
The Calcium/Calmodulin-Dependent Kinases II and IV as Therapeutic Targets in Neurodegenerative and Neuropsychiatric Disorders


Abstract
:
1. Introduction
2. CaMKII
2.1. Genetics
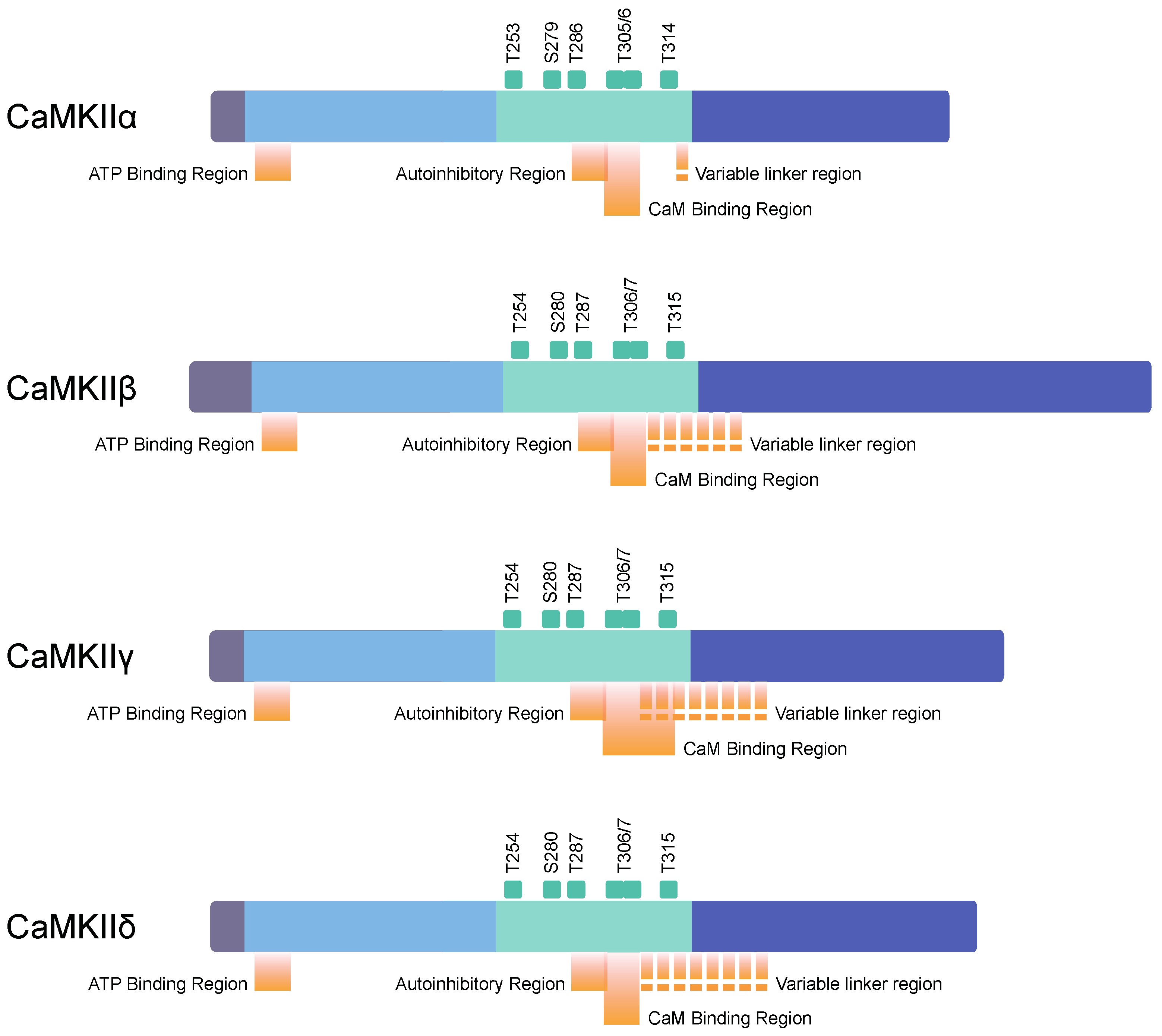
2.2. Structure of Subunits
2.3. Structure of the Holoenzyme
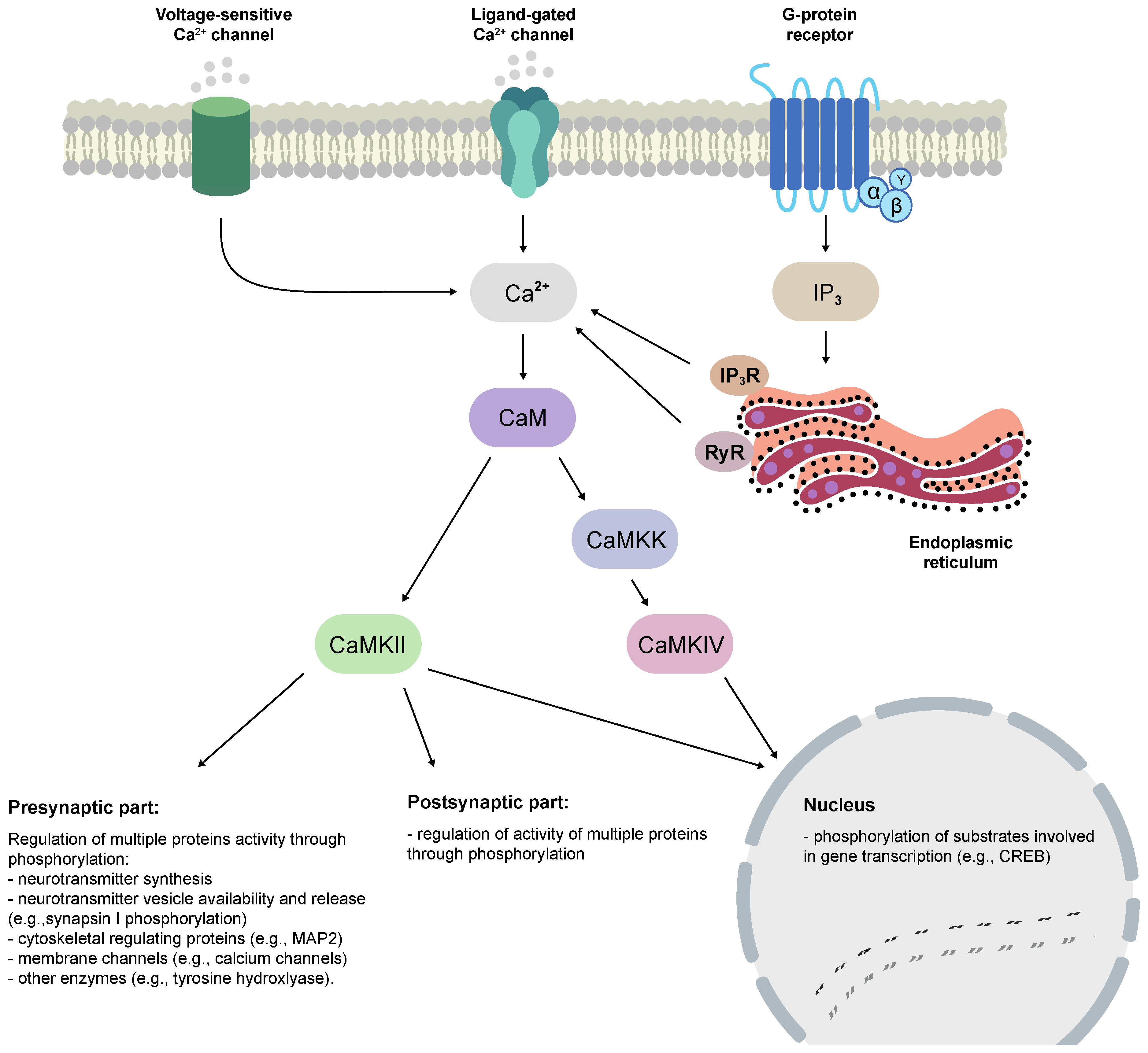
2.4. Mechanism of Activation and Autoregulation
2.5. CaM Trapping
2.6. Other Regulatory Factors
2.7. Function
3. CaMKIV
3.1. Genetics
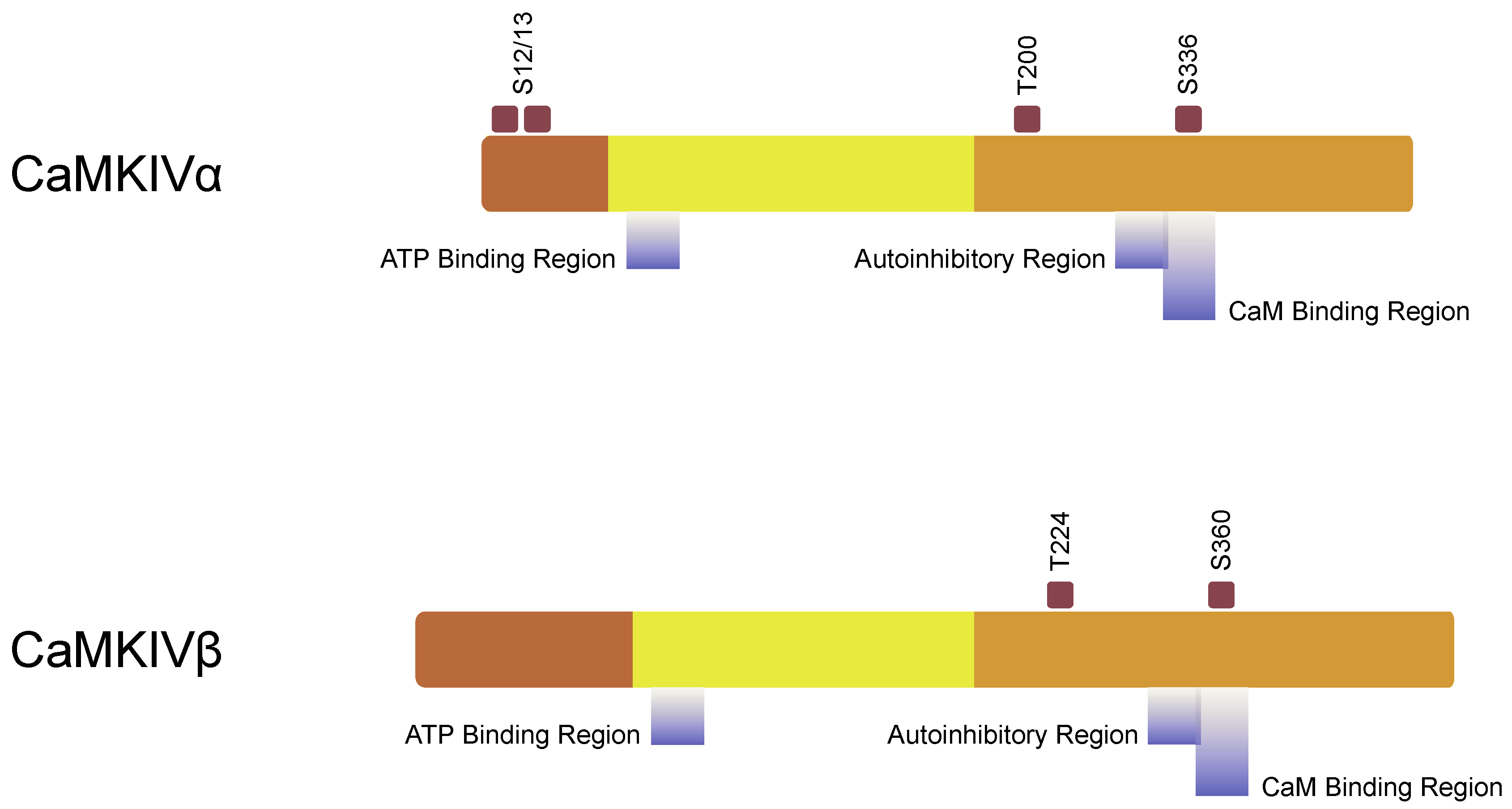
3.2. Structure of Subunit
3.3. Mechanism of Activation and Autoregulation
3.4. Other Regulatory Factors
3.5. Function
4. Role in Neurodegenerative and Neuropsychiatric Disorders
5. Depression-Like State in Rodents
5.1. CaMKII
5.2. CaMKIV
6. Anxiety-Like State in Rodents
6.1. CaMKII
6.2. CaMKIV
7. Memory and Its Impairment—Preclinical Studies
7.1. CaMKII
7.2. CaMKIV
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brzozowski, J.S.; Skelding, K.A. The multi-functional calcium/calmodulin stimulated protein kinase (CaMK) family: Emerging targets for anti-cancer therapeutic intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Soderling, T.R. Ca2+-calmodulin-dependent protein kinase cascade. Trends Biochem. Sci. 1999, 24, 232–236. [Google Scholar] [CrossRef]
- Hudmon, A.; Schulman, H. Calcium/Calmodulin-Dependent Protein Kinase II. In Encyclopedia of Biological Chemistry; Lennarz, W.J., Lane, M.D., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2004; pp. 274–280. ISBN 978-0-12-443710-4. [Google Scholar]
- Robison, J.; Colbran, R.J. Calcium/Calmodulin-Dependent Protein Kinases. In Encyclopedia of Biological Chemistry; Lennarz, W.J., Lane, M.D., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2004; pp. 281–286. ISBN 978-0-12-443710-4. [Google Scholar]
- Takemoto-Kimura, S.; Suzuki, K.; Horigane, S.I.; Kamijo, S.; Inoue, M.; Sakamoto, M.; Fujii, H.; Bito, H. Calmodulin kinases: Essential regulators in health and disease. J. Neurochem. 2017, 141, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Swulius, M.T.; Waxham, M.N. Ca2+/calmodulin-dependent protein kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hell, J.W. CaMKII: Claiming center stage in postsynaptic function and organization. Neuron 2014, 81, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Hatano, N.; Inuzuka, H.; Yokokura, S.; Nozaki, N.; Kobayashi, R. Mechanism of the generation of autonomous activity of Ca2+/calmodulin-dependent protein kinase IV. J. Biol. Chem. 2004, 279, 40296–40302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celano, E.; Tiraboschi, E.; Consogno, E.; D’Urso, G.; Mbakop, M.P.; Gennarelli, M.; De Bartolomeis, A.; Racagni, G.; Popoli, M. Selective regulation of presynaptic calcium/calmodulin-dependent protein kinase II by psychotropic drugs. Biol. Psychiatry 2003, 53, 442–449. [Google Scholar] [CrossRef]
- Pati, S.; Sood, A.; Mukhopadhyay, S.; Vaidya, V.A. Acute pharmacogenetic activation of medial prefrontal cortex excitatory neurons regulates anxiety-like behaviour. J. Biosci. 2018, 43, 85–95. [Google Scholar] [CrossRef]
- Iacono, L.L.; Gross, C. α-Ca2+/calmodulin-dependent protein kinase II contributes to the developmental programming of anxiety in serotonin receptor 1A knock-out mice. J. Neurosci. 2008, 28, 6250–6257. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, N.; Yamasaki, N.; Ohira, K.; Takao, K.; Toyama, K.; Eguchi, M.; Yamaguchi, S.; Miyakawa, T. Neural activity changes underlying the working memory defi cit in alpha-CaMKII heterozygous knockout mice. Front. Behav. Neurosci. 2009, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Gustin, R.M.; Shonesy, B.C.; Robinson, S.L.; Rentz, T.J.; Baucum, A.J.; Jalan-Sakrikar, N.; Winder, D.G.; Stanwood, G.D.; Colbran, R.J. Loss of Thr286 phosphorylation disrupts synaptic CaMKIIα targeting, NMDAR activity and behavior in pre-adolescent mice. Mol. Cell. Neurosci. 2011, 47, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Zhou, T.; Liao, L.; Yang, Z.; Wong, C.; Henn, F.; Malinow, R.; Yates, J.R.; Hu, H. βCaMKII in lateral habenula mediates core symptoms of depression. Science 2013, 341, 1016–1020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Feng, J.J.; Yang, S.; Liu, X.F.; Li, J.C.; Zhao, H. Lateral habenula as a link between thyroid and serotoninergic system modiates depressive symptoms in hypothyroidism rats. Brain Res. Bull. 2016, 124, 198–205. [Google Scholar] [CrossRef]
- Patki, G.; Solanki, N.; Atrooz, F.; Allam, F.; Salim, S. Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res. 2013, 1539, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Spanagel, R.; Zhang, S.J.; Bading, H.; Klugmann, M. Adeno-associated virus (AAV)-mediated suppression of Ca2+/calmodulin kinase IV activity in the nucleus accumbens modulates emotional behaviour in mice. BMC Neurosci. 2007, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Tobimatsu, T.; Kameshita, I.; Fujisawa, H. Molecular cloning of the cDNA encoding the third polypeptide (γ) of brain calmodulin-dependent protein kinase II. J. Biol. Chem. 1988, 263, 16082–16086. [Google Scholar] [CrossRef]
- Kanaseki, T.; Ikeuchi, Y.; Sugiura, H.; Yamauchi, T. Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy. J. Cell Biol. 1991, 115, 1049–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmstede, C.A.; Jensen, K.F.; Sahyoun, N.E. Ca2+/Calmodulin-dependent protein kinase enriched in cerebellar granule cells. Identification of a novel neuronal calmodulin-dependent protein kinase. J. Biol. Chem. 1989, 264, 5866–5875. [Google Scholar] [CrossRef]
- Means, A.R.; Cruzalegui, F.; LeMagueresse, B.; Needleman, D.S.; Slaughter, G.R.; Ono, T. A novel Ca2+/calmodulin-dependent protein kinase and a male germ cell-specific calmodulin-binding protein are derived from the same gene. Mol. Cell. Biol. 1991, 11, 3960–3971. [Google Scholar] [CrossRef] [Green Version]
- Boubali, S.; Liopeta, K.; Virgilio, L.; Thyphronitis, G.; Mavrothalassitis, G.; Dimitracopoulos, G.; Paliogianni, F. Calcium/calmodulin-dependent protein kinase II regulates IL-10 production by human T lymphocytes: A distinct target in the calcium dependent pathway. Mol. Immunol. 2012, 52, 51–60. [Google Scholar] [CrossRef]
- Bui, J.D.; Calbo, S.; Hayden-Martinez, K.; Kane, L.P.; Gardner, P.; Hedrick, S.M. A role for CaMKII in T cell memory. Cell 2000, 100, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yao, M.; Li, N.; Wang, C.; Zheng, Y.; Cao, X. CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 2008, 112, 4961–4970. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Brown, J.H. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 2004, 63, 476–486. [Google Scholar] [CrossRef]
- Maier, L.S.; Bers, D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Beckendorf, J.; van den Hoogenhof, M.M.G.; Backs, J. Physiological and unappreciated roles of CaMKII in the heart. Basic Res. Cardiol. 2018, 113, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pan, B.; Weyer, A.; Wu, H.E.; Meng, J.; Fischer, G.; Vilceanu, D.; Light, A.R.; Stucky, C.; Rice, F.L.; et al. CaMKII controls whether touch is painful. J. Neurosci. 2015, 35, 14086–14102. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaeser, F.; Ho, N.; Prywes, R.; Chatila, T.A. Ca2+-dependent gene expression mediated by MEF2 transcription factors. J. Biol. Chem. 2000, 275, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Lou, L.; Maurer, R.A. Regulation of activating transcription factor-1 and the cAMP response element-binding protein by Ca2+/calmodulin-dependent protein kinases type I, II, and IV. J. Biol. Chem. 1996, 271, 3066–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranti, C.K.; Ginty, D.D.; Huang, G.; Chatila, T.; Greenberg, M.E. Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca2+/calmodulin-dependent kinase. Mol. Cell. Biol. 1995, 15, 3672–3684. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G.; Cipolletta, E.; Sorriento, D.; Del Giudice, C.; Anastasio, A.; Monaco, S.; Maione, A.S.; Condorelli, G.; Puca, A.; Trimarco, B.; et al. CaMK4 gene deletion induces hypertension. J. Am. Heart Assoc. 2012, 1, e001081. [Google Scholar] [CrossRef] [Green Version]
- Krebs, J.; Wilson, A.; Kisielow, P. Calmodulin-dependent protein kinase IV during T-cell development. Biochem. Biophys. Res. Commun. 1997, 241, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Ribar, T.J.; Illario, M.; Means, A.R. Defective survival and activation of thymocytes in transgenic mice expressing a catalytically inactive form of Ca2+/calmodulin-dependent protein kinase IV. Mol. Endocrinol. 1997, 11, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Kitsos, C.M.; Sankar, U.; Illario, M.; Colomer-Font, J.M.; Duncan, A.W.; Ribar, T.J.; Reya, T.; Means, A.R. Calmodulin-dependent protein kinase IV regulates hematopoietic stem cell maintenance. J. Biol. Chem. 2005, 280, 33101–33108. [Google Scholar] [CrossRef] [Green Version]
- Gradin, H.M.; Marklund, U.; Larsson, N.; Chatila, T.A.; Gullberg, M. Regulation of microtubule dynamics by Ca2+/calmodulin-dependent kinase IV/Gr-dependent phosphorylation of oncoprotein 18. Mol. Cell. Biol. 1997, 17, 3459–3467. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, K. Unraveling the mechanisms of Angelman Syndrome. Nat. Neurosci. 2007, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Webster, S.J.; Tu, T.; Goulding, D.S.; Haiech, J.; Watterson, D.M.; Van Eldik, L.J. Generation and behavior characterization of CaMKIIβ knockout mice. PLoS ONE 2014, 9, e105191. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Dong, Y.; Lu, Y.; Tucker, D.; Wang, R.; Zhang, Q. Beneficial Effects of a CaMKIIα Inhibitor TatCN21 Peptide in Global Cerebral Ischemia. J. Mol. Neurosci. 2017, 61, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Naz, H.; Tarique, M.; Suhail, M.; Shankar, H.; Muhammad, N.; Usmani, D.; Ashraf, M.; Zughaibi, T.A. Calcium-/Calmodulin-Dependent Protein Kinase IV (CAMKIV): A Multifunctional Enzyme and Its Role in Various Cancer: An Update. Curr. Mol. Biol. Rep. 2020, 6, 139–147. [Google Scholar] [CrossRef]
- Moriguchi, S.; Inagaki, R.; Yi, L.; Shibata, M.; Sakagami, H.; Fukunaga, K. Nicotine Rescues Depressive-like Behaviors via α7-type Nicotinic Acetylcholine Receptor Activation in CaMKIV Null Mice. Mol. Neurobiol. 2020, 57, 4929–4940. [Google Scholar] [CrossRef] [PubMed]
- Skelding, K.A.; Rostas, J.A.P.; Verrills, N.M. Controlling the cell cycle: The role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle 2011, 10, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobimatsu, T.; Fujisawa, H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J. Biol. Chem. 1989, 264, 17907–17912. [Google Scholar] [CrossRef]
- Yamauchi, T. Neuronal Ca2+/calmodulin-dependent protein kinase II—Discovery, progress in a quarter of a century, and perspective: Implication for learning and memory. Biol. Pharm. Bull. 2005, 28, 1342–1354. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.R.; Kapiloff, M.S.; Durgerian, S.; Tatemoto, K.; Russo, A.F.; Hanson, P.; Schulman, H.; Rosenfeld, M.G. Molecular cloning of a brain-specific calcium/calmodulin-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1987, 84, 5962–5966. [Google Scholar] [CrossRef] [Green Version]
- Nishioka, N.; Shiojiri, M.; Kadota, S.; Morinaga, H.; Kuwahara, J.; Arakawa, T.; Yamamoto, S.; Yamauchi, T. Gene of rat Ca2+/calmodulin-dependent protein kinase TT a isoform—Its cloning and whole structure. FEBS Lett. 1996, 396, 333–336. [Google Scholar] [CrossRef] [Green Version]
- Sunyer, T.; Sahyoun, N. Sequence analysis and DNA-protein interactions within the 5′ flanking region of the Ca2+/calmodulin-dependent protein kinase II α-subunit gene. Proc. Natl. Acad. Sci. USA 1990, 87, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.K.; Kennedy, M.B. Deduced primary structure of the β subunit of brain type II Ca2+/calmodulin-dependent protein kinase determined by molecular cloning. Proc. Natl. Acad. Sci. USA 1987, 84, 1794–1798. [Google Scholar] [CrossRef] [Green Version]
- Donai, H.; Morinaga, H.; Yamauchi, T. Genomic organization and neuronal cell type specific promoter activity of β isoform of Ca2+/calmodulin dependent protein kinase II of rat brain. Mol. Brain Res. 2001, 94, 35–47. [Google Scholar] [CrossRef]
- Chao, L.H.; Pellicena, P.; Deindl, S.; Barclay, L.A.; Schulman, H.; Kuriyan, J. Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat. Struct. Mol. Biol. 2010, 17, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, O.S.; Deindl, S.; Sung, R.J.; Nairn, A.C.; Kuriyan, J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 2005, 123, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colbran, R.J.; Smith, M.K.; Schworer, C.M.; Fong, Y.L.; Soderling, T.R. Regulatory domain of calcium/calmodulin-dependent protein kinase II. Mechanism of inhibition and regulation by phosphorylaton. J. Biol. Chem. 1989, 264, 4800–4804. [Google Scholar] [CrossRef]
- Myers, J.B.; Zaegel, V.; Coultrap, S.J.; Miller, A.P.; Bayer, K.U.; Reichow, S.L. The CaMKII holoenzyme structure in activation-competent conformations. Nat. Commun. 2017, 8, 15742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rellos, P.; Pike, A.C.W.; Niesen, F.H.; Salah, E.; Lee, W.H.; von Delft, F.; Knapp, S. Structure of the CaMKIIδ/calmodulin complex reveals the molecular mechanism of CamKII kinase activation. PLoS Biol. 2010, 8, e1000426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodgett, J.R.; Davison, M.T.; Cohen, P. The calmodulin-dependent glycogen synthase kinase from rabbit skeletal muscle: Purification, subunit structure and substrate specificity. Eur. J. Biochem. 1983, 136, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Singla, S.I.; Hudmon, A.; Goldberg, J.M.; Smith, J.L.; Schulman, H. Molecular Characterization of Calmodulin Trapping by Calcium/Calmodulin-dependent Protein Kinase II. J. Biol. Chem. 2001, 276, 29353–29360. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.; Schulman, H. Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 1999, 274, 26199–26208. [Google Scholar] [CrossRef] [Green Version]
- Dupont, G.; Houart, G.; De Koninck, P. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations: A simple model. Cell Calcium 2003, 34, 485–497. [Google Scholar] [CrossRef]
- De Koninck, P.; Schulman, H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 1998, 279, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Migues, P.V.; Lehmann, I.T.; Fluechter, L.; Cammarota, M.; Gurd, J.W.; Sim, A.T.R.; Dickson, P.W.; Rostas, J.A.P. Phosphorylation of CaMKII at Thr253 occurs in vivo and enhances binding to isolated postsynaptic densities. J. Neurochem. 2006, 98, 289–299. [Google Scholar] [CrossRef]
- Hunter, T.; Schulman, H. CaMKII structure—An elegant design. Cell 2005, 123, 765–767. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, L.; Wong, C.C.L.; Li, G.; Xu, T.; Pajvani, U.; Park, S.K.R.; Wronska, A.; Chen, B.X.; Marks, A.R.; Fukamizu, A.; et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012, 15, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, L.; de Souza, J.C.; Harari, A.A.; Backs, J.; Olson, E.N.; Tabas, I. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab. 2013, 18, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toussaint, F.; Charbel, C.; Allen, B.G.; Ledoux, J. Vascular CaMKII: Heart and brain in your arteries. Am. J. Physiol. Cell Physiol. 2016, 311, C462–C478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahl, C.R.; Means, A.R. Regulation of Cell Cycle Progression by Calcium/Calmodulin-Dependent Pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backs, J.; Stein, P.; Backs, T.; Duncan, F.E.; Grueter, C.E.; McAnally, J.; Qi, X.; Schultz, R.M.; Olson, E.N. The γ isoform of CaM kinase II controls mouse egg activation by regulating cell cycle resumption. Proc. Natl. Acad. Sci. USA 2010, 107, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.Y.; Huo, L.J.; Meng, X.Q.; Zhong, Z.S.; Hou, Y.; Chen, D.Y.; Sun, Q.Y. Involvement of Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII) in Meiotic Maturation and Activation of Pig Oocytes. Biol. Reprod. 2003, 69, 1552–1564. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Xia, T.; Cui, Y.; Chu, S.; Ma, Z.; Gu, X. The role of CaMKII in neuropathic pain and fear memory in chronic constriction injury in rats. Int. J. Neurosci. 2019, 129, 146–154. [Google Scholar] [CrossRef]
- Wang, C.; Li, N.; Liu, X.; Zheng, Y.; Cao, X. A novel endogenous human CaMKII inhibitory protein suppresses tumor growth by inducing cell cycle arrest via p27 stabilization. J. Biol. Chem. 2008, 283, 11565–11574. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; An, P.; Quan, X.J.; Zhang, J.; Zhou, Z.Y.; Zou, L.P.; Luo, H.S. Ca2+/calmodulin-dependent protein kinase II regulates colon cancer proliferation and migration via ERK1/2 and p38 pathways. World J. Gastroenterol. 2017, 23, 6111–6118. [Google Scholar] [CrossRef]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K.; et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Guo, S.; Liu, Z.; Wu, L.; Li, M.; Yang, J.; Chen, R.; Liu, X.; Xu, H.; Cai, S.; et al. CAMK2N1 inhibits prostate cancer progression through androgen receptor-dependent signaling. Oncotarget 2014, 5, 10293–10306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, S.; Qian, Y.; Tang, J.; Liang, Z.; Zhang, M.; Si, J.; Li, X.; Huang, W.; Xu, R.; Wang, K. Ca2+/calmodulin-dependent protein kinase IIγ, a critical mediator of the NF-κB network, is a novel therapeutic target in non-small cell lung cancer. Cancer Lett. 2014, 344, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.W. Regulation of synaptic transmission by presynaptic CAMKII and BK channels. Mol. Neurobiol. 2008, 38, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, H.J.; Otmakhov, N.; El Gaamouch, F.; Lemelin, D.; De Koninck, P.; Lisman, J. CaMKII control of spine size and synaptic strength: Role of phosphorylation states and nonenzymatic action. Proc. Natl. Acad. Sci. USA 2010, 107, 14437–14442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taha, S.; Hanover, J.L.; Silva, A.J.; Stryker, M.P. Autophosphorylation of αCaMKII Is required for ocular dominance plasticity. Neuron 2002, 36, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Zybura, A.S.; Baucum, A.J.; Rush, A.M.; Cummins, T.R.; Hudmon, A. CaMKII enhances voltage-gated sodium channel Nav1.6 activity and neuronal excitability. J. Biol. Chem. 2020, 295, 11845–11865. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, B.; Ge, Q.; Wang, Z.W. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J. Neurosci. 2007, 27, 10404–10413. [Google Scholar] [CrossRef] [Green Version]
- Hinds, H.L.; Goussakov, I.; Nakazawa, K.; Tonegawa, S.; Bolshakov, V.Y. Essential function of α-calcium/calmodulin-dependent protein kinase II in neurotransmitter release at a glutamatergic central synapse. Proc. Natl. Acad. Sci. USA 2003, 100, 4275–4280. [Google Scholar] [CrossRef] [Green Version]
- He, X.P.; Yang, F.; Xie, Z.P.; Lu, B. Intracellular Ca2+ and Ca2+/calmodulin-dependent kinase II mediate acute potentiation of neurotransmitter release by neurotrophin-3. J. Cell Biol. 2000, 149, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Ashpole, N.M.; Song, W.; Brustovetsky, T.; Engleman, E.A.; Brustovetsky, N.; Cummins, T.R.; Hudmon, A. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J. Biol. Chem. 2012, 287, 8495–8506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bers, D.M.; Grandi, E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cardiovasc. Pharmacol. 2009, 54, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Lautermilch, N.J.; Watari, H.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Modulation of CaV2.1 channels by Ca2+/calmodulin- dependent protein kinase II bound to the C-terminal domain. Proc. Natl. Acad. Sci. USA 2008, 105, 341–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicole, O.; Bell, D.M.; Leste-Lasserre, T.; Doat, H.; Guillemot, F.; Pacary, E. A novel role for CAMKIIβ in the regulation of cortical neuron migration: Implications for neurodevelopmental disorders. Mol. Psychiatry 2018, 23, 2209–2226. [Google Scholar] [CrossRef]
- Puram, S.V.; Kim, A.H.; Ikeuchi, Y.; Wilson-Grady, J.T.; Merdes, A.; Gygi, S.P.; Azad, B. A Unique CaMKIIβ Signaling Pathway at the Centrosome Regulates Dendrite Patterning in the Brain. Nat. Neurosci. 2011, 14, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Xi, F.; Xu, R.-J.; Xu, J.-H.; Wang, W.-H.; Ma, J.-J.; Wang, F.; Ma, Y.-X.; Qi, S.-B.; Zhang, J.-C.; Zhang, H.-N.; et al. Calcium/calmodulin-dependent protein kinase II regulates mammalian axon growth by affecting F-actin length in growth cone. J. Cell. Physiol. 2019, 234, 23053–23065. [Google Scholar] [CrossRef]
- Fink, C.C.; Bayer, K.U.; Myers, J.W.; Ferrell, J.E.; Schulman, H.; Meyer, T. Selective regulation of neurite extension and synapse formation by the β but not the α isoform of CaMKII. Neuron 2003, 39, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, T.; Hisatsune, C.; Miyamoto, H.; Ogawa, N.; Mikoshiba, K. Regulation of spinogenesis in mature Purkinje cells via mGluR/PKC-mediated phosphorylation of CaMKII. Proc. Natl. Acad. Sci. USA 2017, 114, E5256–E5265. [Google Scholar]
- Giese, K.; Fedorov, N.; Filipowski, R.; Silva, A. Autophosphorylation of Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 1998, 279, 870–873. [Google Scholar] [CrossRef]
- Soderling, T.R.; Derkach, V.A. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000, 23, 75–80. [Google Scholar] [CrossRef]
- Miller, S.; Yasuda, M.; Coats, J.K.; Jones, Y.; Martone, M.E.; Mayford, M. Disruption of dendritic translation of CaMKIIα impairs stabilization of synaptic plasticity and memory consolidation. Neuron 2002, 36, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Ohmstede, C.A.; Bland, M.M.; Merrill, B.M.; Sahyoun, N. Relationship of genes encoding Ca2+/calmodulin-dependent protein kinase Gr and calspermin: A gene within a gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5784–5788. [Google Scholar] [CrossRef] [Green Version]
- Krebs, J. Calmodulin-dependent protein kinase IV: Regulation of function and expression. Biochim. Biophys. Acta 1998, 1448, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Kitani, T.; Okuno, S.; Fujisawa, H. cDNA cloning and expression of human calmodulin-dependent protein kinase IV. J. Biochem. 1994, 115, 637–640. [Google Scholar] [CrossRef]
- Calcium/Calmodulin-Dependent Protein Kinase Type IV. Available online: https://www.uniprot.org/uniprot/P13234 (accessed on 1 March 2021).
- Naz, H.; Islam, A.; Ahmad, F.; Hassan, M.I. Calcium/calmodulin-dependent protein kinase IV: A multifunctional enzyme and potential therapeutic target. Prog. Biophys. Mol. Biol. 2016, 121, 54–65. [Google Scholar] [CrossRef]
- Selbert, M.A.; Anderson, K.A.; Huang, Q.H.; Goldstein, E.G.; Means, A.R.; Edelmani, A.M. Phosphorylation and activation of Ca2+-calmodulin-dependent protein kinase IV by Ca2+-calmodulin-dependent protein kinase Ia kinase: Phosphorylation of threonine 196 is essential for activation. J. Biol. Chem. 1995, 270, 17616–17621. [Google Scholar] [CrossRef] [Green Version]
- Tokumitsu, H.; Brickey, D.A.; Glod, J.; Hidaka, H.; Sikela, J.; Soderling, T.R. Activation mechanisms for Ca2+/calmodulin-dependent protein kinase IV. Identification of a brain CaM-kinase IV kinase. J. Biol. Chem. 1994, 269, 28640–28647. [Google Scholar] [CrossRef]
- Bias, W.B.; Cheung, W.D.; Wang, Z.; Hart, G.W. Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J. Biol. Chem. 2009, 284, 21327–21337. [Google Scholar]
- Guan, Y.; Chen, Q.; Yang, X.; Haines, P.; Pei, M.; Terek, R.; Wei, X.; Zhao, T.; Wei, L. Subcellular relocation of histone deacetylase 4 regulates growth plate chondrocyte differentiation through Ca2+/calmodulin-dependent kinase IV. Am. J. Physiol. Cell Physiol. 2012, 303, 33–41. [Google Scholar] [CrossRef]
- Sato, K.; Suematsu, A.; Nakashima, T.; Takemoto-Kimura, S.; Aoki, K.; Morishita, Y.; Asahara, H.; Ohya, K.; Yamaguchi, A.; Takai, T.; et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat. Med. 2006, 12, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Barbosa-Sampaio, H.; Jones, P.M.; Persaud, S.J.; Muller, D.S. The CaMK4/CREB/IRS-2 Cascade Stimulates Proliferation and Inhibits Apoptosis of β-Cells. PLoS ONE 2012, 7, e45711. [Google Scholar]
- Illario, M.; Giardino-Torchia, M.L.; Sankar, U.; Ribar, T.J.; Galgani, M.; Vitiello, L.; Masci, A.M.; Bertani, F.R.; Ciaglia, E.; Astone, D.; et al. Calmodulin-dependent kinase IV links Toll-like receptor 4 signaling with survival pathway of activated dendritic cells. Blood 2008, 111, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Takai, N.; Miyazaki, T.; Nishida, M.; Nasu, K.; Miyakawa, I. Ca2+/calmodulin-dependent protein kinase IV expression in epithelial ovarian cancer. Cancer Lett. 2002, 183, 185–193. [Google Scholar] [CrossRef]
- Marín-Briggiler, C.I.; Jha, K.N.; Chertihin, O.; Buffone, M.G.; Herr, J.C.; Vazquez-Levin, M.H.; Visconti, P.E. Evidence of the presence of calcium/calmodulin-dependent protein kinase IV in human sperm and its involvement in motility regulation. J. Cell Sci. 2005, 118, 2013–2022. [Google Scholar] [CrossRef] [Green Version]
- Bhoumik, A.; Saha, S.; Majumder, G.C.; Dungdung, S.R. Optimum calcium concentration: A crucial factor in regulating sperm motility in vitro. Cell Biochem. Biophys. 2014, 70, 1177–1183. [Google Scholar] [CrossRef]
- Nagendran, T.; Hardy, L.R. CaMKIV mediates distinct features of basal and activity-dependent dendrite complexity. Neuroscience 2011, 199, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Heiser, J.H.; Schuwald, A.M.; Sillani, G.; Ye, L.; Müller, W.E.; Leuner, K. TRPC6 channel-mediated neurite outgrowth in PC12 cells and hippocampal neurons involves activation of RAS/MEK/ERK, PI3K, and CAMKIV signaling. J. Neurochem. 2013, 127, 303–313. [Google Scholar] [CrossRef]
- Tai, Y.; Feng, S.; Ge, R.; Du, W.; Zhang, X.; He, Z.; Wang, Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J. Cell Sci. 2008, 121, 2301–2307. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, H.; Maeda, R.; Suzuki, R.; Suzuki, A.; Nomoto, M.; Toyoda, H.; Wu, L.J.; Xu, H.; Zhao, M.G.; Ueda, K.; et al. Upregulation of calcium/calmodulin-dependent protein kinase IV improves memory formation and rescues memory loss with aging. J. Neurosci. 2008, 28, 9910–9919. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Sun, L.D.; Atkins, C.M.; Soderling, T.R.; Wilson, M.A.; Tonegawa, S. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell 2001, 106, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Browne, C.A.; Hammack, R.; Lucki, I. Dysregulation of the Lateral Habenula in Major Depressive Disorder. Front. Synaptic Neurosci. 2018, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Robison, A.J. Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci. 2014, 37, 653–662. [Google Scholar] [CrossRef]
- Arruda-Carvalho, M.; Restivo, L.; Guskjolen, A.; Epp, J.R.; Elgersma, Y.; Josselyn, S.A.; Frankland, P.W. Conditional Deletion of α-CaMKII Impairs Integration of Adult-Generated Granule Cells into Dentate Gyrus Circuits and Hippocampus-Dependent Learning. J. Neurosci. 2014, 34, 11919–11928. [Google Scholar] [CrossRef] [Green Version]
- Salvi, S.S.; Pati, S.; Chaudhari, P.R.; Tiwari, P.; Banerjee, T.; Vaidya, V.A. Acute Chemogenetic Activation of CamKIIα-Positive Forebrain Excitatory Neurons Regulates Anxiety-Like Behaviour in Mice. Front. Behav. Neurosci. 2019, 13, 249. [Google Scholar] [CrossRef]
- Yamasaki, N.; Maekawa, M.; Kobayashi, K.; Kajii, Y.; Maeda, J.; Soma, M.; Takao, K.; Tanda, K.; Ohira, K.; Toyama, K.; et al. Alpha-CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol. Brain 2008, 1, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.; Fan, C.; Wang, P.; Li, Y.; Yang, M.; Yu, S.Y. Hippocampal CA1 βcaMKII mediates neuroinflammatory responses via COX-2/PGE2 signaling pathways in depression. J. Neuroinflamm. 2018, 15, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, S.; Tanaka, T.; Tagashira, H.; Narahashi, T.; Fukunaga, K. Novel nootropic drug sunifiram improves cognitive deficits via CaM kinase II and protein kinase C activation in olfactory bulbectomized mice. Behav. Brain Res. 2013, 242, 150–157. [Google Scholar] [CrossRef]
- Moriguchi, S.; Han, F.; Nakagawasai, O.; Tadano, T.; Fukunaga, K. Decreased calcium/calmodulin-dependent protein kinase II and protein kinase C activities mediate impairment of hippocampal long-term potentiation in the olfactory bulbectomized mice. J. Neurochem. 2006, 97, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Guan, S.; Yang, L.; Yang, L.; Wang, L. Cucurbitacin IIa exerts antidepressant-like effects on mice exposed to chronic unpredictable mild stress. Neuroreport 2017, 28, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Rhee, J.; Park, K.; Han, J.S.; Malinow, R.; Chung, C.H. Exposure to stressors facilitates long-term synaptic potentiation in the lateral habenula. J. Neurosci. 2017, 37, 6021–6030. [Google Scholar] [CrossRef] [Green Version]
- Suenaga, T.; Morinobu, S.; Kawano, K.I.; Sawada, T.; Yamawaki, S. Influence of immobilization stress on the levels of CaMKII and phospho-CaMKII in the rat hippocampus. Int. J. Neuropsychopharmacol. 2004, 7, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Suda, S.; Segi-Nishida, E.; Newton, S.S.; Duman, R.S. A Postpartum Model in Rat: Behavioral and Gene Expression Changes Induced by Ovarian Steroid Deprivation. Biol. Psychiatry 2018, 64, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kang, S.; Fu, R.; Wu, L.; Wu, W.; Liu, H.; Gregor, D.; Zuo, W.; Bekker, A.; Ye, J.H. Inhibition of AMPA receptor and CaMKII activity in the lateral habenula reduces depressive-like behavior and alcohol intake in rats. Neuropharmacology 2017, 126, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Popoli, M.; Brunello, N.; Perez, J.; Racagni, G. Second messenger-regulated protein kinases in the brain: Their functional role and the action of antidepressant drugs. J. Neurochem. 2000, 74, 21–33. [Google Scholar] [CrossRef]
- Robison, A.J.; Vialou, V.; Sun, H.S.; Labonte, B.; Golden, S.A.; Dias, C.; Turecki, G.; Tamminga, C.; Russo, S.; Mazei-Robison, M.; et al. Fluoxetine epigenetically alters the CaMKII promoter in nucleus accumbens to regulate δfosb binding and antidepressant effects. Neuropsychopharmacology 2014, 39, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, V.S.; Giambelli, R.; Musazzi, L.; Tiraboschi, E.; Tardito, D.; Perez, J.; Drago, F.; Racagni, G.; Popoli, M. Chronic antidepressants induce redistribution and differential activation of αcaM kinase II between presynaptic compartments. Neuropsychopharmacology 2007, 32, 2511–2519. [Google Scholar] [CrossRef] [Green Version]
- Popoli, M.; Mori, S.; Brunello, N.; Perez, J.; Gennarelli, M.; Racagni, G. Serine/threonine kinases as molecular targets of antidepressants: Implications for pharmacological treatment and pathophysiology of affective disorders. Pharmacol. Ther. 2001, 89, 149–170. [Google Scholar] [CrossRef]
- Du, J.; Szabo, S.T.; Gray, N.A.; Manji, H.K. Focus on CaMKII: A molecular switch in the pathophysiology and treatment of mood and anxiety disorders. Int. J. Neuropsychopharmacol. 2004, 7, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Popoli, M.; Gennarelli, M.; Racagni, G. Modulation of synaptic plasticity by stress and antidepressants. Bipolar Disord. 2002, 4, 166–182. [Google Scholar] [CrossRef]
- Consogno, E.; Racagni, G.; Popoli, M. Modifications in brain CaM kinase II after long-term treatment with desmethylimipramine. Neuropsychopharmacology 2001, 24, 21–30. [Google Scholar] [CrossRef]
- Pilc, A.; Branski, P.; Palucha, A.; Aronowski, J. The effect of prolonged imipramine and electroconvulsive shock treatment on calcium/calmodulin-dependent protein kinase II in the hippocampus of rat brain. Neuropharmacology 1999, 38, 597–603. [Google Scholar] [CrossRef]
- Adaikkan, C.; Taha, E.; Barrera, I.; David, O.; Rosenblum, K. Calcium/Calmodulin-Dependent Protein Kinase II and Eukaryotic Elongation Factor 2 Kinase Pathways Mediate the Antidepressant Action of Ketamine. Biol. Psychiatry 2018, 84, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.H.; Zhang, G.F.; Xu, N.; Duan, G.F.; Jia, M.; Liu, R.; Zhou, Z.Q.; Yang, J.J.; Yang, J.J. Extrasynaptic CaMKIIα is involved in the antidepressant effects of ketamine by downregulating GluN2B receptors in an LPS-induced depression model. J. Neuroinflamm. 2020, 17, 1–19. [Google Scholar] [CrossRef]
- Han, F.; Nakano, T.; Yamamoto, Y.; Shioda, N.; Lu, Y.M.; Fukunaga, K. Improvement of depressive behaviors by nefiracetam is associated with activation of CaM kinases in olfactory bulbectomized mice. Brain Res. 2009, 1265, 205–214. [Google Scholar] [CrossRef]
- Xu, J.; Yabuki, Y.; Yu, M.; Fukunaga, K. T-type calcium channel enhancer SAK3 produces anti-depressant-like effects by promoting adult hippocampal neurogenesis in olfactory bulbectomized mice. J. Pharmacol. Sci. 2018, 137, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Meng, X.; Zhai, Y.; Ye, T.; Zhou, P.; Nan, F.; Sun, G.; Sun, X. Antidepressant-like effects of the Guanxin Danshen formula via mediation of the CaMK II-CREB-BDNF signalling pathway in chronic unpredictable mild stress-induced depressive rats. Ann. Transl. Med. 2019, 7, 564. [Google Scholar] [CrossRef]
- Sawamoto, A.; Okuyama, S.; Yamamoto, K.; Amakura, Y.; Yoshimura, M.; Nakajima, M.; Furukawa, Y. 3,5,6,7,8,31,41-Heptamethoxyflavone, a Citrus Flavonoid, Ameliorates Corticosterone-Induced Depression-like Behavior and Restores Brain-Derived Neurotrophic Factor Expression, Neurogenesis, and Neuroplasticity in the Hippocampus. Molecules 2016, 21, 541. [Google Scholar] [CrossRef] [Green Version]
- Cunha, M.P.; Budni, J.; Pazini, F.L.; Oliveira, Á.; Rosa, J.M.; Lopes, M.W.; Leal, R.B.; Rodrigues, A.L.S. Involvement of PKA, PKC, CAMK-II and MEK1/2 in the acute antidepressant-like effect of creatine in mice. Pharmacol. Rep. 2014, 66, 653–659. [Google Scholar] [CrossRef]
- Brod, L.M.P.; Fronza, M.G.; Vargas, J.P.; Lüdtke, D.S.; Brüning, C.A.; Savegnago, L. Modulation of PKA, PKC, CAMKII, ERK 1/2 pathways is involved in the acute antidepressant-like effect of (octylseleno)-xylofuranoside (OSX) in mice. Psychopharmacology 2017, 234, 717–725. [Google Scholar] [CrossRef]
- Ramos-Hryb, A.B.; Cunha, M.P.; Pazini, F.L.; Lieberknecht, V.; Prediger, R.D.S.; Kaster, M.P.; Rodrigues, A.L.S. Ursolic acid affords antidepressant-like effects in mice through the activation of PKA, PKC, CAMK-II and MEK1/2. Pharmacol. Rep. 2017, 69, 1240–1246. [Google Scholar] [CrossRef]
- Pesarico, A.P.; Sampaio, T.B.; Stangherlin, E.C.; Mantovani, A.C.; Zeni, G.; Nogueira, C.W. The antidepressant-like effect of 7-fluoro-1,3-diphenylisoquinoline-1-amine in the mouse forced swimming test is mediated by serotonergic and dopaminergic systems. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 54, 179–186. [Google Scholar] [CrossRef]
- Zeni, A.L.B.; Zomkowski, A.D.E.; Maraschin, M.; Rodrigues, A.L.S.; Tasca, C.I. Involvement of PKA, CaMKII, PKC, MAPK/ERK and PI3K in the acute antidepressant-like effect of ferulic acid in the tail suspension test. Pharmacol. Biochem. Behav. 2012, 103, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, R.C.; Souza, D.G.; Soletti, R.C.; López, M.G.; Rodrigues, A.L.S.; Gabilan, N.H. Involvement of PKA, MAPK/ERK and CaMKII, but not PKC in the acute antidepressant-like effect of memantine in mice. Neurosci. Lett. 2006, 395, 93–97. [Google Scholar] [CrossRef]
- Pesarico, A.P.; Stangherlin, E.C.; Rosa, S.G.; Mantovani, A.C.; Zeni, G.; Nogueira, C.W. Contribution of NMDA, GABAA and GABAB receptors and l-arginine-NO-cGMP, MEK1/2 and CaMK-II pathways in the antidepressant-like effect of 7-fluoro-1,3-diphenylisoquinoline-1-amine in mice. Eur. J. Pharmacol. 2016, 782, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Brod, L.M.P.; Fronza, M.G.; Vargas, J.P.; Lüdtke, D.S.; Luchese, C.; Wilhelm, E.A.; Savegnago, L. Involvement of monoaminergic system in the antidepressant-like effect of (octylseleno)-xylofuranoside in the mouse tail suspension test. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 65, 201–207. [Google Scholar] [CrossRef]
- Zeni, A.L.B.; Zomkowski, A.D.E.; Maraschin, M.; Rodrigues, A.L.S.; Tasca, C.I. Ferulic acid exerts antidepressant-like effect in the tail suspension test in mice: Evidence for the involvement of the serotonergic system. Eur. J. Pharmacol. 2012, 679, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Li, G.; Xu, H.; Wang, Y.; Shi, M. Baicalin ameliorates chronic mild stress-induced depression-like behaviors in mice and attenuates inflammatory cytokines and oxidative stress. Braz. J. Med. Biol. Res. 2019, 52, e8434. [Google Scholar] [CrossRef]
- Kim, Y.; Morath, B.; Hu, C.; Byrne, L.K.; Sutor, S.L.; Frye, M.A.; Tye, S.J. Antidepressant actions of lateral habenula deep brain stimulation differentially correlate with CaMKII/GSK3/AMPK signaling locally and in the infralimbic cortex. Behav. Brain Res. 2016, 306, 170–177. [Google Scholar] [CrossRef]
- Li, X.; Li, W.; Luo, J.; Wei, K.; Li, P.; Liu, X.B.; Min, S. Effects of propofol on the activation of hippocampal CaMKIIα in depressed rats receiving electroconvulsive therapy. J. ECT 2012, 28, 242–247. [Google Scholar] [CrossRef]
- Takao, K.; Tanda, K.; Nakamura, K.; Kasahara, J.; Nakao, K.; Katsuki, M.; Nakanishi, K.; Yamasaki, N.; Toyama, K.; Adachi, M.; et al. Comprehensive behavioral analysis of calcium/calmodulin-dependent protein kinase IV knockout mice. PLoS ONE 2010, 5, e9460. [Google Scholar] [CrossRef]
- Moriguchi, S.; Sakagami, H.; Yabuki, Y.; Sasaki, Y.; Izumi, H.; Zhang, C.; Han, F.; Fukunaga, K. Stimulation of Sigma-1 Receptor Ameliorates Depressive-like Behaviors in CaMKIV Null Mice. Mol. Neurobiol. 2015, 52, 1210–1222. [Google Scholar] [CrossRef]
- Tran, L.; Keele, N.B. CaMKIIα Knockdown Decreases Anxiety in the Open Field and Low Serotonin-Induced Upregulation of GluA1 in the Basolateral Amygdala. Behav. Brain Res. 2016, 303, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Shum, F.W.F.; Ko, S.W.; Lee, Y.S.; Kaang, B.K.; Zhuo, M. Genetic alteration of anxiety and stress-like behavior in mice lacking CaMKIV. Mol. Pain 2005, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Rainnie, D.G.; Greene, R.W.; Tonegawa, S. Abnormal fear response and aggressive behavior in mutant mice deficient for α-calcium-calmodulin kinase II. Science 1994, 266, 291–294. [Google Scholar] [CrossRef]
- Song, Z.X.; Chen, Q.; Ding, Q.; Zheng, F.; Li, C.W.; Xu, L.P.; Wang, H.B. Function of Ca2+-/calmodulin-dependent protein kinase IV in Ca2+-stimulated neuronal signaling and behavior. Sci. China Life Sci. 2015, 58, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Jackson, K.J.; Sanjakdar, S.S.; Chen, X.; Damaj, M.I. Nicotine Reward and Affective Nicotine Withdrawal Signs Are Attenuated in Calcium/Calmodulin-Dependent Protein Kinase IV Knockout Mice. PLoS ONE 2012, 7, e51154. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, S.; Furuichi, T.; Yoshida, T.; Endoh, K.; Kato, K.; Sado, M.; Maeda, R.; Kitamoto, A.; Miyao, T.; Suzuki, R.; et al. Transgenic up-regulation of alpha-CaMKII in forebrain leads to increased anxiety-like behaviors and aggression. Mol. Brain 2009, 2, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Need, A.C.; Giese, K.P. Handling and environmental enrichment do not rescue learning and memory impairments in αCamKIIT286A mutant mice. Genes Brain Behav. 2003, 2, 132–139. [Google Scholar] [CrossRef]
- Wei, F.; Qiu, C.-S.; Liauw, J.; Robinson, D.A.; Ho, N.; Chatila, T.; Zhuo, M. Calcium–calmodulin-dependent protein kinase IV is required for fear memory. Nat. Neurosci. 2002, 5, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Irvine, E.E.; Vernon, J.; Giese, K.P. αCaMKII autophosphorylation contributes to rapid learning but is not necessary for memory. Nat. Neurosci. 2005, 8, 411–412. [Google Scholar] [CrossRef]
- Lee, K.H.; Chatila, T.A.; Ram, R.A.; Thompson, R.F. Impaired Memory of Eyeblink Conditioning in CaMKIV KO Mice. Behav. Neurosci. 2009, 123, 438–442. [Google Scholar] [CrossRef]
- Boyden, E.S.; Katoh, A.; Pyle, J.L.; Chatila, T.A.; Tsien, R.W.; Raymond, J.L.L. Selective Engagement of Plasticity Mechanisms for Motor Memory Storage. Neuron 2006, 51, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.; Liauw, J.A.; Blaeser, F.; Wei, F.; Hanissian, S.; Muglia, L.M.; Wozniak, D.F.; Nardi, A.; Arvin, K.L.; Holtzman, D.M.; et al. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-Deficient mice. J. Neurosci. 2000, 20, 6459–6472. [Google Scholar] [CrossRef] [Green Version]
- Frankland, P.W.; Bontempi, B.; Talton, L.E.; Kaczmarek, L.; Silva, A.J. The Involvement of the Anterior Cingulate Cortex in Remote Contextual Fear Memory. Science 2004, 304, 881–883. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.J.; Paylor, R.; Wehner, J.M.; Tonegawa, S. Impaired spatial learning in α-calcium-calmodulin kinase II mutant mice. Science 1992, 257, 206–211. [Google Scholar] [CrossRef]
- Bach, M.E.; Hawkins, R.D.; Osman, M.; Kandel, E.R.; Mayford, M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal ltp in the range of the θ frequency. Cell 1995, 81, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Frankland, P.W.; O’Brien, C.; Ohno, M.; Kirkwood, A.; Silva, A.J. α-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature 2001, 411, 309–313. [Google Scholar] [CrossRef]
- Wu, L.J.; Zhang, X.H.; Fukushima, H.; Zhang, F.; Wang, H.; Toyoda, H.; Li, B.M.; Kida, S.; Zhuo, M. Genetic enhancement of trace fear memory and cingulate potentiation in mice overexpressing Ca2+/calmodulin-dependent protein kinase IV. Eur. J. Neurosci. 2008, 27, 1923–1932. [Google Scholar] [CrossRef]
- Steenland, H.W.; Wu, V.; Fukushima, H.; Kida, S.; Zhuo, M. CaMKIV over-expression boosts cortical 4-7 Hz oscillations during learning and 1-4 Hz delta oscillations during sleep. Mol. Brain 2010, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Soderling, T.R. CaM-kinases: Modulators of synaptic plasticity. Curr. Opin. Neurobiol. 2000, 10, 375–380. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, D.; Cui, T.T.; Li, J.F.; Gao, Y.Y.; Wang, N.Y.; Jia, P.L.; Zhang, H.Y.; Sun, Z.R.; Zou, W.; et al. Mechanisms Underlying the Antidepressant Effect of Acupuncture via the CaMK Signaling Pathway. Front. Behav. Neurosci. 2020, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Moriguchi, S. Stimulation of the sigma-1 receptor and the effects on neurogenesis and depressive behaviors in mice. Adv. Exp. Med. Biol. 2017, 964, 201–211. [Google Scholar] [PubMed]
- Moriguchi, S.; Inagaki, R.; Shimojo, H.; Sugimura, Y.; Fukunaga, K. Memantine Improves Depressive-Like Behaviors via Kir6.1 Channel Inhibition in Olfactory Bulbectomized Mice; IBRO: Paris, France, 2020; Volume 442, ISBN 8122795455. [Google Scholar]
- Song, N.; Nakagawa, S.; Izumi, T.; Toda, H.; Kato, A.; Boku, S.; Inoue, T.; Sakagami, H.; Li, X.; Koyama, T. Involvement of CaMKIV in neurogenic effect with chronic fluoxetine treatment. Int. J. Neuropsychopharmacol. 2013, 16, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Bandelow, B.; Michaelis, S. Epidemiology of anxiety disorders in the 21st century. Dialogues Clin. Neurosci. 2015, 17, 327–335. [Google Scholar] [PubMed]
- Bleier, J.; Toliver, A. Exploring the role of caMKIV in homeostatic plasticity. J. Neurosci. 2017, 37, 11520–11522. [Google Scholar] [CrossRef]
- Easton, A.C.; Lucchesi, W.; Schumann, G.; Giese, K.P.; Müller, C.P.; Fernandes, C. αcaMKII autophosphorylation controls exploratory activity to threatening novel stimuli. Neuropharmacology 2011, 61, 1424–1431. [Google Scholar] [CrossRef]
- Butler, R.K.; Sharko, A.C.; Oliver, E.M.; Brito-Vargas, P.; Kaigler, K.F.; Fadel, J.R.; Wilson, M.A. Activation of phenotypically-distinct neuronal subpopulations of the rat amygdala following exposure to predator odor. Neuroscience 2011, 175, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Shen, G.; Van Sickle, B.J.; Tietz, E.I. Calcium/calmodulin-dependent protein kinase II mediates hippocampal glutamatergic plasticity during benzodiazepine withdrawal. Neuropsychopharmacology 2010, 35, 1897–1909. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.E.; Das, P.; Gunning, W.T.; Tietz, E.I. Regulation of Ca2+/calmodulin-depenDent protein kinase II signaling within hippocampal glutamatergic postsynapses during flurazepam withdrawal. Neural Plast. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Soltani, H.; Sadat-Shirazi, M.S.; Pakpour, B.; Ashabi, G.; Zarrindast, M.R. Toxic effect of calcium/calmodulin kinase II on anxiety behavior, neuronal firing and plasticity in the male offspring of morphine-abstinent rats. Behav. Brain Res. 2020, 395, 112877. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, K.I.; Lin, H.C.; Lee, T.S. Genetic Deletion of Soluble Epoxide Hydroxylase Causes Anxiety-Like Behaviors in Mice. Mol. Neurobiol. 2019, 56, 2495–2507. [Google Scholar] [CrossRef]
- Zlomuzica, A.; Viggiano, D.; Degen, J.; Binder, S.; Ruocco, L.A.; Sadile, A.G.; Willecke, K.; Huston, J.P.; Dere, E. Behavioral alterations and changes in Ca/calmodulin kinase II levels in the striatum of connexin36 deficient mice. Behav. Brain Res. 2012, 226, 293–300. [Google Scholar] [CrossRef]
- Michalak, A.; Wnorowski, A.; Berardinelli, A.; Zinato, S.; Rusinek, J.; Budzyńska, B. Diazepam and SL-327 synergistically attenuate anxiety-like behaviours in mice—Possible hippocampal MAPKs specificity. Neuropharmacology 2020, 180, 108302. [Google Scholar] [CrossRef]
- Ji, Y.; Zhu, J.; Cuola, D.; Cui, J.; Shi, Y.; Xu, D.; Dang, W.; Zhu, Y. Memantine attenuated alcohol withdrawal-induced anxiety-like behaviors through down-regulating NR1-CaMKII-ERK signaling pathway. Neurosci. Lett. 2018, 686, 133–139. [Google Scholar]
- Wu, Z.M.; Ni, G.L.; Shao, A.M.; Cui, R. Genistein alleviates anxiety-like behaviors in post-traumatic stress disorder model through enhancing serotonergic transmission in the amygdala. Psychiatry Res. 2017, 255, 287–291. [Google Scholar] [CrossRef]
- Barker, A.; Jones, R.; Jennison, C. A prevalence study of age-associated memory impairment. Br. J. Psychiatry 1995, 167, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Stoppini, L.; Miyamoto, E.; Muller, D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1993, 268, 7863–7867. [Google Scholar] [CrossRef]
- Fukunaga, K.; Muller, D.; Miyamoto, E. Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long term potentiation. J. Biol. Chem. 1995, 270, 6119–6124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, C.F.; Tonegawa, S.; Wang, Y. The role of calcium-calmodulin kinase II in three forms of synaptic plasticity. Curr. Biol. 1994, 4, 687–693. [Google Scholar] [CrossRef]
- Sanhueza, M.; McIntyre, C.C.; Lisman, J.E. Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor. J. Neurosci. 2007, 27, 5190–5199. [Google Scholar] [CrossRef] [Green Version]
- Elgersma, Y.; Fedorov, N.B.; Ikonen, S.; Choi, E.S.; Elgersma, M.; Carvalho, O.M.; Giese, K.P.; Silva, A.J. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 2002, 36, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Chávez, A.E.; Mineur, Y.S.; Morimoto-Tomita, M.; Lutzu, S.; Kim, K.S.; Picciotto, M.R.; Castillo, P.E.; Tomita, S. CaMKII Phosphorylation of TARPγ-8 Is a Mediator of LTP and Learning and Memory. Neuron 2016, 92, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Mayford, M.; Bach, M.E.; Huang, Y.Y.; Wang, L.; Hawkins, R.D.; Kandel, E.R. Control of memory formation through regulated expression of a CaMKII transgene. Science 1996, 274, 1678–1683. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Kasbi, K.; Rothe, S.; Aziz, W.; Giese, K.P. Age-dependent changes in autophosphorylation of alpha calcium/calmodulin dependent kinase II in hippocampus and amygdala after contextual fear conditioning. Brain Res. Bull. 2017, 134, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.E. Roles of hippocampal nitric oxide and calcium/calmodulin-dependent protein kinase II in inhibitory avoidance learning in rats. Behav. Pharmacol. 2007, 18, 29–38. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Farb, C.R.; Bauer, E.P.; LeDoux, J.E.; Schafe, G.E. Pavlovian Fear Conditioning Regulates Thr286 Autophosphorylation of Ca2+/Calmodulin-Dependent Protein Kinase II at Lateral Amygdala Synapses. J. Neurosci. 2004, 24, 3281–3288. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, R.; Moriguchi, S.; Fukunaga, K. Aberrant Amygdala-dependent Fear Memory in Corticosterone-treated Mice. Neuroscience 2018, 388, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, Y.; Yanagawa, Y.; Imoto, K. Differential involvement of kinase activity of Ca2+/ calmodulin-dependent protein kinase IIα in hippocampus-and amygdala-dependent memory revealed by kinase-dead knock-in mouse. eNeuro 2018, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.R.; Escobedo-Lozoya, Y.; Szatmari, E.M.; Yasuda, R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 2009, 458, 299–304. [Google Scholar] [CrossRef]
- Tinsley, C.J.; Narduzzo, K.E.; Ho, J.W.; Barker, G.R.; Brown, M.W.; Warburton, E.C. A role for calcium-calmodulin-dependent protein kinase II in the consolidation of visual object recognition memory. Eur. J. Neurosci. 2009, 30, 1128–1139. [Google Scholar] [CrossRef]
- Wang, H.; Shimizu, E.; Tang, Y.P.; Cho, M.; Kyin, M.; Zuo, W.; Robinson, D.A.; Alaimo, P.J.; Zhang, C.; Morimoto, H.; et al. Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc. Natl. Acad. Sci. USA 2003, 100, 4287–4292. [Google Scholar] [CrossRef] [Green Version]
- Von Hertzen, L.S.J.; Giese, K.P. Alpha-isoform of Ca2+/calmodulin-dependent kinase II autophosphorylation is required for memory consolidation-specific transcription. Neuroreport 2005, 16, 1411–1414. [Google Scholar] [CrossRef]
- McGlade-McCulloh, E.; Yamamoto, H.; Tan, S.E.; Brickey, D.A.; Soderling, T.R. Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature 1993, 362, 640–642. [Google Scholar] [CrossRef]
- Raka, F.; Di Sebastiano, A.R.; Kulhawy, S.C.; Ribeiro, F.M.; Godin, C.M.; Caetano, F.A.; Angers, S.; Ferguson, S.S.G. Ca2+/Calmodulin-dependent protein Kinase II interacts with group i Metabotropic Glutamate and facilitates Receptor Endocytosis and ERK1/2 signaling: Role of β-Amyloid. Mol. Brain 2015, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Liu, W.; Yan, Z. Β-amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 2009, 284, 10639–10649. [Google Scholar] [CrossRef] [Green Version]
- Min, D.; Guo, F.; Zhu, S.; Xu, X.; Mao, X.; Cao, Y.; Lv, X.; Gao, Q.; Wang, L.; Chen, T.; et al. The alterations of Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of Alzheimer’s disease and vascular dementia. Neurosci. Lett. 2013, 538, 60–65. [Google Scholar] [CrossRef]
- Zhao, D.; Watson, J.B.; Xie, C.W. Amyloid β prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J. Neurophysiol. 2004, 92, 2853–2858. [Google Scholar] [CrossRef]
- Jiang, X.; Chai, G.S.; Wang, Z.H.; Hu, Y.; Li, X.G.; Ma, Z.W.; Wang, Q.; Wang, J.Z.; Liu, G.P. Spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice. Sci. Rep. 2015, 5, 9488. [Google Scholar] [CrossRef] [Green Version]
- Reese, L.C.; Laezza, F.; Woltjer, R. Dysregulated phosphorylation of Ca2+/calmodulin-dependent protein kinase II-α in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s disease. J. Neurochem. 2014, 119, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.J.; Chen, G.H.; Hu, X.Y.; Lu, Y.P.; Zhou, J.N.; Liu, R.Y. The expression of calcium/calmodulin-dependent protein kinase II-α in the hippocampus of patients with Alzheimer’s disease and its links with AD-related pathology. Brain Res. 2005, 1031, 101–108. [Google Scholar] [CrossRef]
- Gandy, S.; Czernik, A.J.; Greengard, P. Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. USA 1988, 85, 6218–6221. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Hiragami, Y.; Murayama, M.; Ishizuka, K.; Kawahara, M.; Takashima, A. Phosphorylation of tau at serine 416 by Ca2+/calmodulin- dependent protein kinase II in neuronal soma in brain. J. Neurochem. 2005, 94, 1438–1447. [Google Scholar] [CrossRef]
- Mah, V.H.; Eskin, T.A.; Kazee, A.M.; Lapham, L.; Higgins, G.A. In situ hybridization of calcium/calmodulin dependent protein kinase II and tau mRNAs: Species differences and relative preservation in Alzheimer’s disease. Mol. Brain Res. 1992, 12, 85–94. [Google Scholar] [CrossRef]
- Simonian, N.A.; Elvhage, T.; Czernik, A.J.; Greengard, P.; Hyman, B.T. Calcium/calmodulin-dependent protein kinase II immunostaining is preserved in Alzheimer’s disease hippocampal neurons. Brain Res. 1994, 657, 294–299. [Google Scholar] [CrossRef]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007, 25, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.R.; Cheng, X.R.; Zhou, W.X.; Zhang, Y.X. Age-related expression of calcium/calmodulin-dependent protein kinase II A in the hippocampus and cerebral cortex of senescence accelerated mouse prone/8 mice is modulated by anti-Alzheimer’s disease drugs. Neuroscience 2009, 159, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S. Pharmacological study on Alzheimer’s drugs targeting calcium/calmodulin- dependent protein kinase II. J. Pharmacol. Sci. 2011, 117, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, S.; Tagashira, H.; Sasaki, Y.; Yeh, J.Z.; Sakagami, H.; Narahashi, T.; Fukunaga, K. CaMKII activity is essential for improvement of memory-related behaviors by chronic rivastigmine treatment. J. Neurochem. 2014, 128, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhao, D.; Xie, C.W. Neurotrophins enhance CaMKII activity and rescue amyloid-β-induced deficits in hippocampal synaptic plasticity. J. Alzheimer’s Dis. 2010, 21, 823–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.M.; Yang, Y.J.; Zhang, L.; Zhang, X.; Guan, F.F.; Zhang, L.F. Naringin enhances CaMKII activity and improves long-term memory in a mouse model of Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 5576–5586. [Google Scholar] [CrossRef] [Green Version]
- Nihei, M.K.; Desmond, N.L.; McGlothan, J.L.; Kuhlmann, A.C.; Guilarte, T.R. N-methyl-D-aspartate receptor subunit changes are associated with lead-induced deficits of long-term potentiation and spatial learning. Neuroscience 2000, 99, 233–242. [Google Scholar] [CrossRef]
- Toscano, C.D.; O’Callaghan, J.P.; Guilarte, T.R. Calcium/calmodulin-dependent protein kinase II activity and expression are altered in the hippocampus of Pb2+-exposed rats. Brain Res. 2005, 1044, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Farahmandfar, M.; Kadivar, M.; Naghdi, N. Possible interaction of hippocampal nitric oxide and calcium/calmodulin-dependent protein kinase II on reversal of spatial memory impairment induced by morphine. Eur. J. Pharmacol. 2015, 751, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Zhao, M.G.; Mercaldo, V.; Chen, T.; Descalzi, G.; Kida, S.; Zhuo, M. Calcium/calmodulin-dependent kinase IV contributes to translation-dependent early synaptic potentiation in the anterior cingulate cortex of adult mice. Mol. Brain 2010, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, S.; Piao, F.; Hong, Y.; Liu, P.; Zhao, Y. Arsenic down-regulates the expression of Camk4, an important gene related to cerebellar LTD in mice. Neurotoxicol. Teratol. 2009, 31, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Li, S.; Guo, Y.; Liu, X.; Yang, Y.; Guo, J.; Li, S.; Zhang, C.; Shang, L.; Piao, F. Subchronic exposure to arsenic represses the TH/TRβ1-CaMK IV signaling pathway in mouse cerebellum. Int. J. Mol. Sci. 2016, 17, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkadhi, K.A.; Srivareerat, M.; Tran, T.T. Intensification of long-term memory deficit by chronic stress and prevention by nicotine in a rat model of Alzheimer’s disease. Mol. Cell. Neurosci. 2010, 45, 289–296. [Google Scholar] [CrossRef]
- Yin, Y.; Gao, D.; Wang, Y.; Wang, Z.H.; Wang, X.; Ye, J.; Wu, D.; Fang, L.; Pi, G.; Yang, Y.; et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E3773–E3781. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, S.; Kita, S.; Yabuki, Y.; Inagaki, R.; Izumi, H.; Sasaki, Y.; Tagashira, H.; Horie, K.; Takeda, J.; Iwamoto, T.; et al. Reduced CaM Kinase II and CaM Kinase IV Activities Underlie Cognitive Deficits in NCKX2 Heterozygous Mice. Mol. Neurobiol. 2018, 55, 3889–3900. [Google Scholar] [CrossRef]
- Miyano, O.; Kameshita, I.; Fujisawa, H. Purification and characterization of a brain-specific multifunctional calmodulin-dependent protein kinase from rat cerebellum. J. Biol. Chem. 1992, 267, 1198–1203. [Google Scholar] [CrossRef]
- Cai, B.; Ye, S.; Wang, T.-T.; Wang, Y.; Li, J.; Zhan, J.-X.; Shen, G.-M. Genistein protects hippocampal neurons against injury by regulating calcium/calmodulin dependent protein kinase IV protein levels in alzheimer’s disease model rats. Neural Regen. Res. 2017, 12, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Cárdenas, C.; Mei, L.; Cheung, K.H.; Foskett, J.K. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin-driven inositol trisphosphate receptor (InsP 3R) Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 13293–13298. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaMKII | CaMKIV | |
|---|---|---|
| Brain regions | prefrontal cortex [10,11], hippocampus [12,13], amygdala [14], lateral habenula [15,16] | hippocampus [17], nucleus accumbens [18] |
| Cell types | isoforms α and β: brain neurons; isoforms γ and δ: present in every examined tissue [19,20] | nervous tissue (mostly in cerebellum’s granular cells [21]), thymus (especially T-lymphocytes), spleen, and testis [22] |
| Function | regulation of various cellular functions by phosphorylating a wide range of substrates, e.g., regulation of immune system [23,24,25], cardiac function [26,27,28], pain [29] | regulation of various cellular functions by phosphorylating a wide range of substrates, e.g., controlling transcription factors such as CREB, ATF-1, SRF [30,31,32,33], blood pressure [34], immune system [35,36,37], microtubule dynamics [38] |
| Examples of dysfunction and associated disease-like state in rodents | abnormal phosphorylation—Angelman syndrome [39], β isoform overexpression—depressive-like symptoms [15], β isoform knockout—anxiety-related behavior [40], inhibition of α isoform expression—anxiolytic-like effect [41] | overexpression and increased activity—various types of cancer [42], knockout or inhibition—depressive-like state [17,43] |
| CaMKII | CaMKIV | ||||||
|---|---|---|---|---|---|---|---|
| Genetic Manipulation | Behavioral Assay | Effect in Rodents | Reference | Genetic Manipulation | Behavioral Assay | Effect in Rodents | Reference |
| Homozygous CaMKIIα knockout | FST | no changes | [116] | CaMKIV knockout | FST, TST | no changes | [153] |
| Heterozygous CaMKIIα knockout | FST | depressive-like state | [118] | homozygous CaMKIV knockout | FST, TST | depressive-like state | [154] |
| CaMKIIβ overexpression | FST, SPT | antidepressant-like effect | [15,119] | - | - | - | - |
| CaMKIIβ knockdown (hippocampus) in UCMS rats | SPT, FST | antidepressant-like effect | [119] | - | - | - | - |
| CaMKIIα knockdown | OFT | anxiolytic-like effect | [155] | homozygous CaMKIV knockout | EPM, LDT, acoustic startle and prepulse inhibition | anxiolytic-like effect | [153,156] |
| Hetero- and homozygous CaMKIIα knockout | FC | anxiolytic-like effect | [157] | homozygous CaMKIV knockout | marble-burying test, light dark box | anxiolytic-like effect | [158] |
| Hetero- and homozygous CaMKIIβ knockout | EPM, OFT | anxiolytic-like effect | [40] | hetero- and homozygous CaMKIV knockout + nicotine dependence and withdrawal | conditioned place preference test | anxiolytic-like effect (during withdrawal) | [159] |
| CaMKIIα overexpression | OFT, EZM, RT | anxiety-like state | [160] | ||||
| CaMKIIα-T286 mutant mice | Morris water maze | memory impairment | [161] | homozygous CaMKIV knockout | FC | [158,162] | |
| CaMKIIα-T286 mutant mice | PA, cued FC | memory impairment | [163] | homozygous CaMKIV knockout | eyeblink conditioning test, vestibulo-ocular reflex testing | [164,165] | |
| Heterozygous CaMKIIα knockout | RAM, delayed alternation task | working memory impairment | [13,118] | homozygous CaMKIV knockout | Morris water maze, Barnes maze, RAM | [166] | |
| Heterozygous CaMKIIα knockout | FC | memory impairment | [157,167] | CaMKIV knockout | Barnes maze, RAM, PA | no changes | [153] |
| Heterozygous CaMKIIα knockout | Morris water maze | memory impairment | [168] | CaMKIV knockout | FC | memory impairment | [153] |
| CaMKII-Asp-286 | Barnes maze, FC | spatial memory impairnent, no effect on contextual memory | [169] | heterozygous CaMKIVα knockout | Morris water maze, FC | memory impairment | [113] |
| Heterozygous CaMKIIα knockout | Morris water maze, FC | long-term memory impairment | [170] | CaMKIV overexpression | FC | memory enhacement | [171,172] |
| Kinase-dead CaMKIIα (K42R)-knockin | Morris water maze, FC | spatial and contextual memory impairment | CaMKIV overexpression | PA, social recognition test | [112] | ||
| Hetero- and homozygous CaMKIIβ knockout | NOR | memory impairment | [40] | ||||
| Potential Modulator | Action | Animal Models/Tests | Reference |
|---|---|---|---|
| Antidepressant-Like Activity | |||
| KN-62 |
| FST, SPT, and ethanol withdrawal (in rats) | [126,141,142,143,144,146] |
| Paroxetine, venlafaxine, fluvoxamine (long treatment) |
| - | [127,130,131,132] |
| Fluoxetine (chronic) |
| chronic social defeat stress and SPT (in mice) | [128] |
| Fluoxetine, desipramine, Reboxetine (long treatment) |
| - | [129] |
| Imipramine (chronic) |
| congenitally learned helplessness (in rats) | [15,134] |
| Fluoxetine (chronic) |
| FST and SPT during chronic unpredictable mild stress and LPS-induced model of depression (in rats) | [119] |
| Fluvoxamine, desipramine (chronic) |
| - | [10] |
| Desmethylimipramine (chronic) |
| - | [133] |
| Ketamine |
| FST, TST, and NSFT (in mice) | [135] |
| Ketamine |
| OFT, FST, and NSFT during LPS-model of depression (in mice) | [136] |
| Sunifiram (chronic) |
| no effect in TST (in OB mice) | [120] |
| Nefiracetam (chronic) |
| TST and FST (in OB mice) | [137] |
| SAK3 (ethyl 8’-methyl-2’,4-dioxo-2-(piperidin-1-yl)-2’H-spiro[cyclopentane-1,3’-imidazo[1,2-a]pyridine]-2-ene-3-carboxylate) (chronic) |
| TST, FST, and SPT (in OB mice) | [138] |
| Guanxin Danshen formula |
| FST, TST, and SPT during unpredictable chronic mild stress (in rats) | [139] |
| 3,5,6,7,8,3,4′-Heptamethoxyflavone |
| FST and TST during corticosterone-induced depression (in mice) | [140] |
| Baicalin |
| FST, TST, and SPT during mice chronic unpredictable stress (in mice) | [150] |
| Ursolid acid, creatine, ferulic acid, (octylseleno)-xylofuranoside, memantine and 7-fluoro-1,3-diphenylisoquinoline-1-amine |
| FST or TST (in mice) | [141,142,143,144,145,146,147,148,149] |
| Nicotine |
| TST and FST (in CaMKIV knockout mice) | [43] |
| Fluoxetine (chronic) |
| SPT and OFT during chronic unpredictable stress (in rats) | [174] |
| Anxiolytic-like activity | |||
| TatCN21peptide |
| EPM and OFT (in rats) | [41] |
| Diazepam |
| OFT and EPM (in mice) | [187] |
| Memantine |
| OFT and EPM during chronic alcohol exposure (in rats) | [188] |
| Genistein |
| EPM and contextual freezing behavior during posttraumatic stress disorder (in rats) | [189] |
| Procognitive activity | |||
| Rivastigmine (chronic) |
| Y-maze task, NOR, passive avoidance task, and Barnes maze task (in OB mice) | [222] |
| Sunifiram |
| Y-maze task, NOR (in OB mice) | [120] |
| Nefiracetam |
| - | [221] |
| Naringin (chronic) |
| - | [224] |
| Genistein |
| special learning and memory in Morris water maze during Alzheimer ‘s disease model (in rats) | [235] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sałaciak, K.; Koszałka, A.; Żmudzka, E.; Pytka, K. The Calcium/Calmodulin-Dependent Kinases II and IV as Therapeutic Targets in Neurodegenerative and Neuropsychiatric Disorders. Int. J. Mol. Sci. 2021, 22, 4307. https://doi.org/10.3390/ijms22094307
Sałaciak K, Koszałka A, Żmudzka E, Pytka K. The Calcium/Calmodulin-Dependent Kinases II and IV as Therapeutic Targets in Neurodegenerative and Neuropsychiatric Disorders. International Journal of Molecular Sciences. 2021; 22(9):4307. https://doi.org/10.3390/ijms22094307
Chicago/Turabian StyleSałaciak, Kinga, Aleksandra Koszałka, Elżbieta Żmudzka, and Karolina Pytka. 2021. "The Calcium/Calmodulin-Dependent Kinases II and IV as Therapeutic Targets in Neurodegenerative and Neuropsychiatric Disorders" International Journal of Molecular Sciences 22, no. 9: 4307. https://doi.org/10.3390/ijms22094307