
Characterization of the Intramolecular Interactions and Regulatory Mechanisms of the Scaffold Protein Tks4

 , ,
, ,  , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
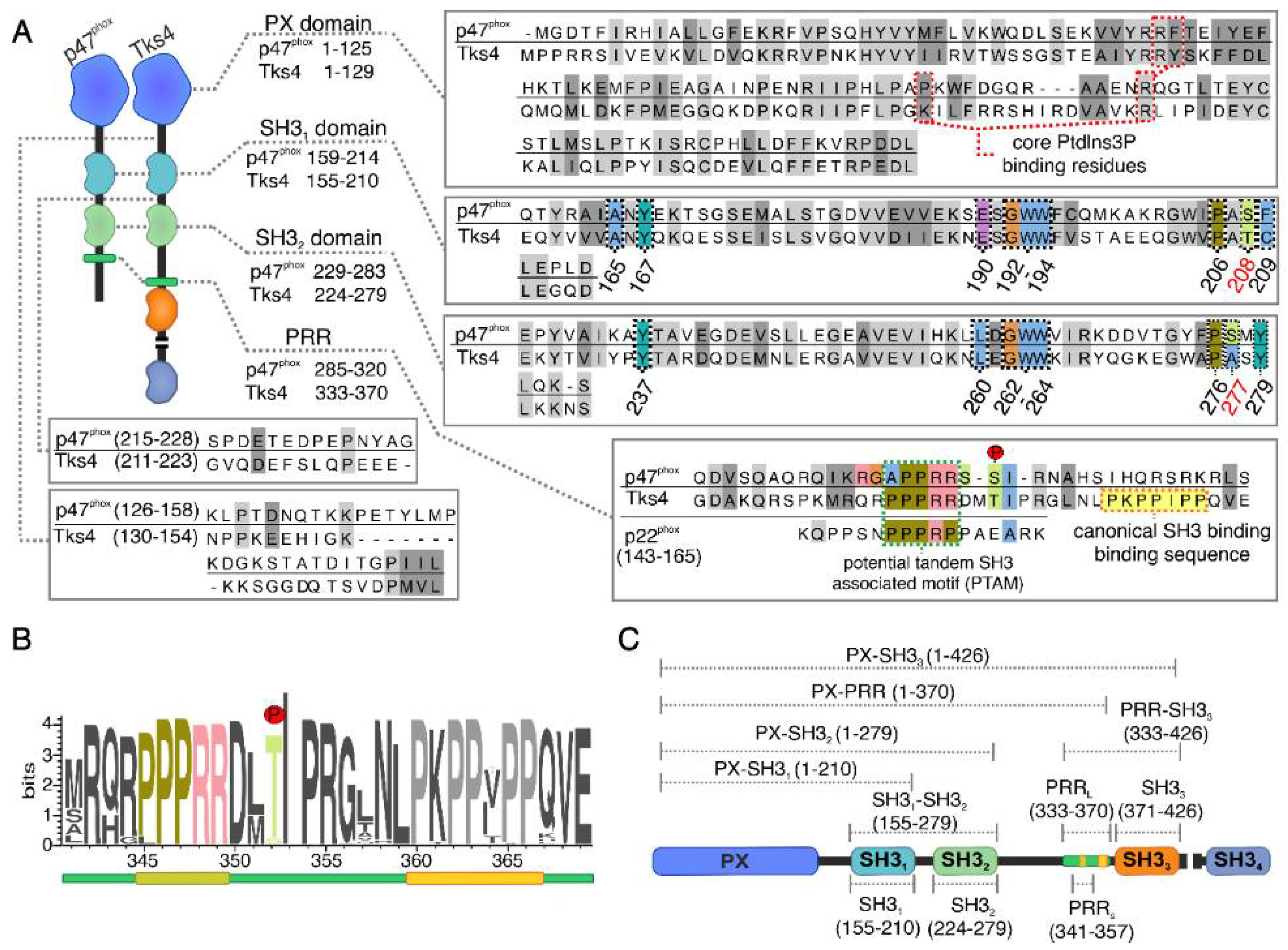
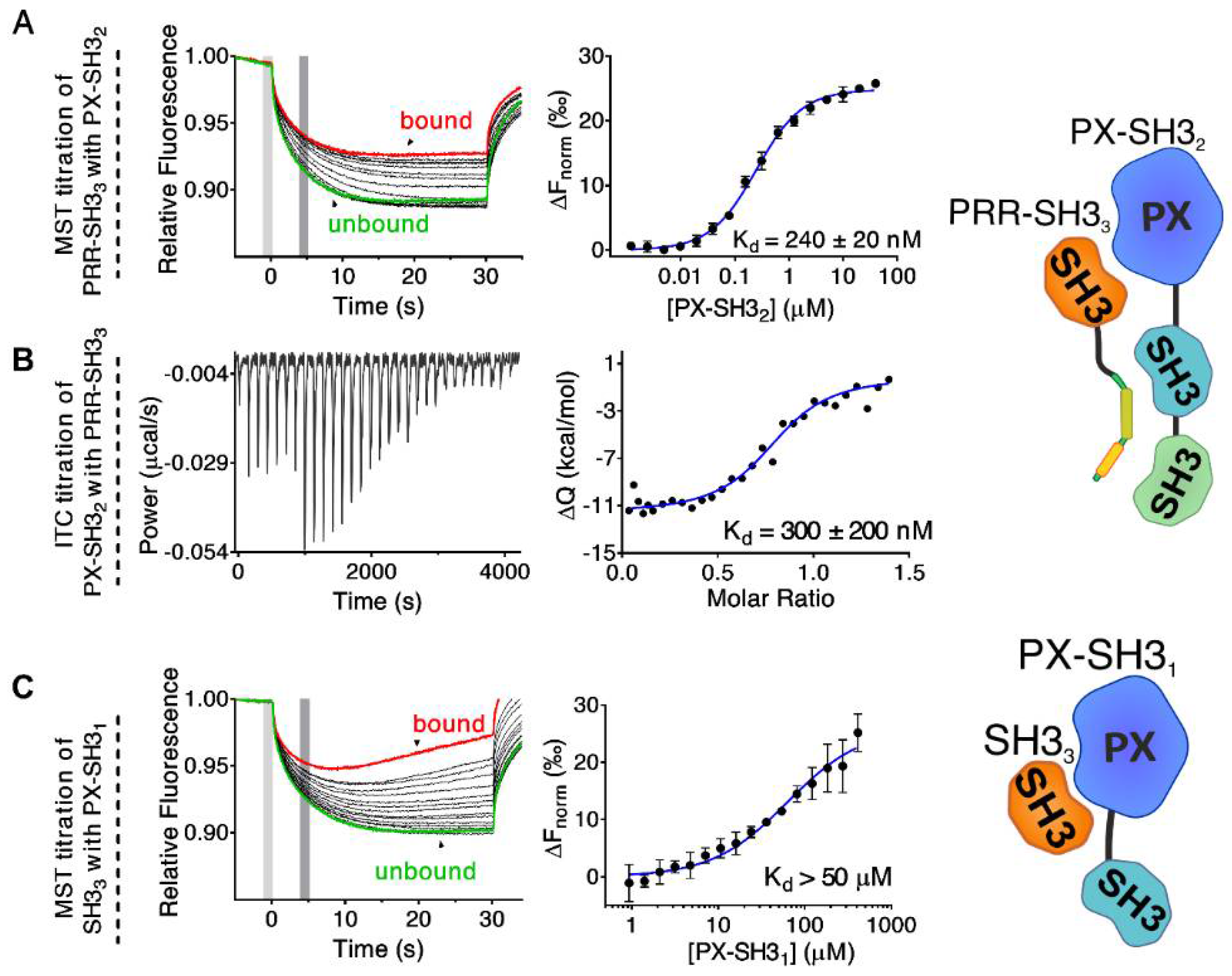
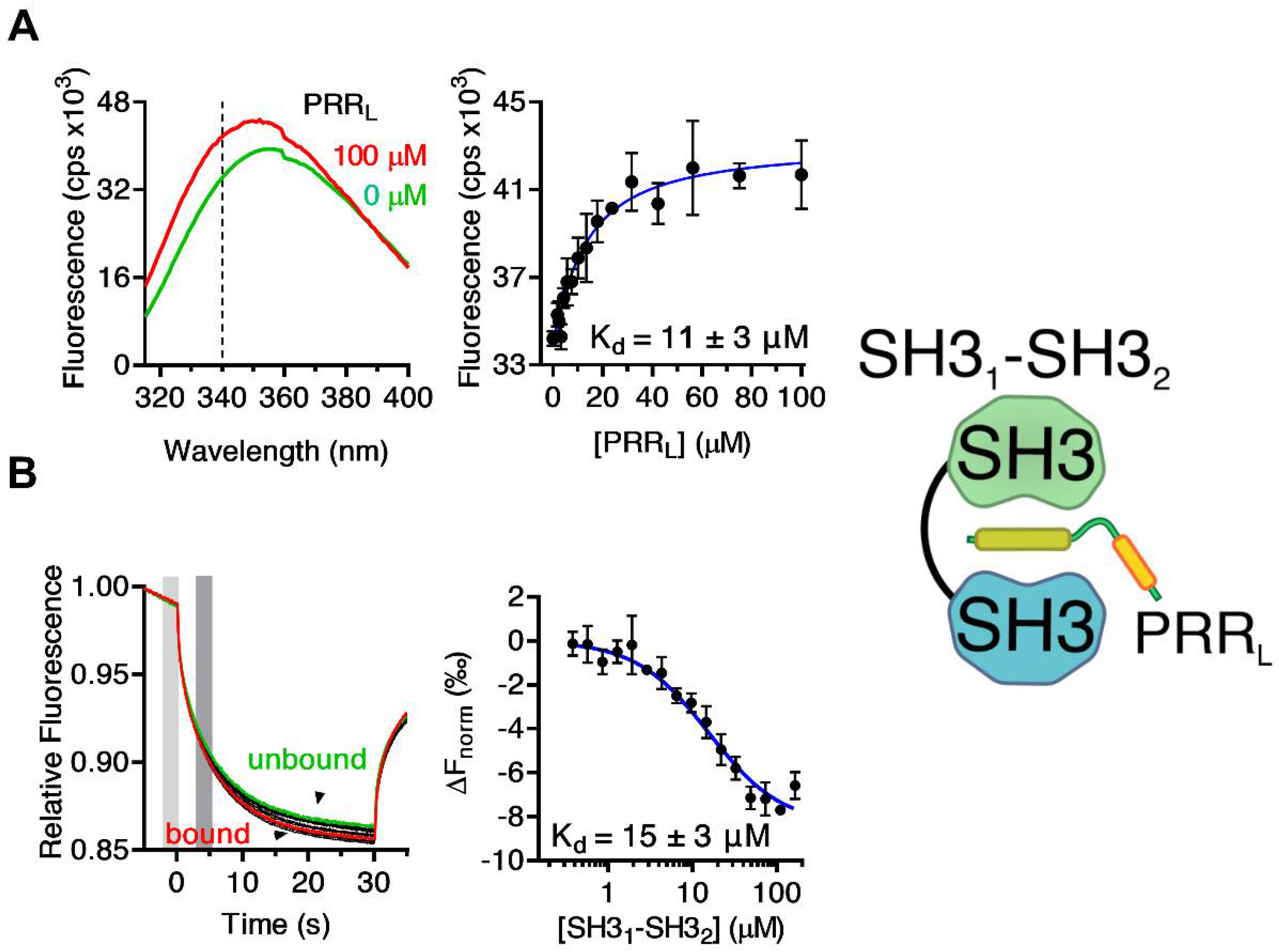
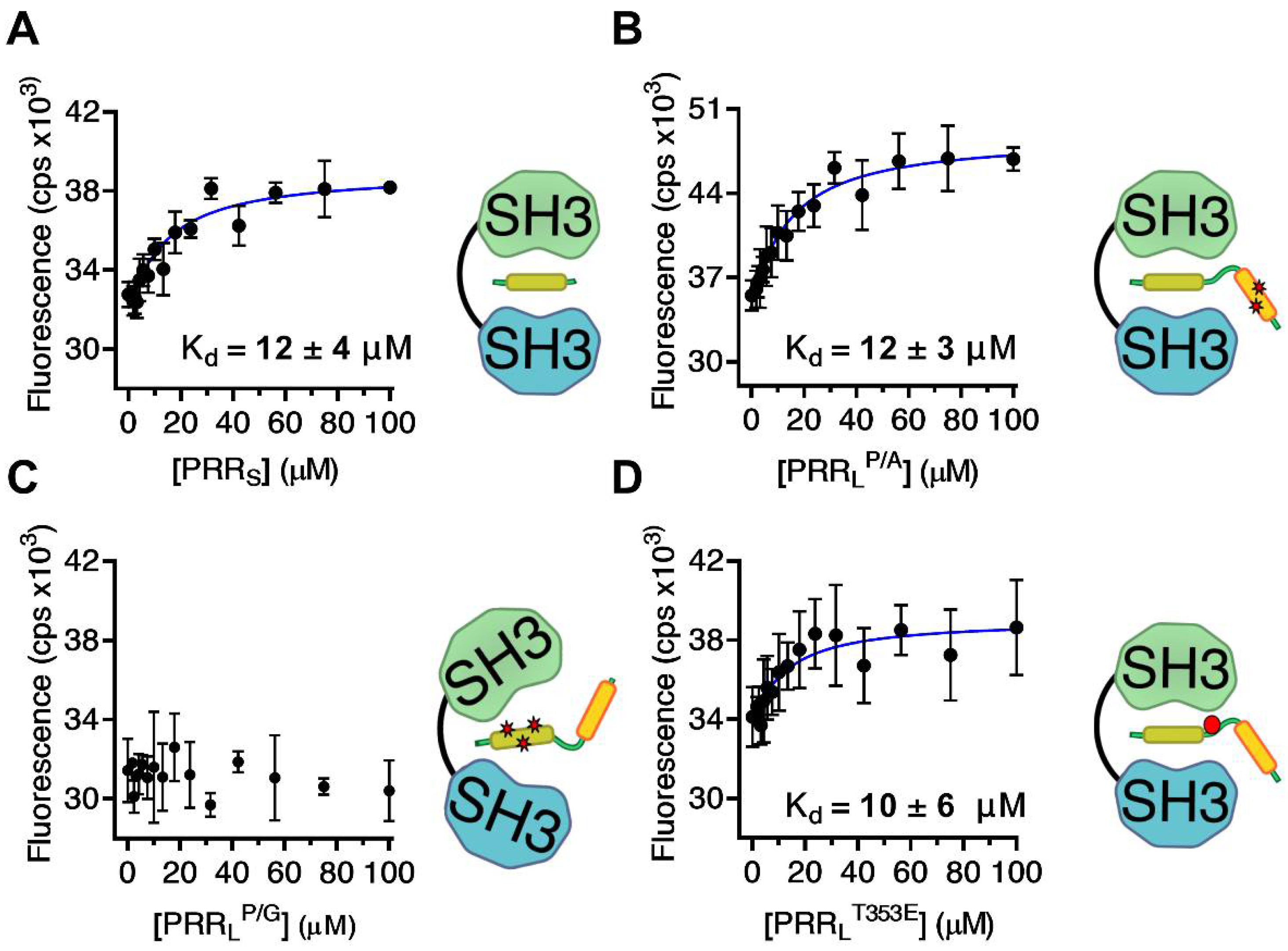
2.1. Mapping the Intermolecular Interactions in the Tks4 N Terminus
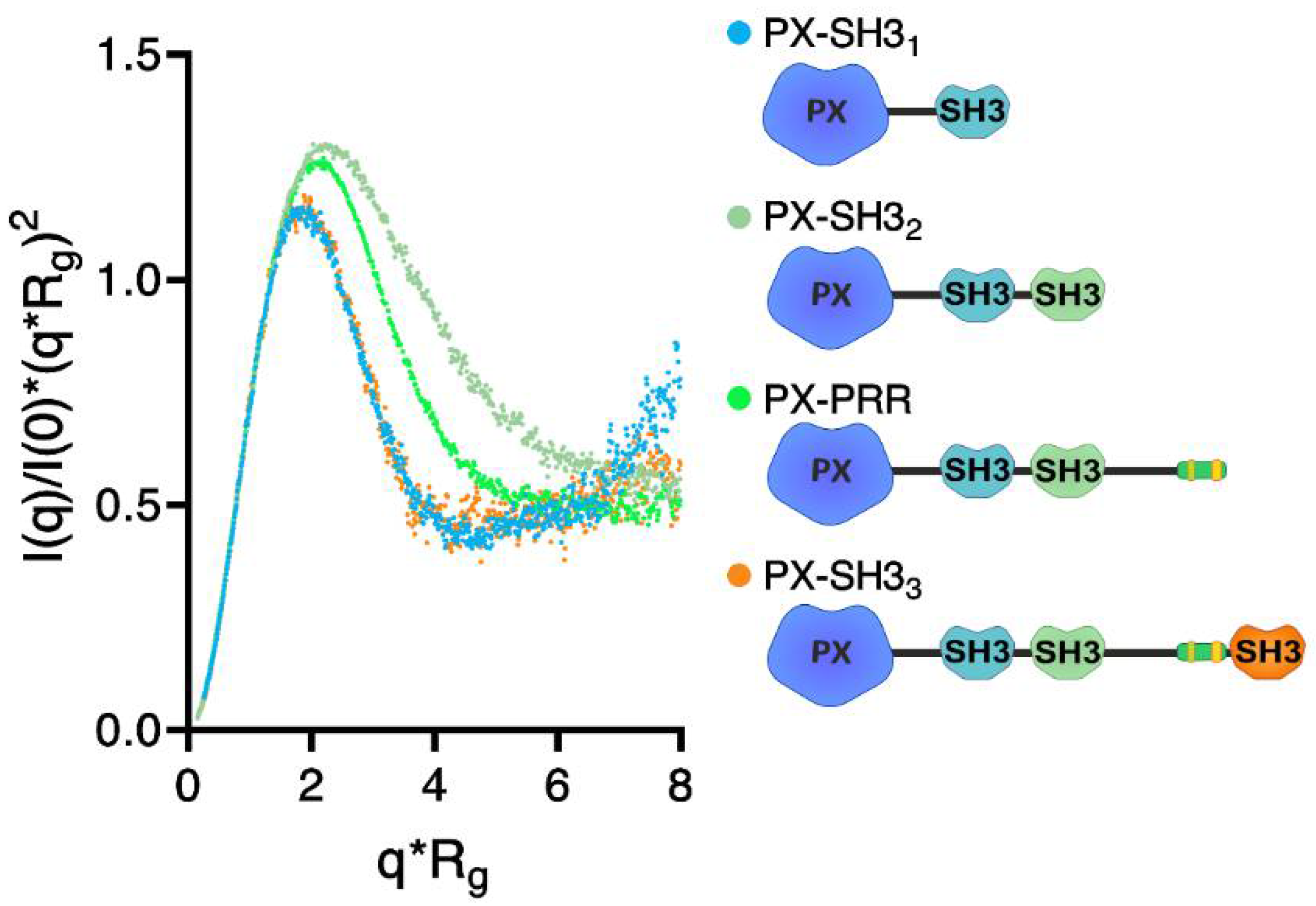
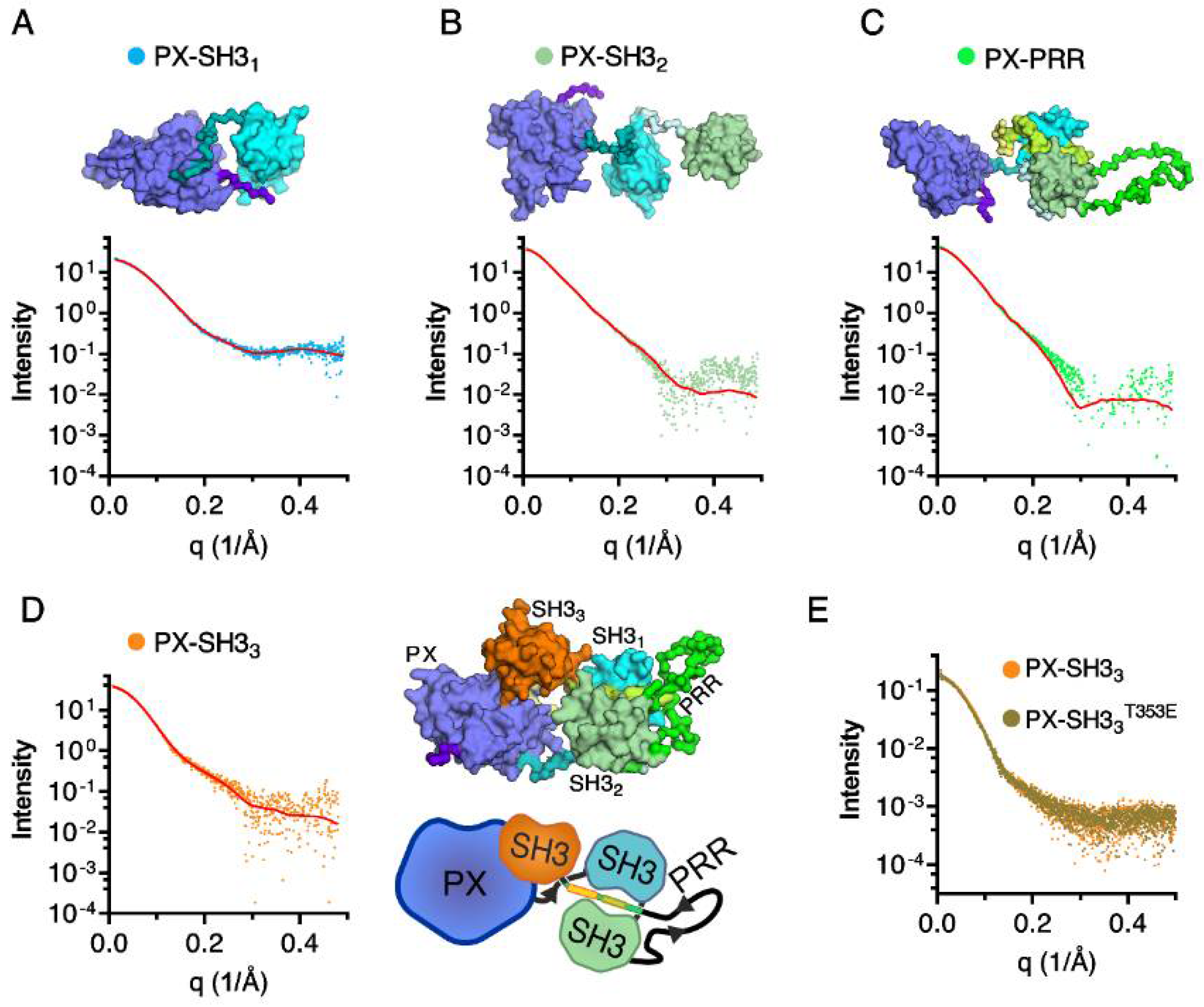
2.2. Confirmation of the Closed Conformation of the Tks4 N-Terminal Region via SAXS Analysis
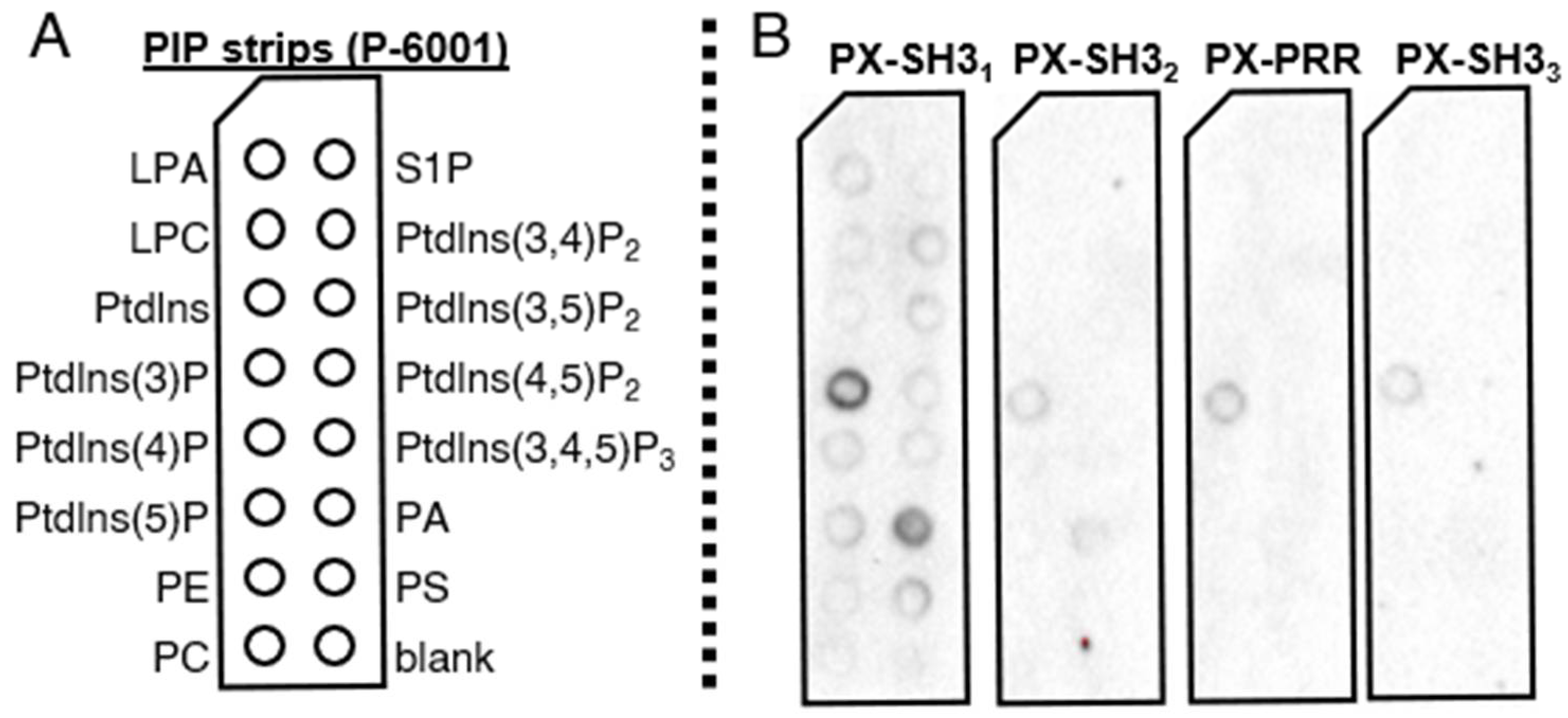
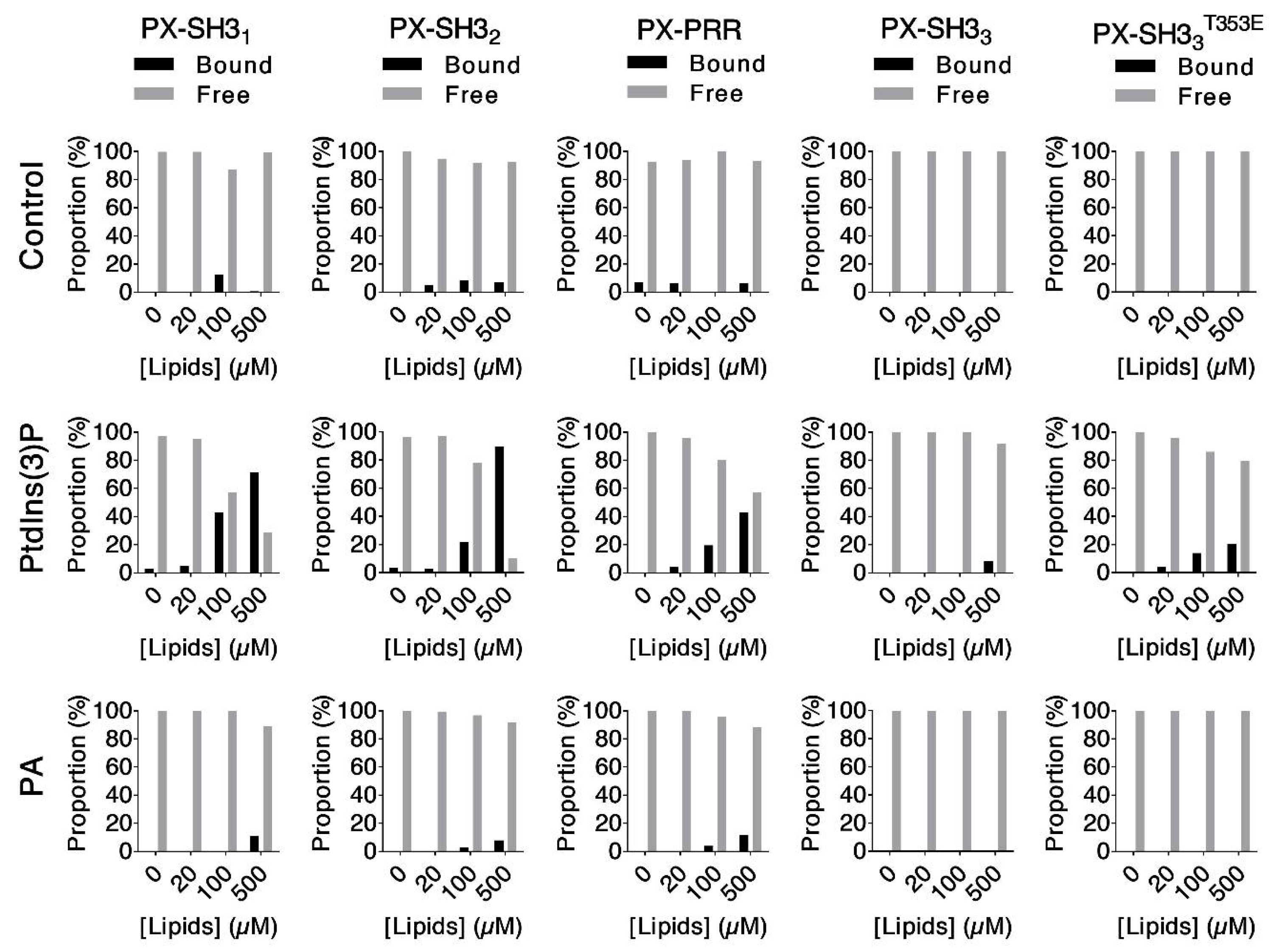
2.3. Lipid Binding Assays
3. Discussion
4. Materials and Methods
4.1. DNA Constructs and Protein Expression
4.2. Synthetic Peptides
4.3. Fluorescence Spectroscopy-Based Titrations
4.4. Isothermal Titration Calorimetry (ITC)
4.5. Microscale Thermophoresis (MST)
4.6. Small Angle X-ray Scattering (SAXS) Experiments
4.7. Protein–Lipid Overlay Assays (PIP Strip)
4.8. Preparation of PM-Mimetic Vesicles
4.9. Liposome Co-Sedimentation Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TKS4 | Tyrosine kinase substrate with 4 SH3 |
| EGF | epidermal growth factor |
| PX | Phox homolog |
| SH3 | Src homolog 3 |
| PRR | proline-rich region |
| MST | Microscale thermophoresis |
| ITC | Isothermal titration calorimetry |
| SAXS | Small Angle X-ray Scattering |
| PtdIns(3)P | Phosphatidylinositol-3-phosphate |
| TKS5 | Tyrosine kinase substrate with 5 SH3 |
| FTHS | Frank-ter Haar Syndrome |
| EMT | epithelial mesenchymal transition |
| AIR | autoinhibitory region |
| PTAM | potential tandem SH3-associated motif |
| PA | Phosphatidic acid |
| TCEP | Tris(2-carboxyethyl)phosphine |
References
- Buday, L.; Tompa, P. Functional classification of scaffold proteins and related molecules. FEBS J. 2010, 277, 4348–4355. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Yaffe, M.B. Protein regulation in signal transduction. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold proteins: Hubs for controlling the flow of cellular information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexa, A.; Varga, J.; Reményi, A. Scaffolds are ‘active’ regulators of signaling modules. FEBS J. 2010, 277, 4376–4382. [Google Scholar] [CrossRef] [Green Version]
- Saksela, K.; Permi, P. SH3 domain ligand binding: What’s the consensus and where’s the specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein-protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Amatya, N.; Lin, D.Y.-W.; Andreotti, A.H. Dynamic regulatory features of the protein tyrosine kinases. Biochem. Soc. Trans. 2019, 47, 1101–1116. [Google Scholar] [CrossRef]
- Chandra, M.; Collins, B.M. The Phox Homology (PX) Domain. Adv. Exp. Med. Biol. 2019, 1111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, R.D.; Collins, B.M. Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: Structures, functions and roles in disease. Biochem. J. 2011, 441, 39–59. [Google Scholar] [CrossRef]
- Gianni, D.; Diaz, B.; Taulet, N.; Fowler, B.; Courtneidge, S.A.; Bokoch, G.M. Novel p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity. Sci. Signal. 2009, 2, ra54. [Google Scholar] [CrossRef] [Green Version]
- Saini, P.; Courtneidge, S.A. Tks adaptor proteins at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Buschman, M.D.; Bromann, P.A.; Cejudo-Martin, P.; Wen, F.; Pass, I.; Courtneidge, S.A. The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol. Biol. Cell 2009, 20, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, P.; Abram, C.L.; Gibson, T.; Courtneidge, S.A. A new method for isolating tyrosine kinase substrates used to identify fish, an SH3 and PX domain-containing protein, and Src substrate. EMBO J. 1998, 17, 4346–4357. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, S.; Abdullah, C.; Buschman, M.D.; Diaz, B.; Courtneidge, S.A. The role of Tks adaptor proteins in invadopodia formation, growth and metastasis of melanoma. Oncotarget 2016, 7, 78473–78486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seals, D.F.; Azucena, E.F.; Pass, I.; Tesfay, L.; Gordon, R.; Woodrow, M.; Resau, J.H.; Courtneidge, S.A. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 2005, 7, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Kudlik, G.; Takács, T.; Radnai, L.; Kurilla, A.; Szeder, B.; Koprivanacz, K.; Merő, B.L.; Buday, L.; Vas, V. Advances in understanding TKS4 and TKS5: Molecular scaffolds regulating cellular processes from podosome and invadopodium formation to differentiation and tissue homeostasis. Int. J. Mol. Sci. 2020, 21, 8117. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Eguchi, T.; Osada, S.; Nishizuka, M.; Imagawa, M. A novel gene, fad49, plays a crucial role in the immediate early stage of adipocyte differentiation via involvement in mitotic clonal expansion. FEBS J. 2008, 275, 5576–5588. [Google Scholar] [CrossRef]
- Vas, V.; Háhner, T.; Kudlik, G.; Ernszt, D.; Kvell, K.; Kuti, D.; Kovács, K.J.; Tóvári, J.; Trexler, M.; Merő, B.L.; et al. Analysis of Tks4 knockout mice suggests a role for Tks4 in adipose tissue homeostasis in the context of beigeing. Cells 2019, 8, 831. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.A.; Diaz, B.; Bromann, P.A.; Tsai, J.H.; Kawakami, Y.; Maurer, J.; Stewart, R.A.; Izpisúa-Belmonte, J.C.; Courtneidge, S.A. A Src-Tks5 pathway is required for neural crest cell migration during embryonic development. PLoS ONE 2011, 6, e22499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejudo-Martin, P.; Yuen, A.; Vlahovich, N.; Lock, P.; Courtneidge, S.A.; Díaz, B. Genetic disruption of the Sh3pxd2a gene reveals an essential role in mouse development and the existence of a novel isoform of Tks5. PLoS ONE 2014, 9, e107674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, Z.; Cejudo-Martin, P.; de Brouwer, A.; van der Zwaag, B.; Ruiz-Lozano, P.; Scimia, M.C.; Lindsey, J.D.; Weinreb, R.; Albrecht, B.; Megarbane, A.; et al. Disruption of the podosome adaptor protein TKS4 (SH3PXD2B) causes the skeletal dysplasia, eye, and cardiac abnormalities of frank-ter haar syndrome. Am. J. Hum. Genet. 2010, 86, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, Y.; Ziprkowski, M.; Romano, A.; Stein, R.; Katznelson, M.B.; Cohen, B.; Goodman, R.M. Megalocornea associated with multiple skeletal anomalies: A new genetic syndrome? J. Genet. Hum. 1973, 21, 67–72. [Google Scholar] [PubMed]
- ter Haar, B.; Hamel, B.; Hendriks, J.; de Jager, J.; Opitz, J.M. Melnick-Needles syndrome: Indication for an autosomal recessive form. Am. J. Med. Genet. 1982, 13, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Vas, V.; Kovács, T.; Körmendi, S.; Bródy, A.; Kudlik, G.; Szeder, B.; Mező, D.; Kállai, D.; Koprivanacz, K.; Merő, B.L.; et al. Significance of the Tks4 scaffold protein in bone tissue homeostasis. Sci. Rep. 2019, 9, 5781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dülk, M.; Kudlik, G.; Fekete, A.; Ernszt, D.; Kvell, K.; Pongrácz, J.E.; Merő, B.L.; Szeder, B.; Radnai, L.; Geiszt, M.; et al. The scaffold protein Tks4 is required for the differentiation of mesenchymal stromal cells (MSCs) into adipogenic and osteogenic lineages. Sci. Rep. 2016, 6, 34280. [Google Scholar] [CrossRef] [Green Version]
- Szeder, B.; Tárnoki-Zách, J.; Lakatos, D.; Vas, V.; Kudlik, G.; Merő, B.; Koprivanacz, K.; Bányai, L.; Hámori, L.; Róna, G.; et al. Absence of the Tks4 scaffold protein induces epithelial-mesenchymal transition-like changes in human colon cancer cells. Cells 2019, 8, 1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dülk, M.; Szeder, B.; Glatz, G.; Merő, B.L.; Koprivanacz, K.; Kudlik, G.; Vas, V.; Sipeki, S.; Cserkaszky, A.; Radnai, L.; et al. EGF regulates the interaction of Tks4 with Src through Its SH2 and SH3 domains. Biochemistry 2018, 57, 4186–4196. [Google Scholar] [CrossRef]
- Bögel, G.; Gujdár, A.; Geiszt, M.; Lányi, Á.; Fekete, A.; Sipeki, S.; Downward, J.; Buday, L. Frank-ter Haar syndrome protein Tks4 regulates epidermal growth factor-dependent cell migration. J. Biol. Chem. 2012, 287, 31321–31329. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Pryciak, P.M. Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Curr. Biol. 2008, 18, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Enkhbat, A.; Yoshioka, K. Role of plasma membrane localization of the scaffold protein JSAP1 during differentiation of cerebellar granule cell precursors. Genes Cells 2011, 16, 58–68. [Google Scholar] [CrossRef] [Green Version]
- El-Benna, J.; Dang, P.M.-C.; Gougerot-Pocidalo, M.-A.; Marie, J.-C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcoux, J.; Man, P.; Petit-Haertlein, I.; Vivès, C.; Forest, E.; Fieschi, F. p47phox molecular activation for assembly of the neutrophil NADPH oxidase complex. J. Biol. Chem. 2010, 285, 28980–28990. [Google Scholar] [CrossRef] [Green Version]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.C.; El-Benna, J. NADPH Oxidase Activation in Neutrophils: Role of the Phosphorylation of Its Subunits; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2018; Volume 48. [Google Scholar] [CrossRef] [Green Version]
- Ogura, K.; Nobuhisa, I.; Yuzawa, S.; Takeya, R.; Torikai, S.; Saikawa, K.; Sumimoto, H.; Inagaki, F. NMR solution structure of the tandem Src homology 3 domains of p47phox complexed with a p22phox-derived proline-rich peptide. J. Biol. Chem. 2006, 281, 3660–3668. [Google Scholar] [CrossRef] [Green Version]
- Finan, P.; Shimizu, Y.; Gout, I.; Hsuan, J.; Truong, O.; Butcher, C.; Bennett, P.; Waterfield, M.D.; Kellie, S. An SH3 domain and proline-rich sequence mediate an interaction between two components of the phagocyte NADPH oxidase complex. J. Biol. Chem. 1994, 269, 13752–13755. [Google Scholar] [CrossRef]
- Yuzawa, S.; Ogura, K.; Horiuchi, M.; Suzuki, N.N.; Fujioka, Y.; Kataoka, M.; Sumimoto, H.; Inagaki, F. Solution structure of the tandem Src homology 3 domains of p47phox in an autoinhibited form. J. Biol. Chem. 2004, 279, 29752–29760. [Google Scholar] [CrossRef] [Green Version]
- Yuzawa, S.; Suzuki, N.N.; Fujioka, Y.; Ogura, K.; Sumimoto, H.; Inagaki, F. A molecular mechanism for autoinhibition of the tandem SH3 domains of p47phox, the regulatory subunit of the phagocyte NADPH oxidase. Genes Cells 2004, 9, 443–456. [Google Scholar] [CrossRef]
- Ago, T.; Nunoi, H.; Ito, T.; Sumimoto, H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH Oxidase Protein p47 phox. J. Biol. Chem. 1999, 274, 33644–33653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ago, T.; Kuribayashi, F.; Hiroaki, H.; Takeya, R.; Ito, T.; Kohda, D.; Sumimoto, H. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc. Natl. Acad. Sci. USA 2003, 100, 4474–4479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groemping, Y.; Lapouge, K.; Smerdon, S.J.; Rittinger, K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 2003, 113, 343–355. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Durand, D.; Cannella, D.; Dubosclard, V.; Pebay-Peyroula, E.; Vachette, P.; Fieschi, F. Small-angle X-ray scattering reveals an extended organization for the autoinhibitory resting state of the p47(phox) modular protein. Biochemistry 2006, 45, 7185–7193. [Google Scholar] [CrossRef] [PubMed]
- Merő, B.; Radnai, L.; Gógl, G.; Tőke, O.; Leveles, I.; Koprivanacz, K.; Szeder, B.; Dülk, M.; Kudlik, G.; Vas, V.; et al. Structural insights into the tyrosine phosphorylation-mediated inhibition of SH3 domain-ligand interactions. J. Biol. Chem. 2019, 294, 4608–4620. [Google Scholar] [CrossRef] [Green Version]
- Pisabarro, M.T.; Serrano, L. Rational design of specific high-affinity peptide ligands for the Abl-SH3 domain. Biochemistry 1996, 35, 10634–10640. [Google Scholar] [CrossRef]
- Viguera, A.R.; Arrondo, J.L.; Musacchio, A.; Saraste, M.; Serrano, L. Characterization of the interaction of natural proline-rich peptides with five different SH3 domains. Biochemistry 1994, 33, 10925–10933. [Google Scholar] [CrossRef]
- Patel, H.V.; Tzeng, S.R.; Liao, C.Y.; Chen, S.H.; Cheng, J.W. SH3 domain of Bruton’s tyrosine kinase can bind to proline-rich peptides of TH domain of the kinase and p120(cbl). Proteins Struct. Funct. Genet. 1997, 29, 545–552. [Google Scholar] [CrossRef]
- Bhatt, V.S.; Zeng, D.; Krieger, I.; Sacchettini, J.C.; Cho, J.-H. Binding mechanism of the n-terminal SH3 Domain of CrkII and Proline-Rich Motifs in cAbl. Biophys. J. 2016, 110, 2630–2641. [Google Scholar] [CrossRef] [Green Version]
- Karathanassis, D.; Stahelin, R.V.; Bravo, J.; Perisic, O.; Pacold, C.M.; Cho, W.; Williams, R.L. Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 2002, 21, 5057–5068. [Google Scholar] [CrossRef]
- Marcoux, J.; Man, P.; Castellan, M.; Vivès, C.; Forest, E.; Fieschi, F. Conformational changes in p47(phox) upon activation highlighted by mass spectrometry coupled to hydrogen/deuterium exchange and limited proteolysis. FEBS Lett. 2009, 583, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Rufer, A.C.; Rumpf, J.; von Holleben, M.; Beer, S.; Rittinger, K.; Groemping, Y. Isoform-selective interaction of the adaptor protein Tks5/FISH with Sos1 and dynamins. J. Mol. Biol. 2009, 390, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Matsuo, K. Possible role of IRTKS in Tks5-driven osteoclast fusion. Commun. Integr. Biol. 2012, 5, 511–515. [Google Scholar] [CrossRef]
- Diaz, B.; Shani, G.; Pass, I.; Anderson, D.; Quintavalle, M.; Courtneidge, S.A. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci. Signal. 2009, 2, ra53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lányi, Á.; Baráth, M.; Péterfi, Z.; Bőgel, G.; Orient, A.; Simon, T.; Petrovszki, E.E.E.; Kis-Tóth, K.; Sirokmány, G.; Rajnavölgyi, É.; et al. The homolog of the five SH3-domain protein (HOFI/SH3PXD2B) regulates lamellipodia formation and cell spreading. PLoS ONE 2011, 6, e23653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, K.; Lemmon, M.A. Determining selectivity of phosphoinositide-binding domains. Methods 2006, 39, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Chandra, M.; Chin, K.-Y.; Mas, C.; Ryan Feathers, J.; Paul, B.; Datta, S.; Chen, K.-E.E.; Jia, X.; Yang, Z.; Norwood, S.J.; et al. Classification of the human phox homology (PX) domains based on their phosphoinositide binding specificities. Nat. Commun. 2019, 10, 1528. [Google Scholar] [CrossRef] [PubMed]
- Abram, C.L.; Seals, D.F.; Pass, I.; Salinsky, D.; Maurer, L.; Roth, T.M.; Courtneidge, S.A. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J. Biol. Chem. 2003, 278, 16844–16851. [Google Scholar] [CrossRef] [Green Version]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef]
- Marat, A.L.; Haucke, V. Phosphatidylinositol 3-phosphates—at the interface between cell signalling and membrane traffic. EMBO J. 2016, 35, 561–579. [Google Scholar] [CrossRef]
- Fontayne, A.; Dang, P.M.; Gougerot-Pocidalo, M.-A.; El-Benna, J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 2002, 41, 7743–7750. [Google Scholar] [CrossRef]
- Sumimoto, H.; Kage, Y.; Nunoi, H.; Sasaki, H.; Nose, T.; Fukumaki, Y.; Ohno, M.; Minakami, S.; Takeshige, K. Role of Src homology 3 domains in assembly and activation of the phagocyte NADPH oxidase. Proc. Natl. Acad. Sci. USA 1994, 91, 5345–5349. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro, Á.; Muñoz, E.; Sabín, J.; Costas, M.; Bastos, M.; Velázquez-Campoy, A.; Garrido, P.F.; Dumas, P.; Ennifar, E.; García-Río, L.; et al. AFFINImeter: A software to analyze molecular recognition processes from experimental data. Anal. Biochem. 2019, 577, 117–134. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 1650–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tks4 Fragment | Sequence | Kd (µM) | Method |
|---|---|---|---|
| PRRL | GSHMGDAKQRSPKMRQRPPPRRDMTIPRGLNLPKPPIPPQVE | 15 ± 3 | MST |
| 11 ± 3 | FL | ||
| PRRS | KMRQRPPPRRDMTIPRG | 12 ± 4 | FL |
| PRRLP/G | GSHMGDAKQRSPKMRQRGGGRRDMTIPRGLNLPKPPIPPQVE | - | FL |
| PRRLP/A | GSHMGDAKQRSPKMRQRPPPRRDMTIPRGLNLPKAAIPPQVE | 12 ± 3 | FL |
| PRRLT353E | GSHMGDAKQRSPKMRQRPPPRRDMEIPRGLNLPKPPIPPQVE | 10 ± 6 | FL |
| Tks4 Fragment | MW (kDa) | Rg (nm) | Dmax (nm) | VP (nm3) | χ (CORAL fit) |
|---|---|---|---|---|---|
| PX–SH31 | 24.6 | 2.14 ± 0.05 | 6.3 ± 0.3 | 39.1 | 0.74 |
| PX–SH32 | 33 | 3.04 ± 0.16 | 9.7 ± 0.4 | 63.5 | 1.39 |
| PX–PRR | 42.6 | 3.05 ± 0.28 | 10.3 ± 0.2 | 80.1 | 1.8 |
| PX–SH33 | 49 | 2.86 ± 0.05 | 9.2 ± 0.3 | 83.3 | 0.66 |
| PX–SH33T353E | 49 | 2.85 ± 0.09 | 9.3 ± 0.3 | 84.7 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merő, B.; Koprivanacz, K.; Cserkaszky, A.; Radnai, L.; Vas, V.; Kudlik, G.; Gógl, G.; Sok, P.; Póti, Á.L.; Szeder, B.; et al. Characterization of the Intramolecular Interactions and Regulatory Mechanisms of the Scaffold Protein Tks4. Int. J. Mol. Sci. 2021, 22, 8103. https://doi.org/10.3390/ijms22158103
Merő B, Koprivanacz K, Cserkaszky A, Radnai L, Vas V, Kudlik G, Gógl G, Sok P, Póti ÁL, Szeder B, et al. Characterization of the Intramolecular Interactions and Regulatory Mechanisms of the Scaffold Protein Tks4. International Journal of Molecular Sciences. 2021; 22(15):8103. https://doi.org/10.3390/ijms22158103
Chicago/Turabian StyleMerő, Balázs, Kitti Koprivanacz, Anna Cserkaszky, László Radnai, Virag Vas, Gyöngyi Kudlik, Gergő Gógl, Péter Sok, Ádám L. Póti, Bálint Szeder, and et al. 2021. "Characterization of the Intramolecular Interactions and Regulatory Mechanisms of the Scaffold Protein Tks4" International Journal of Molecular Sciences 22, no. 15: 8103. https://doi.org/10.3390/ijms22158103