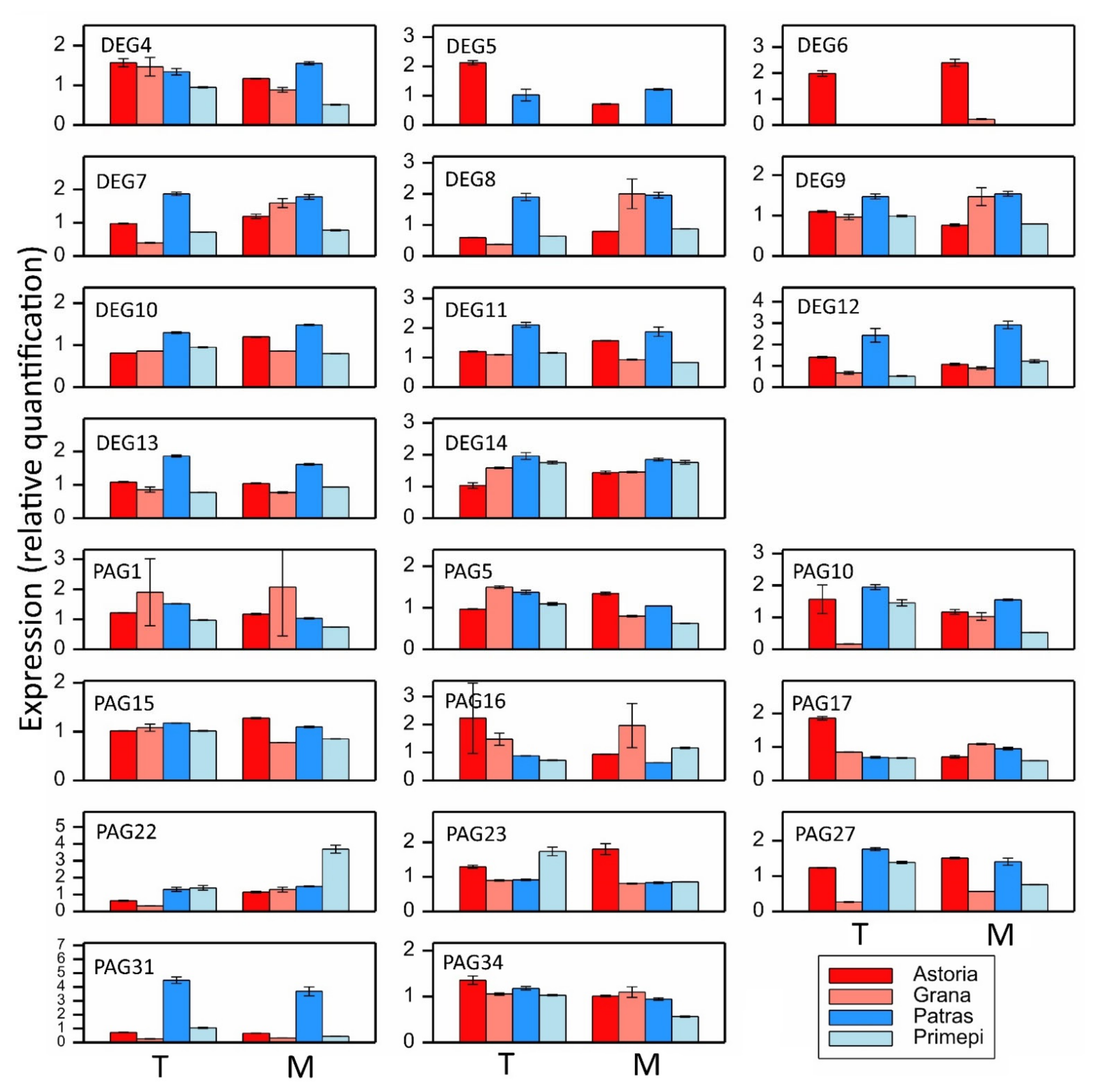
2.5. qRT-PCR Analysis of Selected DEGs and PAGs
2.6. Annotation of PPR Genes
3. Discussion
4. Materials and Methods
4.1. Plant Growth Conditions and Sampling
4.2. RNA Isolation
4.3. RNA-Seq Analysis
4.4. Paralog Analysis
4.5. Mapping DEGs and PAGs in Rf Linkage Groups
4.6. Validation of Transcripts by RT-qPCR Analysis
4.7. Annotation of PPR Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shewry, P.R.; Hey, S.J. The contribution of wheat to human diet and health. Food Energy Secur. 2015, 4, 178–202. [Google Scholar] [CrossRef]
- Giraldo, P.; Benavente, E.; Manzano-Agugliaro, F.; Gimenez, E. Worldwide Research Trends on Wheat and Barley: A Bibliometric Comparative Analysis. Agronomy 2019, 9, 352. [Google Scholar] [CrossRef] [Green Version]
- FAOSAT, F.A.O. Food and Agriculture Organization of the United Nations. 2020. Available online: http://www.fao.org/faostat/en/#home (accessed on 16 August 2021).
- Święcicki, W.K.; Surma, M.; Koziara, W.; Skrzypczak, G.; Szukała, J.; Bartkowiak-Broda, I.; Zimny, J.; Banaszak, Z.; Marciniak, K. Modern technologies in plant production—Friendly for human and environment. Pol. J. Agron. 2011, 7, 102–112. [Google Scholar]
- Longin, C.F.H.; Gowda, M.; Mühleisen, J.; Ebmeyer, E.; Kazman, E.; Schachschneider, R.; Schacht, J.; Kirchhoff, M.; Zhao, Y.; Reif, J.C. Hybrid wheat: Quantitative genetic parameters and consequences for the design of breeding programs. Theor. Appl. Genet. 2013, 126, 2791–2801. [Google Scholar] [CrossRef]
- Muhleisen, J.; Piepho, H.P.; Maurer, H.P.; Longin, C.F.; Reif, J.C. Yield stability of hybrids versus lines in wheat, barley, and triticale. Theor. Appl. Genet. 2014, 127, 309–316. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.P. Flowering biology of wheat, particularly in view of hybrid seed production—A review. Euphytica 1971, 20, 152–170. [Google Scholar] [CrossRef]
- Waddington, S.R.; Cartwright, P.M.; Wall, P.C. A quantitative scale of spike initial and pistil development in barley and wheat. Ann. Bot. 1983, 51, 119–130. [Google Scholar] [CrossRef]
- Langer, S.M.; Longin, C.F.H.; Würschum, T. Phenotypic evaluation of floral and flowering traits with relevance for hybrid breeding in wheat (Triticum aestivum L.). Plant Breed. 2014, 133, 433–441. [Google Scholar] [CrossRef]
- Whitford, R.; Fleury, D.; Reif, J.C.; Garcia, M.; Okada, T.; Korzun, V.; Langridge, P. Hybrid breeding in wheat: Technologies to improve hybrid wheat seed production. J. Experimantal Bot. 2013, 64, 5411–5428. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.J.; Baumann, U.; Kouidri, A.; Suchecki, R.; Baes, M.; Garcia, M.; Okada, T.; Dong, C.; Wu, Y.; Sandhu, A.; et al. Molecular identification of the wheat male fertility gene Ms1 and its prospects for hybrid breeding. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gowda, M.; Longin, C.F.H.; Lein, V.; Reif, J.C. Relevance of specific versus general combining ability in wheat (Triticum aestivum L.). Crop Sci. 2012, 52, 2494–2500. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wei, G.; Xia, Y.; Liu, X.; Tang, J.; Lu, Y.; Lan, H.; Zhang, S.; Li, C.; Cao, M. Comparative transcriptome analysis reveals that tricarboxylic acid cycle-related genes are associated with maize CMS-C fertility restoration. BMC Plant Biol. 2018, 18, 190. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Srivastava, R.; Kumar, J. Male sterility systems in wheat and opportunities for hybrid wheat development. Acta. Physiol. Plant. 2015, 37, 1713. [Google Scholar] [CrossRef]
- Kumar, S. Heterosis in wheat hybrids derived from Triticum timopheevi based cytoplasmic male sterility system. Indian J. Agric. Sci. 2013, 83, 1392–1395. [Google Scholar]
- Lukaszewski, A.J. Chromosomes 1BS and 1RS for control of male fertility in wheats and triticales with cytoplasms of Aegilops kotschyi, Ae. mutica and Ae. uniaristata. Theor. Appl. Genet. 2017, 130, 2521–2526. [Google Scholar] [CrossRef] [PubMed]
- Tomar, S.M.S.; Pallavi, S.; Asad, A.; Bhanwar, S.; Balyan, H.S. Assessment of agro-morphological and molecular diversity among fertility restorer lines in wheat (Triticum aestivum L.). Indian J. Genet. Plant Breed. 2009, 69, 183–190. [Google Scholar]
- Yang, T.Z. Investigation on the intraspecifically cytoplasmic variability in Triticum aestivum. Acta Agron. Sin. 1983, 9, 217–222. [Google Scholar]
- Murai, K.; Tsunewaki, K. Photoperiod-sensitive cytoplasmic male sterility in wheat with Aegilops crassa cytoplasm. Euphytica 1993, 67, 41–48. [Google Scholar] [CrossRef]
- Wang, L.Q. Study on the cytoplasmic male sterility (CMS) lines of 85EA and 89AR in common wheat. In Recent Advances in Crop Male Sterility and Heterosis; Li, J.X., Zhou, H.S., Eds.; China Agricultural Press: Beijing, China, 1996; pp. 136–144. [Google Scholar]
- Yang, C.Y.; He, P.R. Studies on photoperiod-thermo sensitive male sterility and status of heterosis in wheat. Tritical Crop. 1997, 17, 25–27. [Google Scholar]
- Luo, H.B.; He, J.M.; Dai, J.T.; Liu, X.L.; Yang, Y.C. Studies on the characteristics of seed production of two ecological male sterile lines in wheat. J. Hunan Agric. Univ. 1998, 24, 83–89. [Google Scholar]
- Itabashi, E.; Iwata, N.; Fujii, S.; Kazama, T.; Toriyama, K. The fertility restorer gene, Rf2, for Lead Rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J. 2011, 65, 359–367. [Google Scholar] [CrossRef]
- Fujii, S.; Toriyama, K. Suppressed expression of retrograde-regulated male sterility restores pollen fertility in cytoplasmic male sterile rice plants. Proc. Natl. Acad. Sci. USA 2009, 106, 9513–9518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Wise, R.P.; Schnable, P.S. The rf2 nuclear restorer gene of male-sterile t-cytoplasm maize. Science 1996, 272, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cui, X.; Horner, H.T.; Weiner, H.; Schnable, P.S. Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 2001, 13, 1063–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazama, T.; Nakamura, T.; Watanabe, M.; Sugita, M.; Toriyama, K. Suppression mechanism of mitochondrial ORF79 accumulation by Rf1 protein in BT-type cytoplasmic male sterile rice. Plant J. 2008, 55, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Livers, R.W. Fertility restoration and its inheritance in cytoplasmic male sterile wheat. Science 1964, 144, 420. [Google Scholar] [CrossRef]
- Robertson, L.D.; Curtis, B.C. Monosomic Analysis of Fertility-Restoration in Common Wheat (Triticum aestivum L.) 1. Crop Sci. 1967, 7, 493–495. [Google Scholar] [CrossRef]
- Tahir, C.M.; Tsunewaki, K. Monosomic analysis of Triticum spelta var. duhamelianum, a fertility restorer for T. timopheevi cytoplasm. Jpn. J. Genet. 1969, 44, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Talaat, E.H. North Dakota State University, Fargo, ND, USA, 1969.
- Yen, F.S.; Evans, L.E.; Larter, E.N. Monosomic analysis of fertility restoration in three restorer lines of wheat. Can. J. Genet. Cytol. 1969, 11, 531–546. [Google Scholar] [CrossRef]
- Snape, J.W.; Flavell, R.B.; O’dell, M.; Hughes, W.G.; Payne, P.I. Intrachromosomal mapping of the nucleolar organiser region relative to three marker loci on chromosome 1B of wheat (Triticum aestivum). Appl. Genet. 1985, 69, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Q.; Sorrells, M.E.; Zhao, Y.H. Inheritance and chromosomal locations of male fertility restoring gene transferred from Aegilops umbellulata Zhuk. to Triticum aestivum L. Mol. Gen. Genet. 1995, 247, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Tomar, S.M.S.; Singh, V.K.; Balyan, H.S. Genetic analysis and molecular mapping of a new fertility restorer gene Rf8 for Triticum timopheevi cytoplasm in wheat (Triticum aestivum L.) using SSR markers. Genetica 2013, 141, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosch, R.A.; Yamazaki, Y.; Devos, K.M.; Dubcovsky, J.; Rogers, W.J.; Appels, R. Catalogue of gene symbols for wheat. In Proceedings of the 10th International Wheat Genetics Symposium, Paestrum, Italy, 1–6 September 2003; Pogna, N.E., Romano, N., Pogna, E.A., Galterio, G., Eds.; Instituto Sperimentale per la Cerealicoltura: Rome, Italy, 2003; pp. 1–34. [Google Scholar]
- Castillo, A.; Atienza, S.G.; Martín, A.C. Fertility of CMS wheat is restored by two Rf loci located on a recombined acrocentric chromosome. J. Exp. Bot. 2014, 65, 6667–6677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahinnia, F.; Geyer, M.; Block, A.; Mohler, V.; Hartl, L. Identification of Rf9, a gene contributing to the genetic complexity of fertility restoration in hybrid wheat. Front. Plant Sci. 2020, 11, 577475. [Google Scholar] [CrossRef]
- Geyer, M.; Bund, A.; Albrecht, T.; Hartl, L.; Mohler, V. Distribution of the fertility-restoring gene Rf3 in common and spelt wheat determined by an informative SNP marker. Mol. Breed. 2016, 36, 167. [Google Scholar] [CrossRef]
- Ma, Z.Q.; Sorrells, M.E. Genetic analysis of fertility restoration in wheat using restriction fragment length ploymorphisms. Crop Sci. 1995, 35, 1137–1143. [Google Scholar] [CrossRef]
- Kojima, T.; Tsujimoto, H.; Ogihara, Y. High resolution RFLP mapping of the fertility restoration (Rf3) gene against Triticum timopheevii cytoplasm located on chromosome 1BS of common wheat. Genes Genet. Syst. 1997, 72, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Bahl, P.N.; Maan, S.S. Chromosomal location of fertility restoring genes in six lines of common wheat. Crop Sci. 1973, 13, 317–320. [Google Scholar] [CrossRef]
- Maan, S.S.; Lucken, K.A.; Bravo, J.M. Genetic analysis of male fertility restoration in wheat I. Chromosome location of Rf genes. Crop Sci. 1984, 24, 17–20. [Google Scholar] [CrossRef]
- Maan, S.S. A gene for embryo–endosperm compatibility and seed viability in alloplasmic Triticum turgidum. Genome 1992, 35, 772–779. [Google Scholar] [CrossRef]
- Geyer, M.; Albrecht, T.; Hartl, L.; Mohler, V. Exploring the genetics of fertility restoration controlled by Rf1 in common wheat (Triticum aestivum L.) using high-density linkage maps. Mol. Genet. Genom. 2018, 293, 451–462. [Google Scholar] [CrossRef]
- Melonek, J.; Duarte, J.; Martin, J.; Beuf, L.; Murigneux, A.; Varenne, P.; Comadran, J.; Specel, S.; Levadoux, S.; Bernath-Levin, K.; et al. The genetic basis of cytoplasmic male sterility and fertility restoration in wheat. Nat. Commun. 2021, 12, 1036. [Google Scholar] [CrossRef]
- Curtis, C.A.; Lukaszewski, A.J. Localization of genes in rye that restore male fertility to hexaploid wheat with timopheevii cytoplasm. Plant Breed. 1993, 111, 106–112. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Tsujimoto, H.; Sasakuma, T. QTL analysis of fertility-restoration against cytoplasmic male sterility in wheat. Genes Genet. Syst. 2001, 76, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Frederic, L.K.; Leslie, L.D.; Wang, S. SSR markers associated with fertility restoration genes against Triticum timopheevii cytoplasm in Triticum aestivum. Euphytica 2005, 141, 33–40. [Google Scholar] [CrossRef]
- Dou, B.; Hou, B.; Xu, H.; Lou, X.; Chi, X.; Yang, F.W.; Ni, Z.; Sun, Q. Efficient mapping of a female sterile gene in wheat (Triticum aestivum L.). Genet. Res. 2009, 91, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.H.; Bai, J.F.; Guo, H.Y.; Duan, W.J.; Liu, Z.H.; Zhang, F.T.; Ma, J.X.; Zhao, C.P.; Zhang, L.P. QTL mapping of male sterility-related traits in a photoperiod and temperature-sensitive genic male sterile wheat line BS366. Plant Breed. 2020, 139, 498–507. [Google Scholar] [CrossRef]
- Mukai, Y.; Tsunewaki, K. Basic studies on hybrid wheat breeding VIII. A new male sterility-fertility restoration system in common wheat utilizing the cytoplasms of Aegilops kotschyi and Ae. Var. Theor. Appl. Genet. 1979, 54, 153–160. [Google Scholar] [CrossRef]
- Mukai, Y.; Endo, T.R. Physical mapping of a fertility restoring gene against Aegilops kotschyi cytoplasm in wheat. Jpn. J. Genet. 1992, 67, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Hamawaki, H.; Mukai, Y. Telocentric mapping of the fertility-restoring gene Rfv1 against Aegilops variabilis cytoplasm in wheat. Jpn. J. Genet. 1980, 55, 453. [Google Scholar]
- Mukai, Y. Determination of the chromosome arm carrying a male fertility-restoring gene against the cytoplasm of Aegilops uniaristata in wheat. Mem. Osaka Kyoiku Univ. Ser. III Nat. Sci. Appl. Sci. 1983, 32, 43–53. [Google Scholar]
- Tsujimoto, H.; Tsunewaki, K. Chromosome location of a fertility-restoring gene of a common wheat Chinese Spring for the Ae. mutica cytoplasm. Wheat Inf. Serv. 1984, 58, 4–8. [Google Scholar]
- Chen, Y.; Jia, Y.; Niu, F.; Wu, Y.; Ye, J.; Yang, X.; Zhang, L.; Song, X. Identification and validation of genetic locus Rfk1 for wheat fertility restoration in the presence of Aegilops kotschyi cytoplasm. Theor. Appl. Genet. 2021, 134, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Tsunewaki, K. Monosomic analysis of two restorers to Ae. caudate and Ae. umbellulata cytoplasms. Jpn. J. Genet. 1974, 49, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Tsunewaki, K. Fine mapping of the first multi-fertility-restoring gene, Rf multi, of wheat for three Aegilops plasmons, using 1BS-1RS recombinant lines. Theor. Appl. Genet. 2015, 128, 723–732. [Google Scholar] [CrossRef]
- Murai, K.; Tsunewaki, K. Genetic analysis on the fertility restoration by Triticum aestivum cv. Chinese Spring against photoperiod-sensitive cytoplasmic male sterility. Jpn. J. Genet. 1994, 69, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Xia, C.; Zhao, G.; Jia, J.; Kong, X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genom. 2012, 13, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Pellny, T.; Lovegrove, A.; Freeman, J.; Tosi, P.; Love, C.; Knox, P.; Shewry, P.; Mitchell, R. Cell Walls of Developing Wheat Starchy Endosperm: Comparison of Composition and RNA-Seq Transcriptome. Plant Physiol. 2011, 158, 612–627. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Liu, Z.; Rocheleau, H.; Fauteux, F.; Wang, Y.; McCartney, C.; Ouellet, T. Transcriptome dynamics associated with resistance and susceptibility against fusarium head blight in four wheat genotypes. BMC Genom. 2018, 19, 642. [Google Scholar] [CrossRef]
- Iquebal, M.A.; Sharma, P.; Jasrotia, R.S.; Jaiswal, S.; Kaur, A.; Saroha, M.; Angadi, U.B.; Sheoran, S.; Singh, R.; Singh, G.P.; et al. RNAseq analysis reveals drought-responsive molecular pathways with candidate genes and putative molecular markers in root tissue of wheat. Sci. Rep. 2019, 9, 13917. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, S.; Li, W.; Liu, Q.; Zhang, L.; Song, X. Comparative transcriptome analysis indicates that a core transcriptional network mediates isonuclear alloplasmic male sterility in wheat (Triticum aestivum L.). BMC Plant Biol. 2020, 20, 10. [Google Scholar] [CrossRef]
- Zhu, T.; Wu, L.; He, H.; Song, J.; Jia, M.; Liu, L.; Wang, X.; Han, R.; Niu, L.; Du, W.; et al. Bulked Segregant RNA-Seq Reveals Distinct Expression Profiling in Chinese Wheat Cultivar Jimai 23 Responding to Powdery Mildew. Front. Genet. 2020, 11, 474. [Google Scholar] [CrossRef]
- Bellin, D.; Ferrarini, A.; Chimento, A.; Kaiser, O.; Levenkova, N.; Bouffard, P.; Delledonne, M. Combining next-generation pyrosequencing with microarray for large scale expression analysis in non-model species. BMC Genom. 2009, 10, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Duan, Y.; Hu, G.; Geng, X.; Zhan, G.; Yan, P.; Liu, Z.; Zhang, L.; Song, X. Identification of candidate genes and biosynthesis pathways related to fertility conversion by wheat KTM3315A transcriptome profiling. Front. Plant Sci. 2017, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Kazama, T.; Toriyama, K. A fertility restorer gene, Rf4, widely used for hybrid rice breeding encodes a pentatricopeptide repeat protein. Rice 2014, 7, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Du, Z.; Qi, X.; Wu, H.; Guo, W.; Zhao, Z. RNA-sequencing analysis reveals transcriptional changes in the roots of low-cadmium-accumulating winter wheat under cadmium stress. Acta. Physiol. Plantaru 2019, 41, 13. [Google Scholar] [CrossRef]
- Xiong, H.; Guo, H.; Xie, Y.; Zhao, L.; Gu, J.; Zhao, S.; Li, J.; Liu, L. RNAseq analysis reveals pathways and candidate genes associated with salinity tolerance in a spaceflight-induced wheat mutant. Sci. Rep. 2017, 7, 2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz-Linneweber, C.; Small, I. Pentatricopeptide repeat proteins: A socket set for organelle gene expression. Trends Plant Sci. 2008, 13, 663–670. [Google Scholar] [CrossRef]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef]
- Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 2015, 113, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Arakawa, T.; Honma, Y.; Kitazaki, K. What Does the Molecular Genetics of Different Types of Restorer-of-Fertility Genes Imply? Plants 2020, 9, 361. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Zou, Y.; Hu, J.; Ding, Y. Genome-wide analysis of the rice PPR gene family and their expression profiles under different stress treatments. BMC Genom. 2018, 19, 720. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [Green Version]
- International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [Green Version]
- Prakash, V.; Tiwari, S.; Kumar, S.; Shukla, R.S.; Tripathi, N. Validation of male sterile, fertility restorer and hybrid lines in wheat (Triticum aestivum L.) with linked SSR markers. J. Mol. Biol. Biotechnol. 2012, 20, 65–71. [Google Scholar]
- Würschum, T.; Leiser, W.L.; Weissmann, S.; Maurer, H.P. Genetic architecture of male fertility restoration of Triticum timopheevii cytoplasm and fine-mapping of the major restorer locus Rf3 on chromosome 1B. Theor. Appl. Genet. 2017, 130, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yang, Y. Wheat Fertile Activity Recovery Gene RF6 Molecular Mark and Its Obtaining Method. CN1312282C, 25 April 2007. [Google Scholar]
- Yim, A.K.Y.; Wong, J.W.H.; Ku, Y.S.; Qin, H.; Chan, T.F.; Lam, H.M. Using RNA-Seq Data to Evaluate Reference Genes Suitable for Gene Expression Studies in Soybean. PLoS ONE 2015, 10, e0136343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everaert, C.; Luypaert, M.; Maag, J.L.V.; Cheng, Q.X.; Dinger, M.E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nong, Q.; Yang, Y.; Zhang, M.; Zhang, M.; Chen, J.; Jian, S.; Lu, H.; Xia, K. RNA-seq-based selection of reference genes for RT-qPCR analysis of pitaya. FEBS Open Bio 2019, 9, 1403–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pombo, M.; Zheng, Y.; Fei, Z.; Martin, G.B.; Rosli, H.G. Use of RNA-seq data to identify and validate RT-qPCR reference genes for studying the tomato-Pseudomonas pathosystem. Sci. Rep. 2017, 7, 44905. [Google Scholar] [CrossRef] [PubMed]
- Pombo, M.A.; Ramos, R.N.; Zheng, Y.; Fei, Z.; Martin, G.B.; Rosli, H.G. Transcriptome-based identification and validation of reference genes for plant-bacteria interaction studies using Nicotiana benthamiana. Sci. Rep. 2019, 9, 1632. [Google Scholar] [CrossRef] [Green Version]
- Garrido, J.; Aguilar, M.; Prieto, P. Identification and validation of reference genes for RT-qPCR normalization in wheat meiosis. Sci. Rep. 2020, 10, 2726. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, B.; Royan, S.; Schallenberg-Rüdinger, M.; Lenz, H.; Castleden, I.R.; McDowell, R.; Vacher, M.A.; Tonti-Filippini, J.; Bond, C.S.; Knoop, V.; et al. The Expansion and Diversification of Pentatricopeptide Repeat RNA-Editing Factors in Plants. Mol. Plant 2020, 13, 215–230. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Gogg, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with highthroughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Bond, C.S.; Small, I.D. Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc. Natl. Acad. Sci. USA 2011, 108, 1723–1728. [Google Scholar] [CrossRef] [Green Version]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van der Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison No. | Stage and Compared Cultivars | Number of DEGs | |
|---|---|---|---|
| Upregulated | Downregulated | ||
| C1 | T: Astoria/Primépi | 2372 | 2317 |
| C2 | T: Grana/Primépi | 43 | 590 |
| C3 | T: Astoria/Patras | 1729 | 1574 |
| C4 | T: Grana/ Patras | 2188 | 2330 |
| C5 | M: Astoria/Primépi | 1555 | 1592 |
| C6 | M: Grana/Primépi | 21 | 66 |
| C7 | M: Astoria/Patras | 951 | 1131 |
| C8 | M: Grana/Patras | 3014 | 3874 |
| C9 | T: Patras/Primépi | 1912 | 2129 |
| C10 | M: Patras/Primépi | 3520 | 3235 |
| C11 | T: non-restoring/restoring | 287 | 295 |
| C12 | M: non-restoring/restoring | 87 | 100 |
| DEG | Gene Symbol | No. of Exons | Variant Alleles | Comparison | Domain * | Reference Sequence with the Highest Similarity ** | Score (%) | E-Value |
|---|---|---|---|---|---|---|---|---|
| DEG1 | TraesCS1A02G031600 | 1 | 1 | C7 C8 | PPR | BrachypodiumRf1 | 59.19 | 0.0 |
| DEG2 | TraesCS1A02G057400 | 3 | 138 | C1 C3 | PPR | Ae. taushii Rf1 | 62.61 | 0.0 |
| DEG3 | TraesCS1B02G038300 | 2 | 266 | C1 C4 C8 | PPR | BrachypodiumRf1 | 57.24 | 2e-159 |
| DEG4 | TraesCS1B02G038400 | 2 | 85 | C1 C4 C9 C10 | PPR | BrachypodiumRf1 | 61.07 | 0.0 |
| DEG5 | TraesCS1B02G038500 | 2 | 160 | C1 C4 C8 | PPR | BrachypodiumRf1 | 59.95 | 0.0 |
| DEG6 | TraesCS1B02G039200 | 1 | 238 | C1 C3 C5 C7 | PPR | BrachypodiumRf1 | 59.69 | 0.0 |
| DEG7 | TraesCS1B02G075000 | 1 | 20 | C3 C9 | PPR | Ae. taushii Rf1 | 76.38 | 0.0 |
| DEG8 | TraesCS2A02G530300 | 2 | 60 | C3 | PPR | PPR-814a | 54.18 | 0.0 |
| DEG9 | TraesCS2A02G530600 | 1 | 52 | C4 C7 C8 | PPR | PPR-814a | 57.66 | 0.0 |
| DEG10 | TraesCS2B02G560000 | 1 | 132 | C3 C4 C8 | PPR | Ae. taushii Rf1 | 55.64 | 3e-174 |
| DEG11 | TraesCS2B02G560700 | 1 | 239 | C3 C4 C8 | PPR | BrachypodiumRf1 | 56.76 | 0.0 |
| DEG12 | TraesCS6A02G099318 | 1 | 7 | C3 C4 | PPR | BrachypodiumRf1 | 57.75 | 7e-106 |
| DEG13 | TraesCS6A02G099384 | 1 | 9 | C7 C8 | PPR | PPR-814a | 49.18 | 1e-38 |
| DEG14 | TraesCS6B02G129200 | 2 | 208 | C1 | PPR | BrachypodiumRf1 | 58.26 | 0.0 |
| DEG15 | TraesCS6B02G321400 | 7 | 38 | C1 C4 C5 C8 C9 C10 | ADD | ZmRf2 | 76.22 | 0.0 |
| DEG16 | TraesCS7A02G369300 | 10 | 285 | C5 | ADD | ZmRf2 | 55.67 | 0.0 |
| DEG17 | TraesCS7A02G369400 | 9 | 250 | C8 | ADD | ZmRf2 | 55.05 | 0.0 |
| DEG18 | TraesCS7B02G116800 | 11 | 290 | C1 C3 C5 C7 | ADD | ZmRf2 | 87.39 | 0.0 |
| DEG19 | TraesCS7B02G259100 | 8 | 222 | C8 | ADD | ZmRf2 | 56.01 | 0.0 |
| DEG20 | TraesCS7D02G353900 | 8 | 196 | C7 C8 | ADD | ZmRf2 | 55.26 | 0.0 |
| DEG21 | TraesCS7D02G354000 | 9 | 201 | C12 | ADD | ZmRf2 | 55.26 | 0.0 |
| DEG/ PAG | Chromo-Some | Position in CS Genome (bp) | Class/ Domain | Missing cre Indicating Rf Function | Candidate for the Gene: | Overexpression Pattern * |
|---|---|---|---|---|---|---|
| DEG4 | 1BS | 18093637–18090927 | PPR | none | Rf3 ** | M: Pa > A,G |
| DEG7 | 1B | 58116320–58114326 | PPR | ARE, MYB | Rf_a *** | T,M: Pa > A,G; T: Pr > G |
| DEG11 | 2B | 753931307–753928992 | PPR | ARE, CGTCA-motif, TGACG | Rf_b (RFL2018) **** | T,M: Pa > A,G |
| PAG1 | 1B | 56620540–56618322 | PPR | ARE | Rf_c *** | T:Pa > A,G; M: Pa > G; T: Pr > G |
| PAG10 | 5B | 36720490–36722627 | PPR | none | Rf_d *** | T,M: Pa > A,G; T: Pr > G |
| PAG25 ***** | 5A | 546692034–546697035 | PPR | ARE | Rf_e*** | T,M: Pa > A,G; T,M: Pr > A,G |
| PAG52 ***** | 7B | 3846939–3849534 | PPR | none | Rf_f*** | M: Pa > A,T; T:Pr > A; M: Pr > A,G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyrka, M.; Bakera, B.; Szeliga, M.; Święcicka, M.; Krajewski, P.; Mokrzycka, M.; Rakoczy-Trojanowska, M. Identification of Rf Genes in Hexaploid Wheat (Triticumaestivum L.) by RNA-Seq and Paralog Analyses. Int. J. Mol. Sci. 2021, 22, 9146. https://doi.org/10.3390/ijms22179146
Tyrka M, Bakera B, Szeliga M, Święcicka M, Krajewski P, Mokrzycka M, Rakoczy-Trojanowska M. Identification of Rf Genes in Hexaploid Wheat (Triticumaestivum L.) by RNA-Seq and Paralog Analyses. International Journal of Molecular Sciences. 2021; 22(17):9146. https://doi.org/10.3390/ijms22179146
Chicago/Turabian StyleTyrka, Mirosław, Beata Bakera, Magdalena Szeliga, Magdalena Święcicka, Paweł Krajewski, Monika Mokrzycka, and Monika Rakoczy-Trojanowska. 2021. "Identification of Rf Genes in Hexaploid Wheat (Triticumaestivum L.) by RNA-Seq and Paralog Analyses" International Journal of Molecular Sciences 22, no. 17: 9146. https://doi.org/10.3390/ijms22179146