
Activation of the Integrated Stress Response and ER Stress Protect from Fluorizoline-Induced Apoptosis in HEK293T and U2OS Cell Lines

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
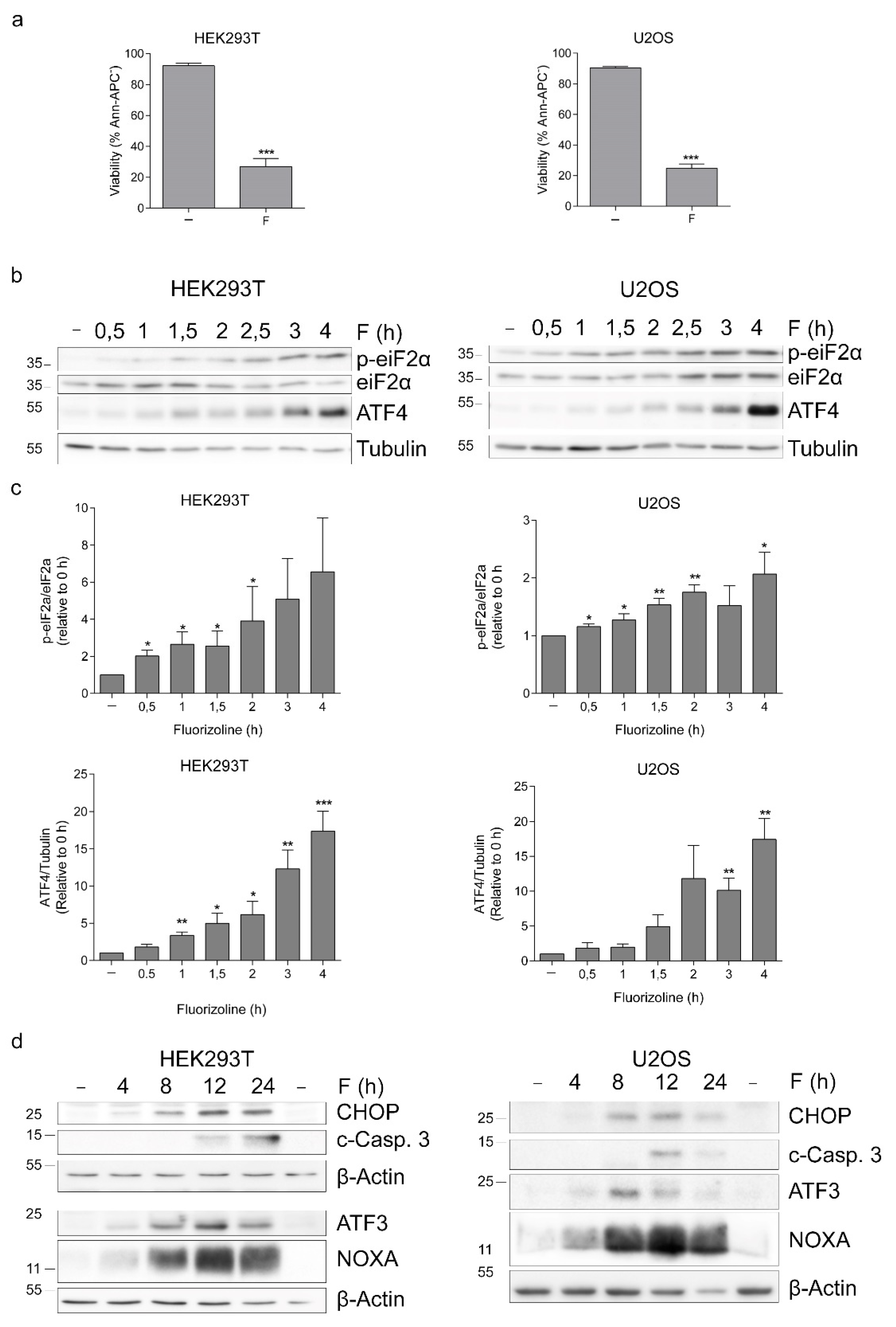
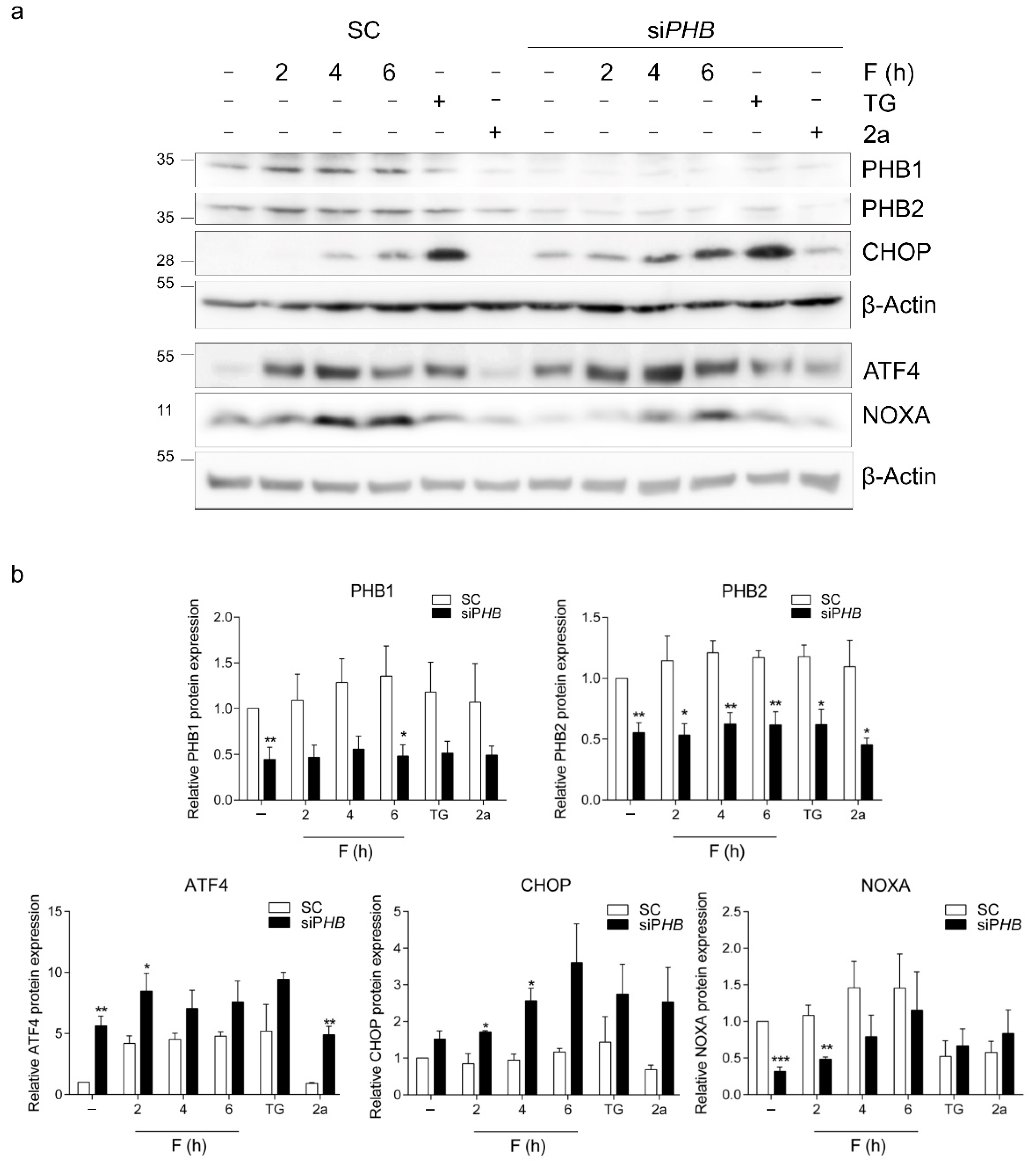
2.1. The ISR Is Induced after Fluorizoline Treatment or PHB Downregulation
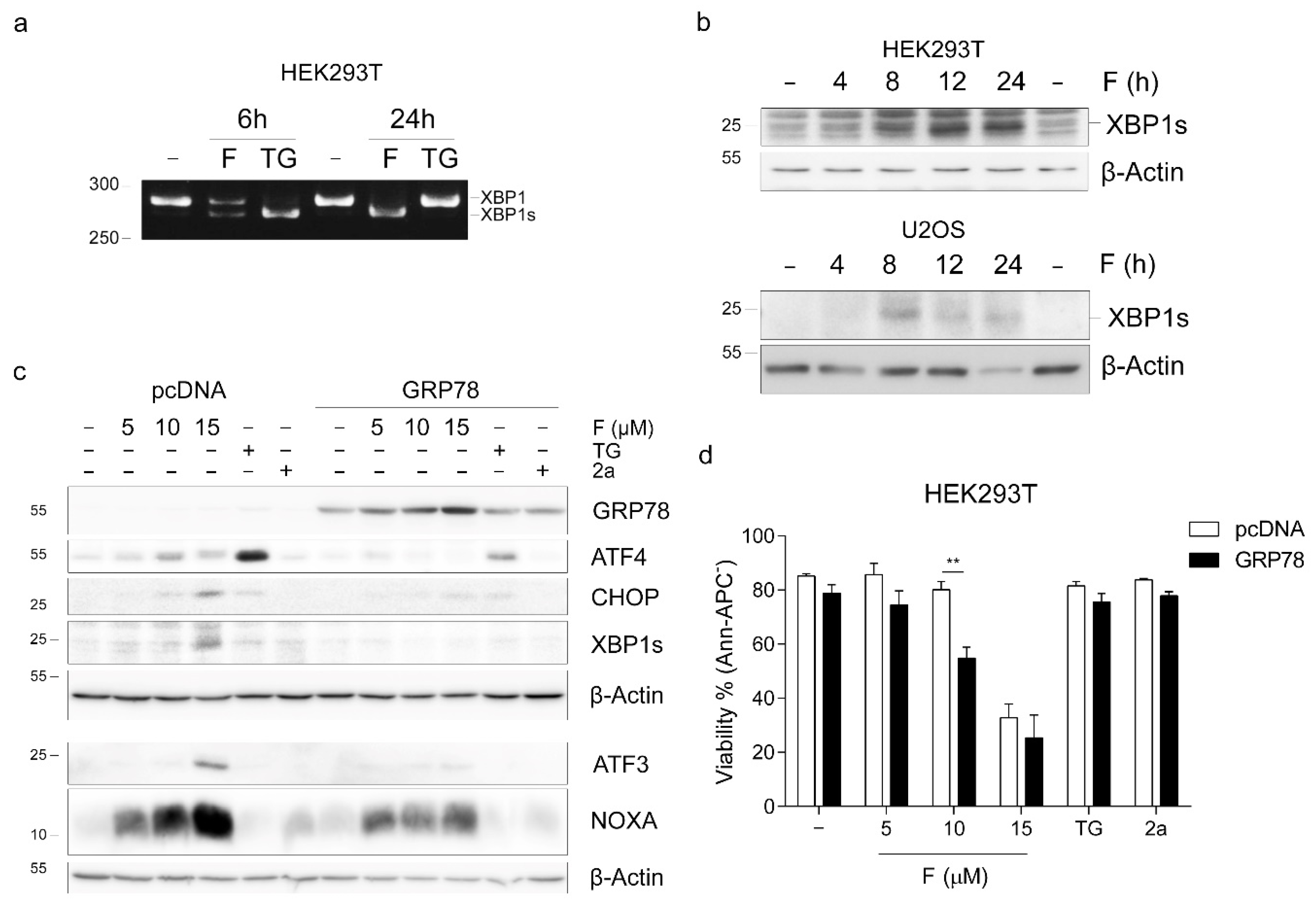
2.2. Fluorizoline Induces ER Stress in HEK293T and U2OS Cells
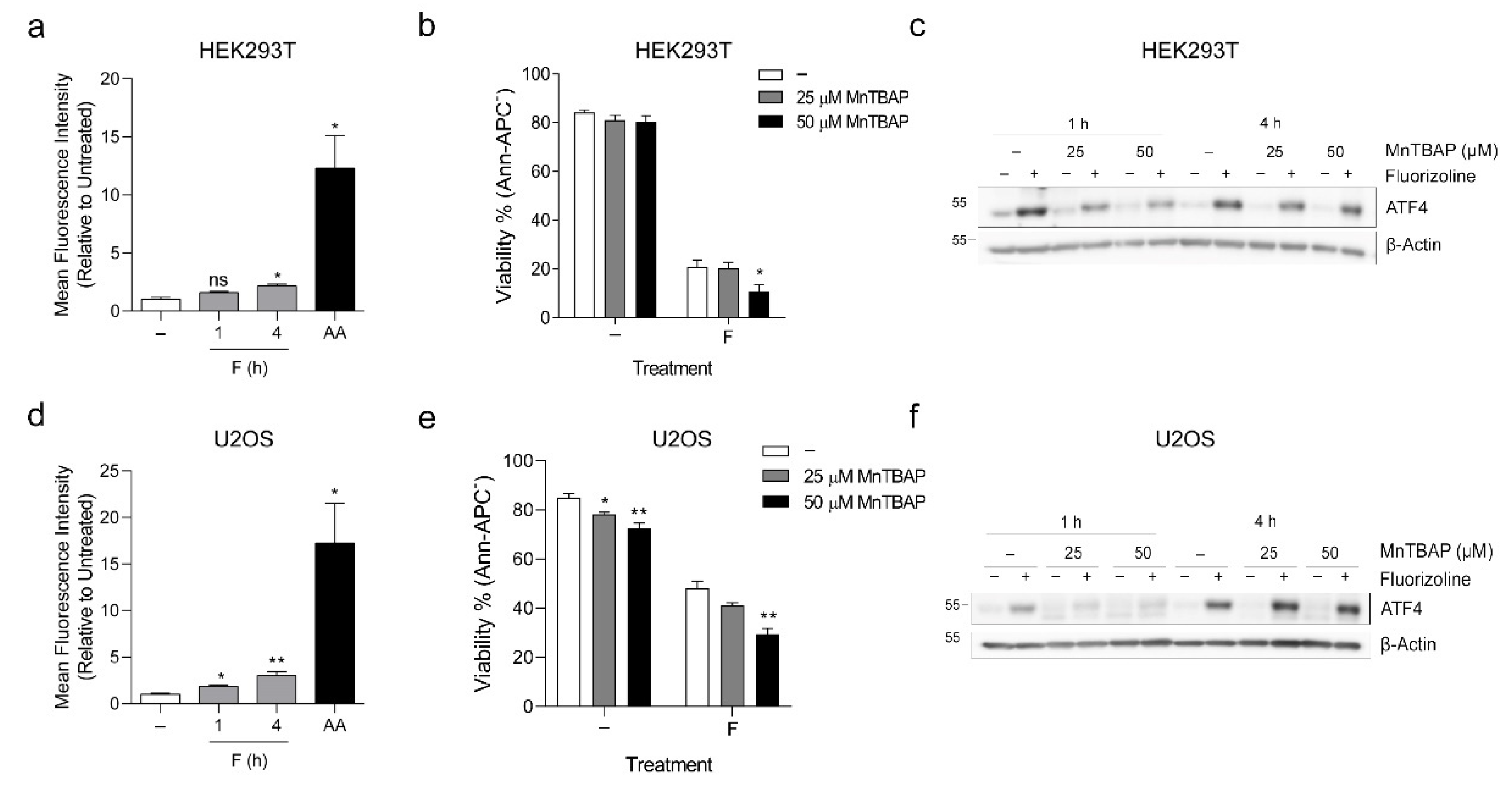
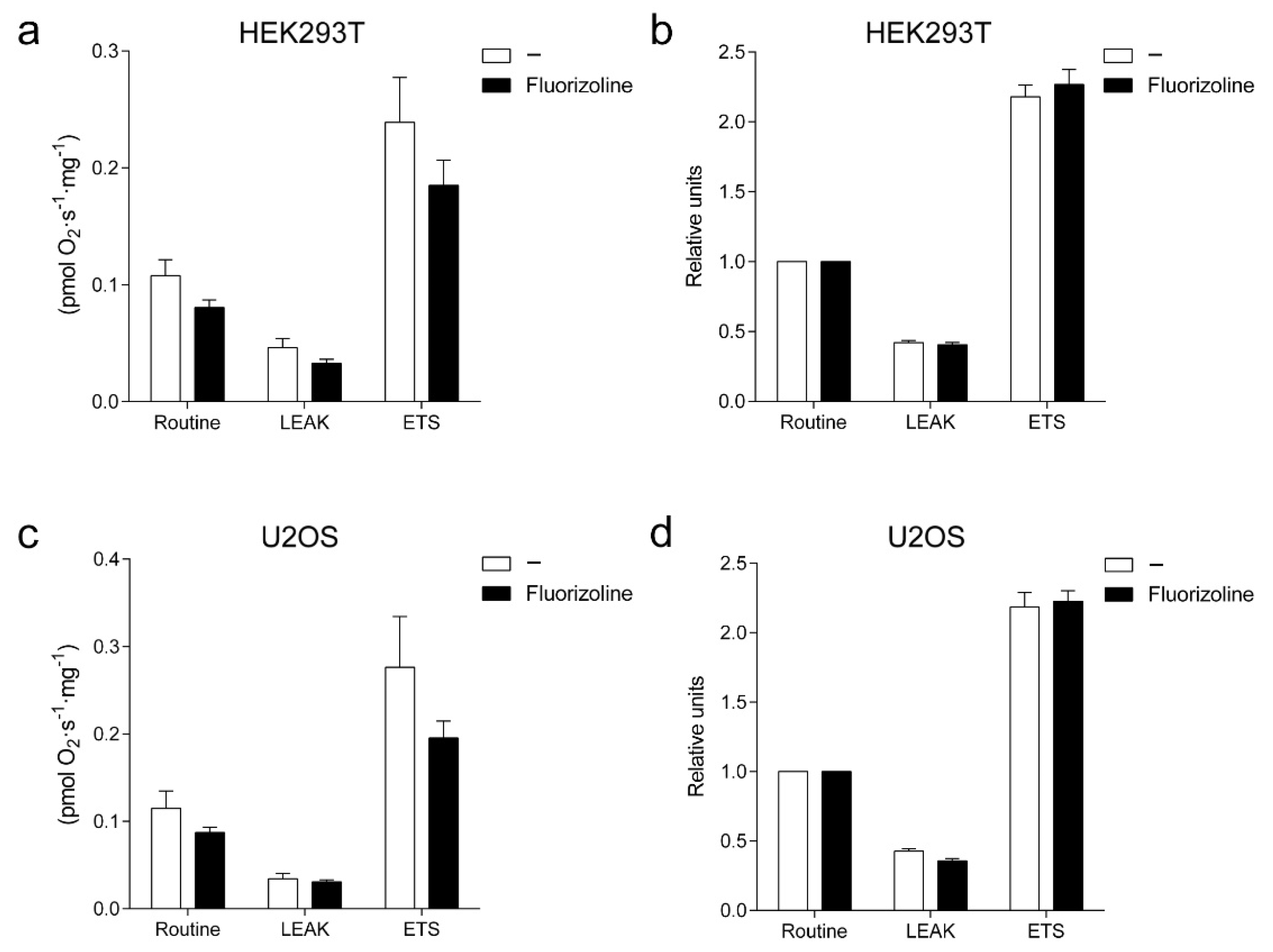
2.3. Fluorizoline Does Not Trigger Apoptosis through Altered Mitochondrial Respiration or ROS Production
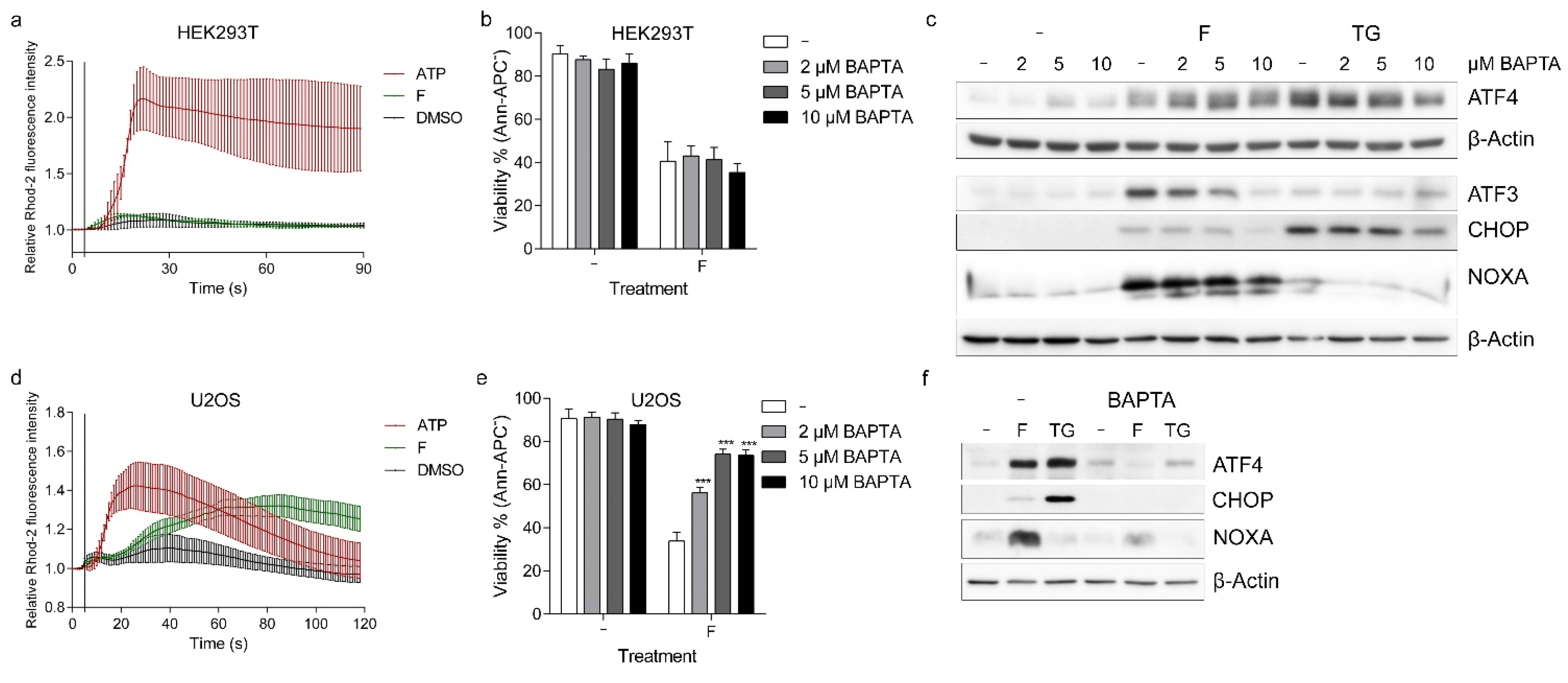
2.4. Fluorizoline Alters Calcium Homeostasis to Trigger Apoptosis in U2OS Cells
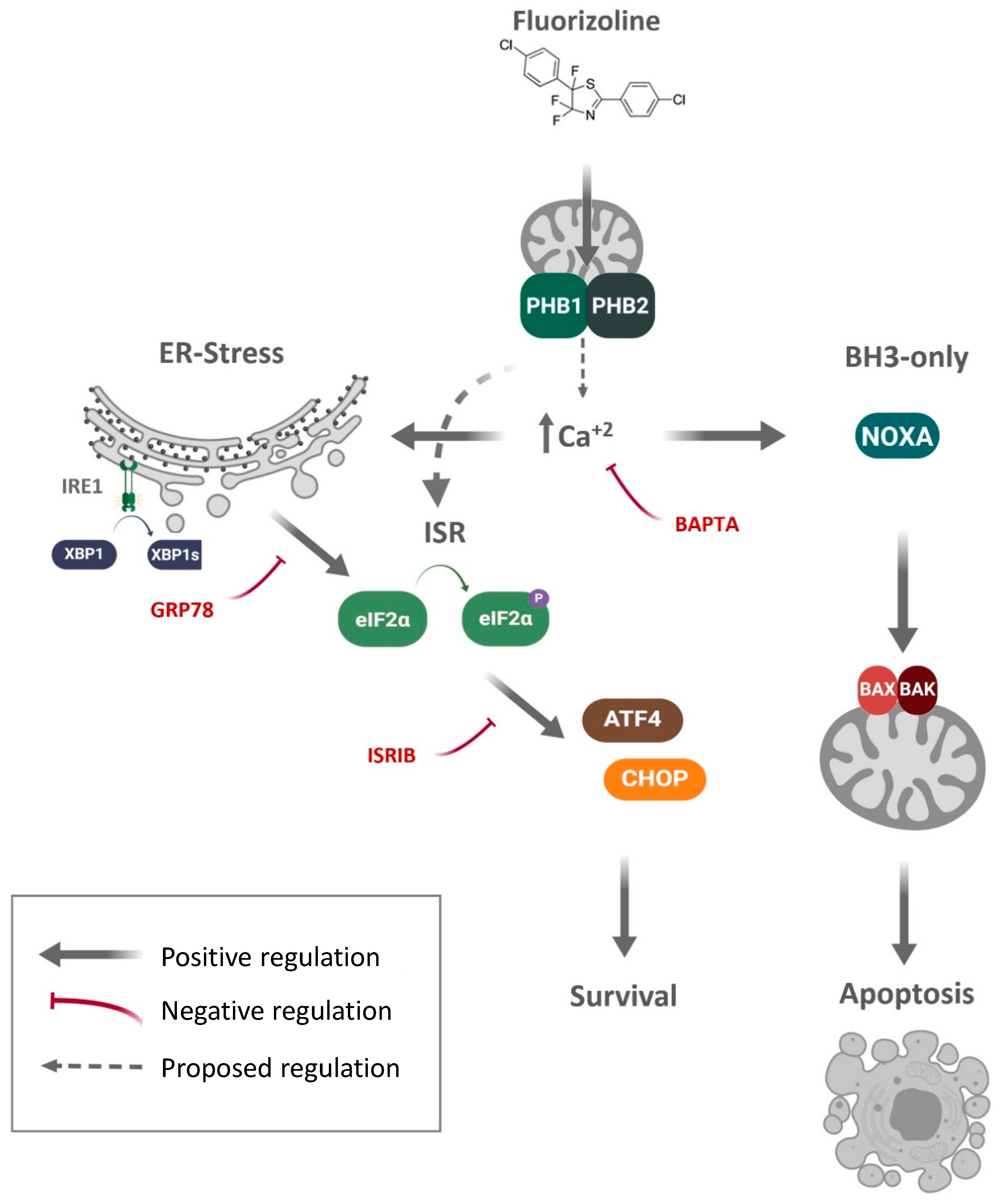
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines Culture
4.3. Transient Transfection
4.4. Western Blot
4.5. PCR
4.6. Flow Cytometry
4.7. Live-Cell Imaging
4.8. Calcium Measurement with Aequorin
4.9. Glutathione Measurement
4.10. Measurement of Oxygen Consumption
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF4 | Activating transcription factor 4 |
| CHOP | C/EBP homologous protein |
| eIF2α | eukaryotic initiation factor 2α |
| ER | Endoplasmic reticulum |
| ETS | Electron transfer system capacity |
| GCN2 | General control nonderepressible 2 kinase |
| GRP78 | Glucose-regulated protein 78 |
| HRI | Heme-regulated inhibitor |
| IRE1 | Inositol-requiring enzyme 1 |
| ISR | Integrated stress response |
| OPA1 | Optic atrophy 1 |
| PERK | PKR-like ER kinase |
| PHB | Prohibitin |
| PKR | Protein kinase R |
| ROS | Reactive oxygen species |
| SEM | Standard error of the mean |
References
- Pérez-Perarnau, A.; Preciado, S.; Palmeri, C.M.; Moncunill-Massaguer, C.; Iglesias-Serret, D.; González-Gironès, D.M.; Miguel, M.; Karasawa, S.; Sakamoto, S.; Cosialls, A.M.; et al. A Trifluorinated Thiazoline Scaffold Leading to Pro-apoptotic Agents Targeting Prohibitins. Angew. Chem. Int. Ed. 2014, 53, 10150–10154. [Google Scholar] [CrossRef] [PubMed]
- Pomares, H.; Palmeri, C.M.; Iglesias-Serret, D.; Moncunill-Massaguer, C.; Saura-Esteller, J.; Núñez-Vázquez, S.; Gamundi, E.; Arnan, M.; Preciado, S.; Albericio, F.; et al. Targeting prohibitins induces apoptosis in acute myeloid leukemia cells. Oncotarget 2016, 7, 64987–65000. [Google Scholar] [CrossRef]
- Cosialls, A.M.; Pomares, H.; Iglesias-Serret, D.; Saura-Esteller, J.; Núñez-Vázquez, S.; González-Gironès, D.M.; De La Banda, E.; Preciado, S.; Albericio, F.; Lavilla, R.; et al. The prohibitin-binding compound fluorizoline induces apoptosis in chronic lymphocytic leukemia cells through the upregulation of NOXA and synergizes with ibrutinib, 5-aminoimidazole-4-carboxamide riboside or venetoclax. Haematologica 2017, 102, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Hernando-Rodríguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. [Google Scholar] [CrossRef] [Green Version]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, B.; He, Q.Y. Significance of prohibitin domain family in tumorigenesis and its implication in cancer diagnosis and treatment review-article. Cell Death Dis. 2018, 9, 580. [Google Scholar] [CrossRef]
- Zhou, Z.; Ai, H.; Li, K.; Yao, X.; Zhu, W.; Liu, L.; Yu, C.; Song, Z.; Bao, Y.; Huang, Y.; et al. Prohibitin 2 localizes in nucleolus to regulate ribosomal RNA transcription and facilitate cell proliferation in RD cells. Sci. Rep. 2018, 8, 1479. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.-B.; Qin, Y.-H.; Lei, F.-Y.; Huang, W.-F.; Drummen, G.P.C. Prohibitin is associated with antioxidative protection in hypoxia/reoxygenation-induced renal tubular epithelial cell injury. Sci. Rep. 2013, 3, 3123. [Google Scholar] [CrossRef] [Green Version]
- Osman, C.; Merkwirth, C.; Langer, T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 2009, 122, 3823–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshinaka, T.; Kosako, H.; Yoshizumi, T.; Furukawa, R.; Hirano, Y.; Kuge, O.; Tamada, T.; Koshiba, T. Structural Basis of Mitochondrial Scaffolds by Prohibitin Complexes: Insight into a Role of the Coiled-Coil Region. iScience 2019, 19, 1065–1078. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Tabti, R.; Elderwish, S.; Abou-Hamdan, H.; Djehal, A.; Yu, P.; Yurugi, H.; Rajalingam, K.; Nebigil, C.G.; Désaubry, L. Prohibitin ligands: A growing armamentarium to tackle cancers, osteoporosis, inflammatory, cardiac and neurological diseases. Cell. Mol. Life Sci. 2020, 77, 3525–3546. [Google Scholar] [CrossRef]
- Guyot, A.C.; Leuxe, C.; Disdier, C.; Oumata, N.; Costa, N.; Le Roux, G.; Fernandez-Varela, P.; Duchon, A.; Charbonnier, J.B.; Herault, Y.; et al. A Small Compound Targeting Prohibitin with Potential Interest for Cognitive Deficit Rescue in Aging mice and Tau Pathology Treatment. Sci. Rep. 2020, 10, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimaru, T.; Komatsu, M.; Tashiro, E.; Imoto, M.; Osada, H.; Miyoshi, Y.; Honda, J.; Sasa, M.; Katagiri, T. Xanthohumol suppresses oestrogen-signalling in breast cancer through the inhibition of BIG3-PHB2 interactions. Sci. Rep. 2014, 4, 7355. [Google Scholar] [CrossRef] [Green Version]
- Moncunill-Massaguer, C.; Saura-Esteller, J.; Pérez-Perarnau, A.; Palmeri, C.M.; Núñez-Vázquez, S.; Cosialls, A.M.; González-Gironès, D.M.; Pomares, H.; Korwitz, A.; Preciado, S.; et al. A novel prohibitin-binding compound induces the mitochondrial apoptotic pathway through NOXA and BIM upregulation. Oncotarget 2015, 6, 41750–41765. [Google Scholar] [CrossRef]
- Saura-Esteller, J.; Sánchez-Vera, I.; Núñez-Vázquez, S.; Jabalquinto-Carrasco, J.; Cosialls, A.M.; Mendive-Tapia, L.; Kukhtar, D.; Martínez-Bueno, M.D.; Lavilla, R.; Cerón, J.; et al. Fluorizoline-induced apoptosis requires prohibitins in nematodes and human cells. Apoptosis 2021, 26, 83–95. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Quiles, V.; Santamaría, E.; Segura, V.; Sesma, L.; Prieto, J.; Corrales, F.J. Prohibitin deficiency blocks proliferation and induces apoptosis in human hepatoma cells: Molecular mechanisms and functional implications. Proteomics 2010, 10, 1609–1620. [Google Scholar] [CrossRef]
- Yang, T.-Y.; Wu, Y.-J.; Chang, C.-I.; Chiu, C.-C.; Wu, M.-L. The Effect of Bornyl cis-4-Hydroxycinnamate on Melanoma Cell Apoptosis Is Associated with Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2018, 19, 1370. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Singh, N.; Sengupta, N.; Fatima, M.; Seth, P.; Mahadevan, A.; Shankar, S.K.; Bhattacharyya, A.; Basu, A. Japanese encephalitis virus induces human neural stem/progenitor cell death by elevating GRP78, PHB and hnRNPC through ER stress. Cell Death Dis. 2018, 8, e2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junior, T.C.T.; De Godoy, L.M.F.; de Souza, G.; Bonatto, D.; Otake, A.H.; Saito, R.D.F.; Rosa, J.C.; Greene, L.J.; Chammas, R. Accumulation of prohibitin is a common cellular response to different stressing stimuli and protects melanoma cells from ER stress and chemotherapy-induced cell death. Oncotarget 2017, 8, 43114–43129. [Google Scholar] [CrossRef]
- Núñez-Vázquez, S.; Sánchez-Vera, I.; Saura-Esteller, J.; Cosialls, A.M.; Noisier, A.F.; Albericio, F.; Lavilla, R.; Pons, G.; Iglesias-Serret, D.; Gil, J. NOXA upregulation by the prohibitin-binding compound fluorizoline is transcriptionally regulated by integrated stress response-induced ATF3 and ATF4. FEBS J. 2021, 288, 1271–1285. [Google Scholar] [CrossRef]
- Iwasaki, S.; Iwasaki, W.; Takahashi, M.; Sakamoto, A.; Watanabe, C.; Shichino, Y.; Floor, S.N.; Fujiwara, K.; Mito, M.; Dodo, K.; et al. The Translation Inhibitor Rocaglamide Targets a Bimolecular Cavity between eIF4A and Polypurine RNA. Mol. Cell 2019, 73, 738–748.e9. [Google Scholar] [CrossRef] [Green Version]
- Yiming, L.; Yunshan, G.; Juan, T.; Jianli, J.; Zhinan, C. New Insights Into the Roles of CHOP-induced Apoptosis in ER Stress. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 629–640. [Google Scholar]
- Sekine, Y.; Zyryanova, A.; Crespillo-Casado, A.; Fischer, P.; Harding, H.P.; Ron, D. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 2015, 348, 1027–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife 2015, 4, e05033. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrtash, A.B.; Hochstrasser, M. Ubiquitin-dependent protein degradation at the endoplasmic reticulum and nuclear envelope. Semin. Cell Dev. Biol. 2019, 93, 111–124. [Google Scholar] [CrossRef]
- Costa, R.; Peruzzo, R.; Bachmann, M.; Montà, G.D.; Vicario, M.; Santinon, G.; Mattarei, A.; Moro, E.; Quintana-Cabrera, R.; Scorrano, L.; et al. Impaired Mitochondrial ATP Production Downregulates Wnt Signaling via ER Stress Induction. Cell Rep. 2019, 28, 1949–1960.e6. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, C.; Xu, F.; Hou, T.; Sun, T.; Jinghang, L.; Cheng, H.; Wang, X. Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. J. Cell Sci. 2017, 130, 2620–2630. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, M.; Shepherd, B.R.; Suarez, Y.; Fernandez-Hernando, C.; Yu, J.; Pan, Y.; Acevedo, L.M.; Shadel, G.S.; Sessa, W.C. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 2008, 180, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial Functions and Estrogen Receptor-dependent Nuclear Translocation of Pleiotropic Human Prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Kerkhofs, M.; Giorgi, C.; Marchi, S.; Seitaj, B.; Parys, J.B.; Pinton, P.; Bultynck, G.; Bittremieux, M. Alterations in Ca2+ Signalling via ER-Mitochondria Contact Site Remodelling in Cancer. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2017; Volume 997, pp. 225–254. [Google Scholar]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Cano-González, A.; Mauro-Lizcano, M.; Iglesias-Serret, D.; Gil, J.; López-Rivas, A. Involvement of both caspase-8 and Noxa-activated pathways in endoplasmic reticulum stress-induced apoptosis in triple-negative breast tumor cells. Cell Death Dis. 2018, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Wei, X.; Song, M.; Tao, J.; Yang, Y.; Khatoon, S.; Liu, H.; Jiang, J.; Wu, B. PUMA mediates ER stress-induced apoptosis in portal hypertensive gastropathy. Cell Death Dis. 2014, 5, e1128. [Google Scholar] [CrossRef] [Green Version]
- A Wali, J.; Rondas, D.; McKenzie, M.D.; Zhao, Y.; Elkerbout, L.; Fynch, S.; Gurzov, E.N.; Akira, S.; Mathieu, C.; Kay, T.W.H.; et al. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014, 5, e1124. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Vu, T.; Cook, W.; Naseri, M.; Zhan, K.; Nakajima, W.; Harada, H. p53-independent Noxa induction by cisplatin is regulated by ATF3/ATF4 in head and neck squamous cell carcinoma cells. Mol. Oncol. 2018, 12, 788–798. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.; Hughes, P.D.; Michalak, E.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.P.; Klocke, B.J.; Ballestas, M.E.; Roth, K.A. CHOP Potentially Co-Operates with FOXO3a in Neuronal Cells to Regulate PUMA and BIM Expression in Response to ER Stress. PLoS ONE 2012, 7, e39586. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübner, A.; Barrett, T.; Flavell, R.A.; Davis, R.J. Multisite Phosphorylation Regulates Bim Stability and Apoptotic Activity. Mol. Cell 2008, 30, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, V.; Giglio, P.; Bernardoni, P.; De Zio, D.; Fimia, G.M.; Piacentini, M.; Corazzari, M. Down-regulation of E2F1 during ER stress is required to induce apoptosis. J. Cell Sci. 2015, 128, 1166–1179. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Fu, L.; Balasubramanian, M.N.; Anthony, T.; Kilberg, M.S. ATF4-dependent Regulation of the JMJD3 Gene during Amino Acid Deprivation Can Be Rescued in Atf4-deficient Cells by Inhibition of Deacetylation*. J. Biol. Chem. 2012, 287, 36393–36403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999, 339 Pt 1, 135–141. [Google Scholar] [CrossRef]
- Young, S.K.; Wek, R.C. Upstream open reading frames differentially regulate genespecific translation in the integrated stress response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 Activated by Proteolysis Binds in the Presence of NF-Y (CBF) Directly to the cis-Acting Element Responsible for the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [Green Version]
- Edagawa, M.; Kawauchi, J.; Hirata, M.; Goshima, H.; Inoue, M.; Okamoto, T.; Murakami, A.; Maehara, Y.; Kitajima, S. Role of Activating Transcription Factor 3 (ATF3) in Endoplasmic Reticulum (ER) Stress-induced Sensitization of p53-deficient Human Colon Cancer Cells to Tumor Necrosis Factor (TNF)-related Apoptosis-inducing Ligand (TRAIL)-mediated Apoptosis through Up-regulation of Death Receptor 5 (DR5) by Zerumbone and Celecoxib. J. Biol. Chem. 2014, 289, 21544–21561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Fessler, E.; Eckl, E.-M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nat. Cell Biol. 2020, 579, 433–437. [Google Scholar] [CrossRef]
- Baker, M.J.; A Lampe, P.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.J.; Kahl, A.; Fruitman, H.; Qian, L.; Zhou, P.; Manfredi, G.; Iadecola, C. Prohibitin levels regulate OMA1 activity and turnover in neurons. Cell Death Differ. 2019, 27, 1896–1906. [Google Scholar] [CrossRef]
- Sassano, M.L.; van Vliet, A.; Agostinis, P. Mitochondria-Associated Membranes As Networking Platforms and Regulators of Cancer Cell Fate. Front. Oncol. 2017, 7, 174. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.; Pardo, B.; Szabadkai, G.; Duchen, M.R.; Satrustegui, J. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 2015, 593, 3447–3462. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.R.; Cordero-Nieves, H.M.; Erbe, R.S.; McAlees, J.W.; Bhatia, S.; Hodes, R.J.; Campbell, K.S.; Sanders, V.M. Prohibitins and the Cytoplasmic Domain of CD86 Cooperate To Mediate CD86 Signaling in B Lymphocytes. J. Immunol. 2013, 190, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Ande, S.R.; Mishra, S. Prohibitin interacts with phosphatidylinositol 3,4,5-triphosphate (PIP3) and modulates insulin signaling. Biochem. Biophys. Res. Commun. 2009, 390, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Fonteriz, R.; de la Fuente, S.; Moreno, A.; Lobaton, C.D.; Montero, M.; Alvarez, J. Monitoring mitochondrial [Ca2+] dynamics with rhod-2, ratiometric pericam and aequorin. Cell Calcium 2010, 48, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Naraghi, M.; Neher, E. Linearized Buffered Ca2+ Diffusion in Microdomains and Its Implications for Calculation of [Ca2+] at the Mouth of a Calcium Channel. J. Neurosci. 1997, 17, 6961–6973. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Xie, J.; Zabolocki, M.; Wang, X.; Jiang, T.; Wang, D.; Désaubry, L.; Bardy, C.; Proud, C.G. The prohibitin-binding compound fluorizoline affects multiple components of the translational machinery and inhibits protein synthesis. J. Biol. Chem. 2020, 295, 9855–9867. [Google Scholar] [CrossRef]
- Senft, A.P.; Dalton, T.P.; Shertzer, H.G. Determining Glutathione and Glutathione Disulfide Using the Fluorescence Probe o-Phthalaldehyde. Anal. Biochem. 2000, 280, 80–86. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saura-Esteller, J.; Sánchez-Vera, I.; Núñez-Vázquez, S.; Cosialls, A.M.; Gama-Pérez, P.; Bhosale, G.; Mendive-Tapia, L.; Lavilla, R.; Pons, G.; Garcia-Roves, P.M.; et al. Activation of the Integrated Stress Response and ER Stress Protect from Fluorizoline-Induced Apoptosis in HEK293T and U2OS Cell Lines. Int. J. Mol. Sci. 2021, 22, 6117. https://doi.org/10.3390/ijms22116117
Saura-Esteller J, Sánchez-Vera I, Núñez-Vázquez S, Cosialls AM, Gama-Pérez P, Bhosale G, Mendive-Tapia L, Lavilla R, Pons G, Garcia-Roves PM, et al. Activation of the Integrated Stress Response and ER Stress Protect from Fluorizoline-Induced Apoptosis in HEK293T and U2OS Cell Lines. International Journal of Molecular Sciences. 2021; 22(11):6117. https://doi.org/10.3390/ijms22116117
Chicago/Turabian StyleSaura-Esteller, José, Ismael Sánchez-Vera, Sonia Núñez-Vázquez, Ana M. Cosialls, Pau Gama-Pérez, Gauri Bhosale, Lorena Mendive-Tapia, Rodolfo Lavilla, Gabriel Pons, Pablo M. Garcia-Roves, and et al. 2021. "Activation of the Integrated Stress Response and ER Stress Protect from Fluorizoline-Induced Apoptosis in HEK293T and U2OS Cell Lines" International Journal of Molecular Sciences 22, no. 11: 6117. https://doi.org/10.3390/ijms22116117