
Modulation of Tubular pH by Acetazolamide in a Ca2+ Transport Deficient Mice Facilitates Calcium Nephrolithiasis

 ,
,
Abstract
:
1. Introduction
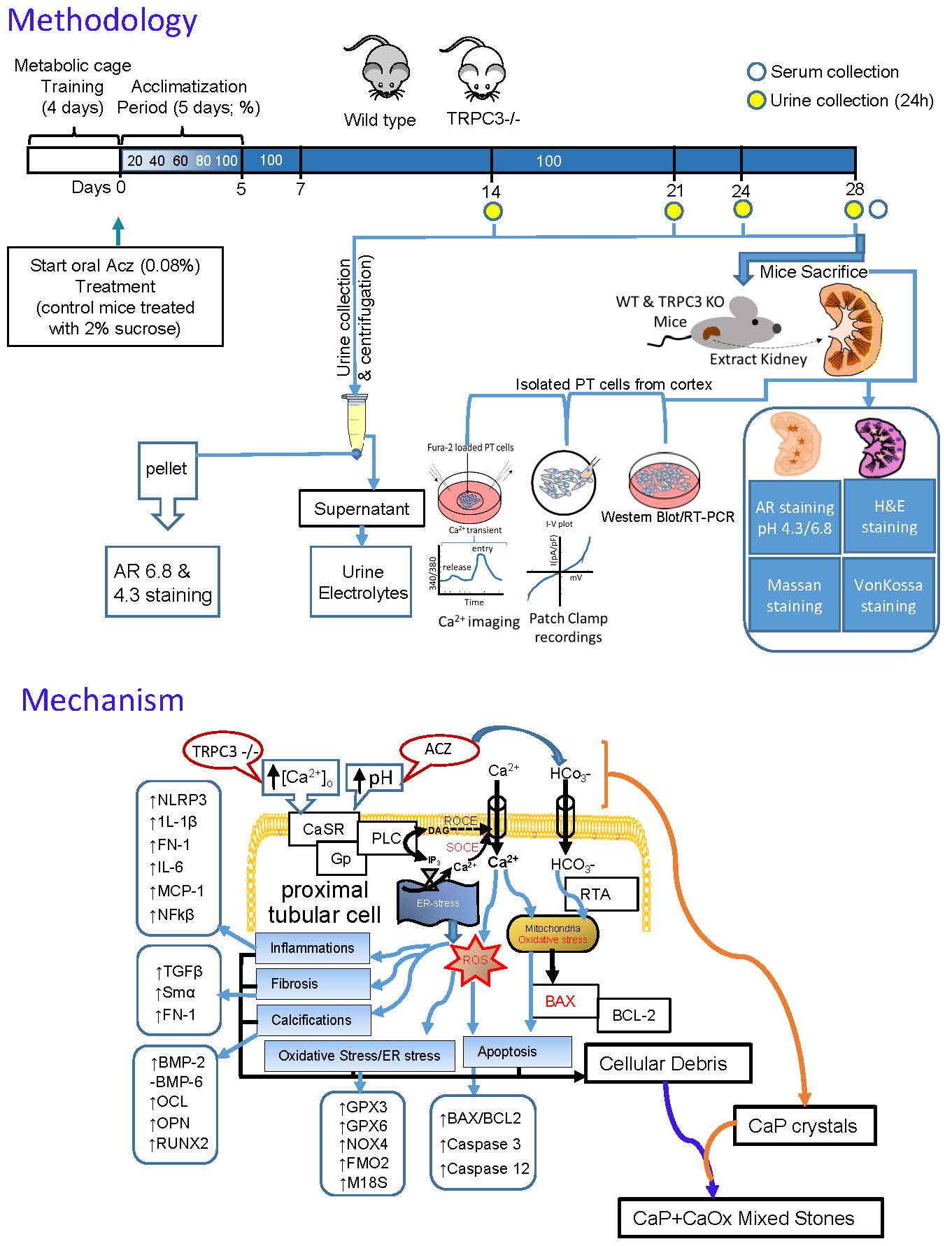
2. Results
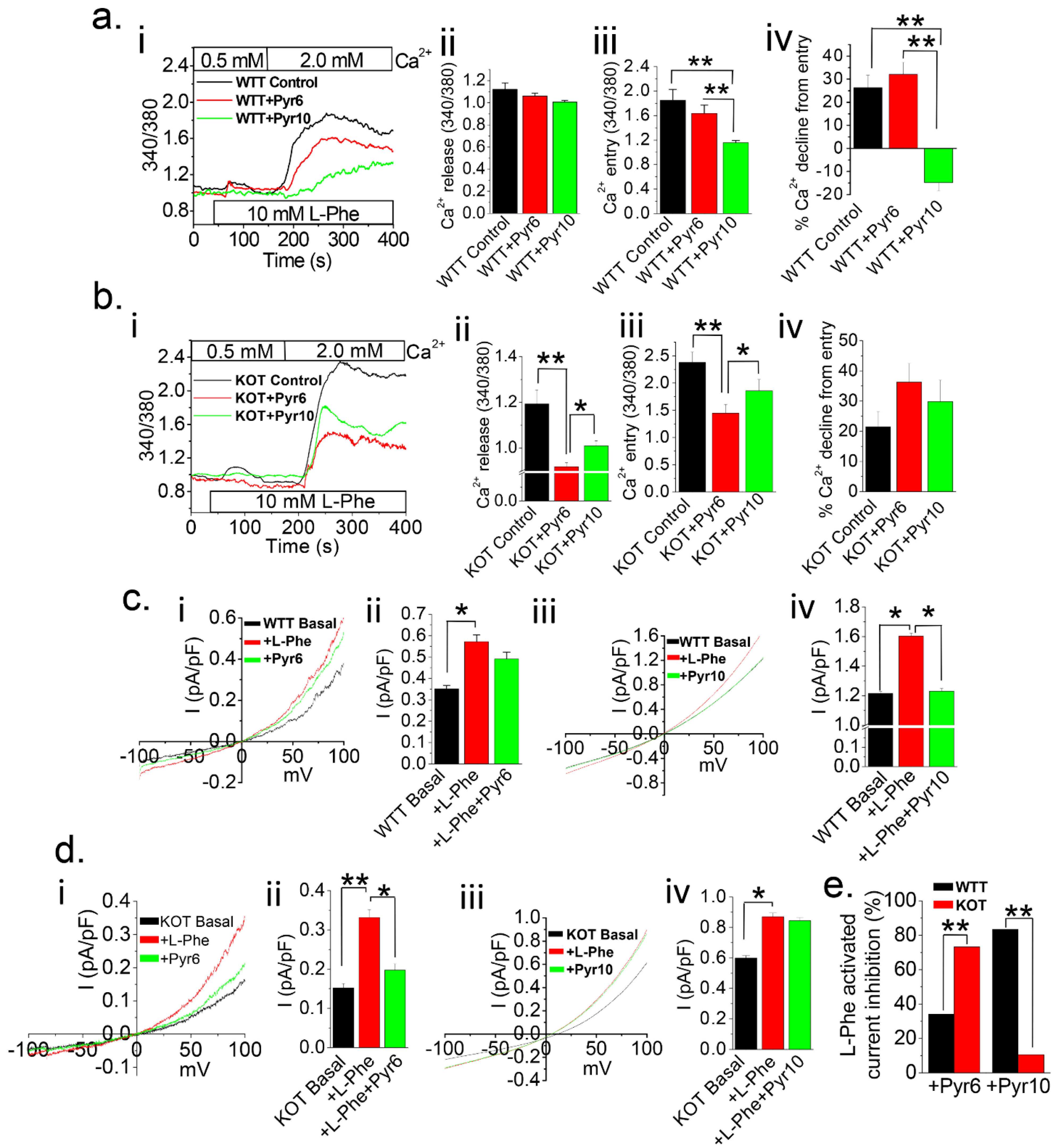
2.1. Urine Alkalization in Mice Increases Ca2+ Entry in PT Cells
2.2. Mode of Ca2+ Entry Switches the from Receptor-Operated Ca2+ Entry (ROCE) to Store-Operated Ca2+ Entry (SOCE) in PT Cells Following Urine Alkalization
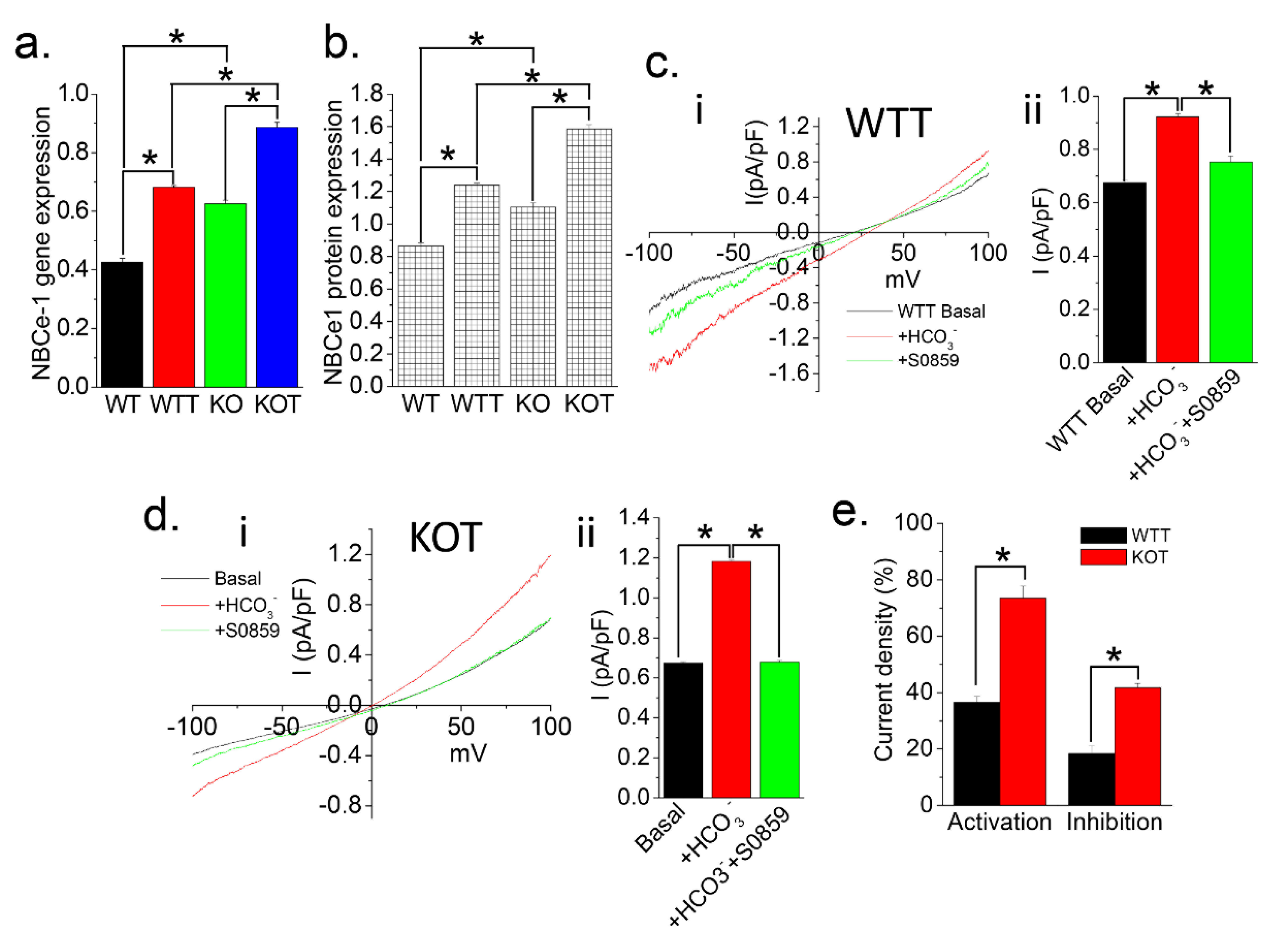
2.3. Urine Alkalization Enhances HCO3− Related Current Activity in PT Cells
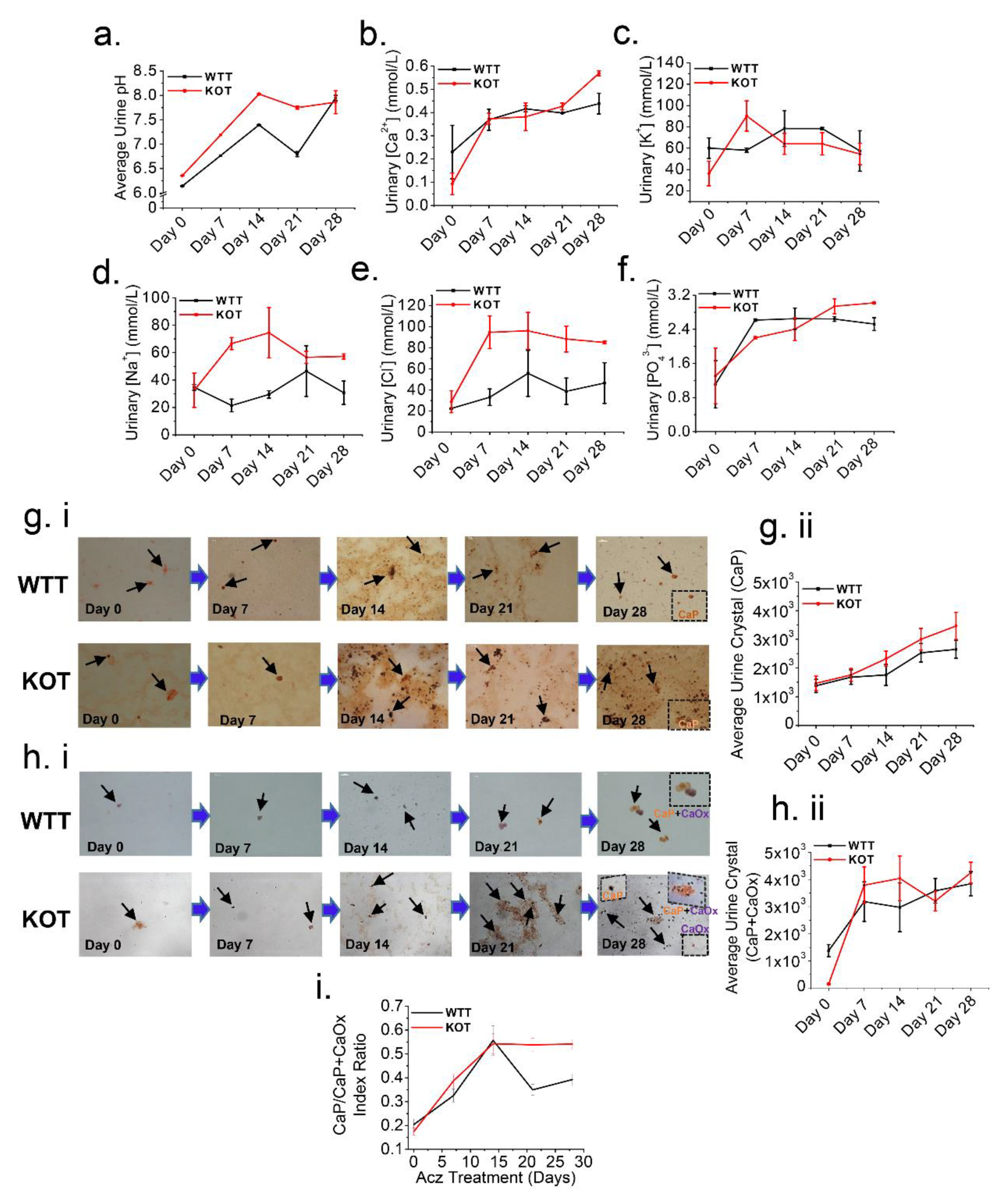
2.4. Acz-Induced Alkaluria Determines the Type (CaP or CaP+CaOx) of Calcium Stone
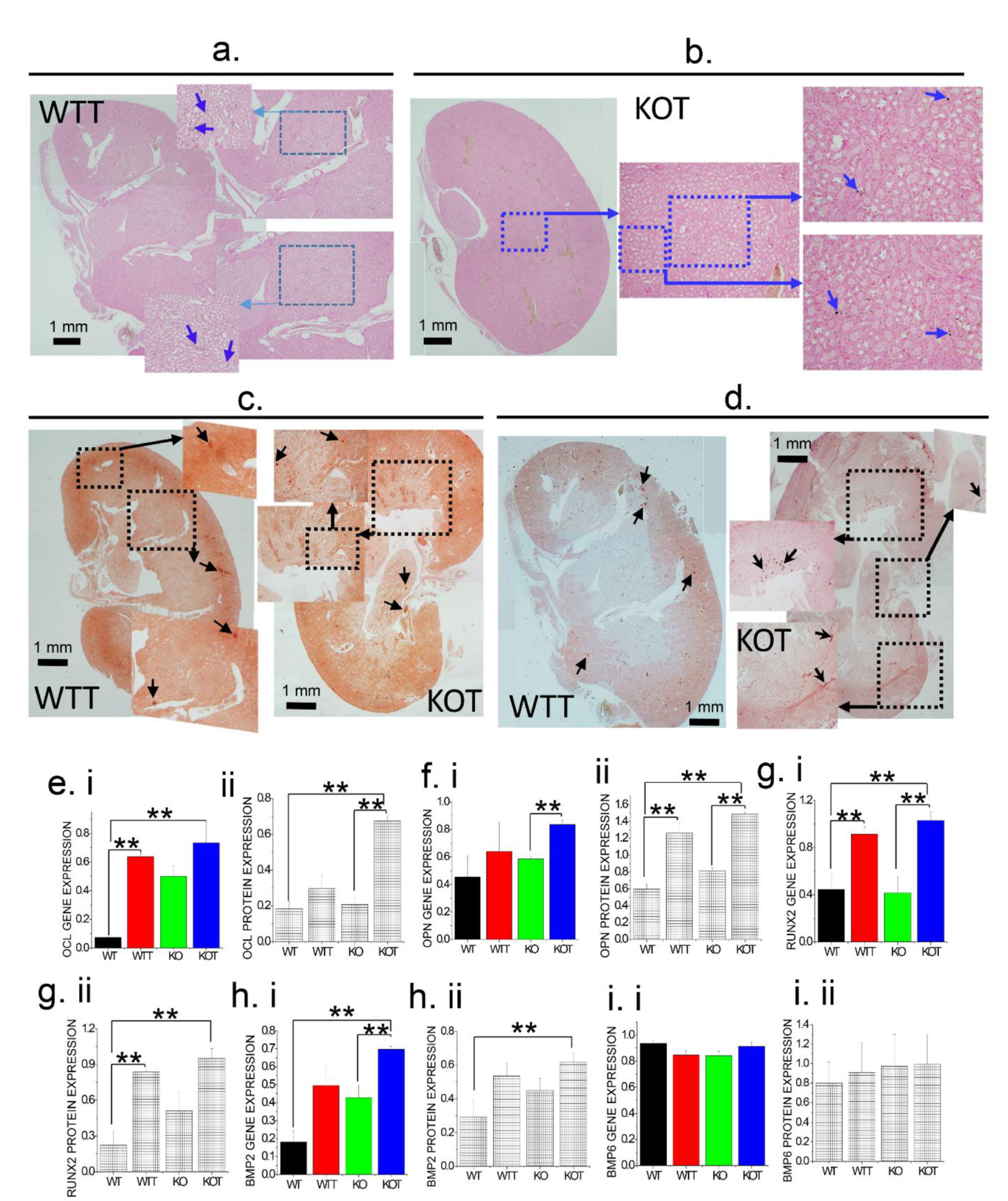
2.5. Alkalization-Induced Calcification Was Upregulated in Absence of TRPC3
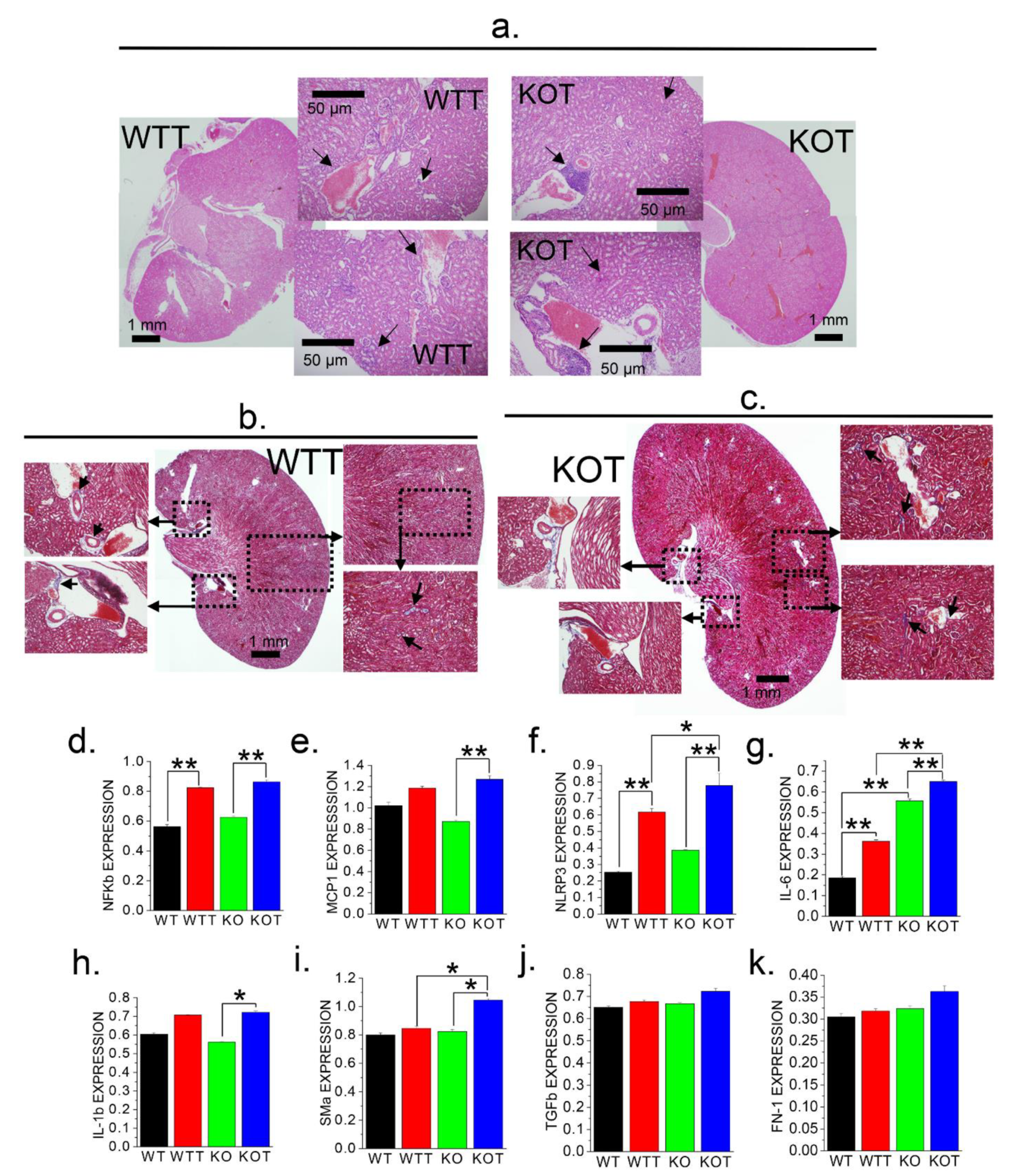
2.6. Alkalization Induced by Acz Results in Inflammatory and Fibrotic Responses in PT Cells
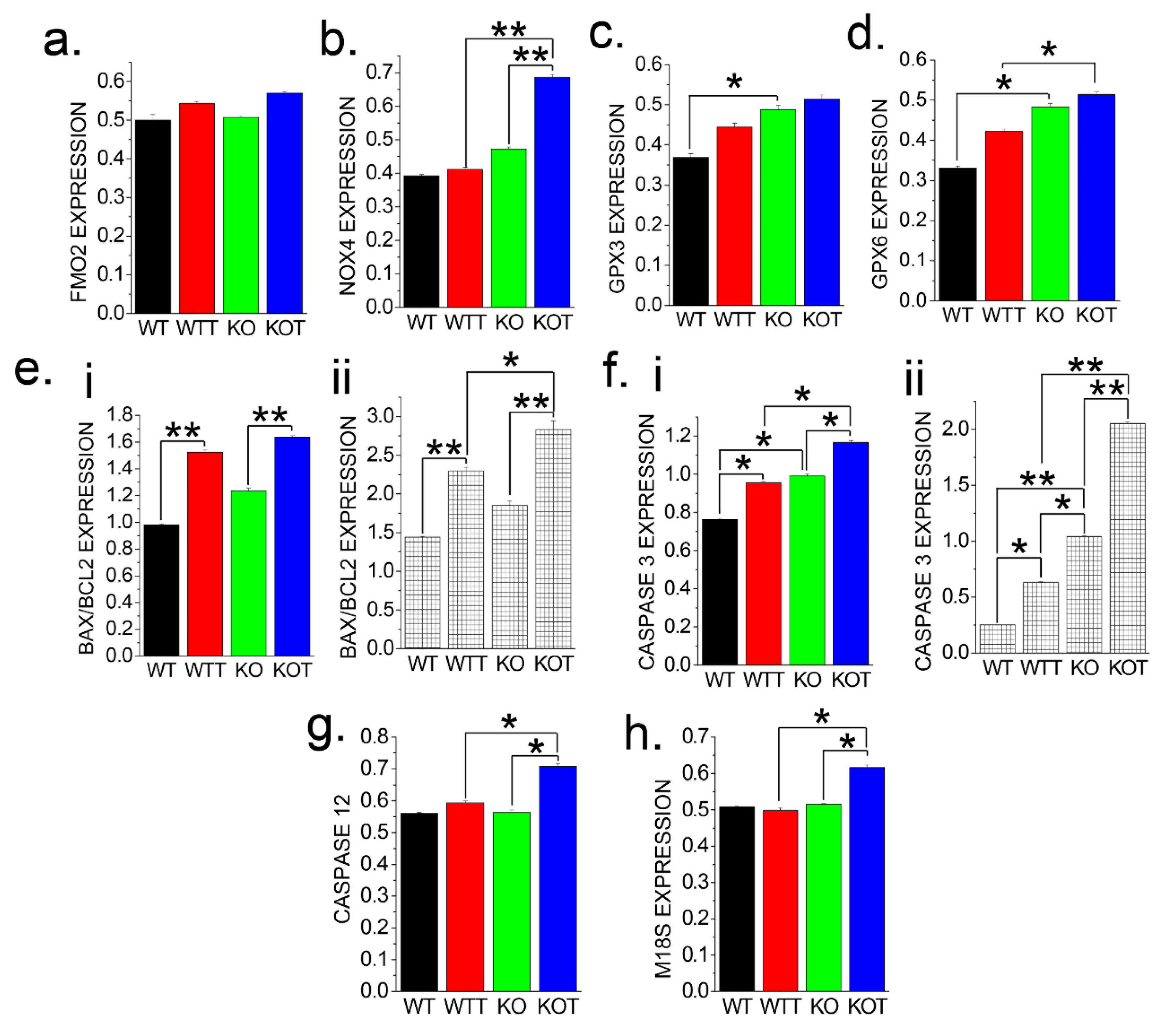
2.7. Oxidative Stress Genes in PT Cells Are Upregulated Due to ALKALIZATION
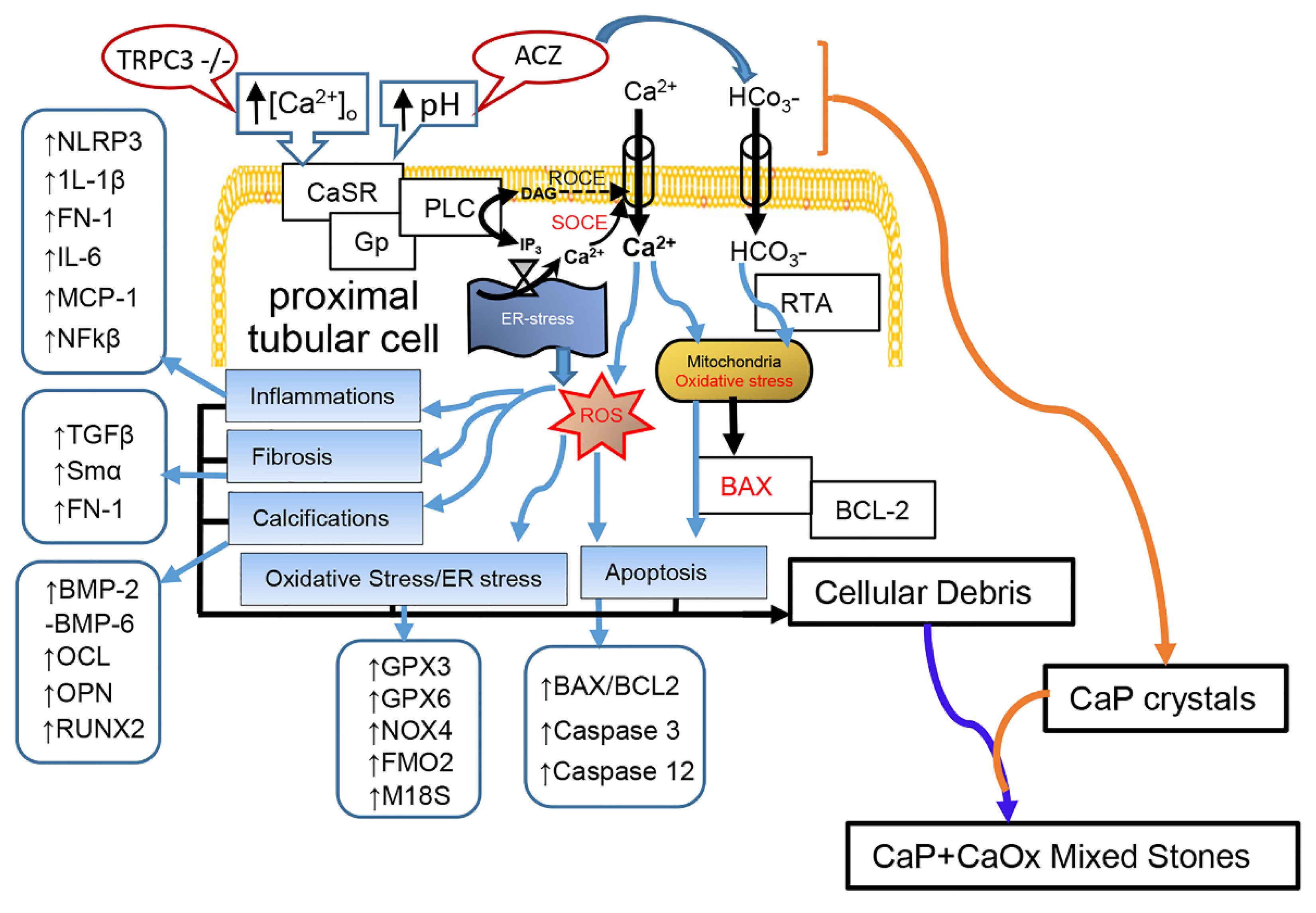
3. Discussion
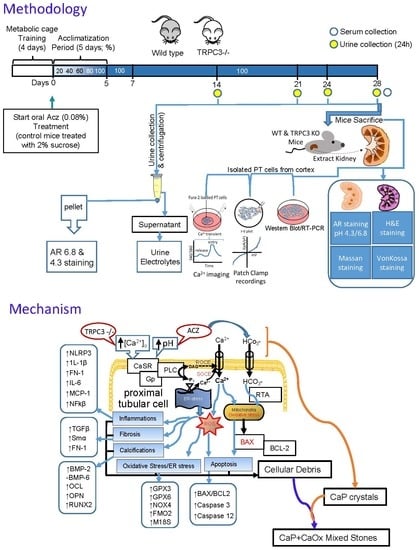
4. Materials and Methods
4.1. Animals
4.2. Materials, Antibodies and Chemicals
4.3. Mice Urine and Serum Electrolyte and pH Measurements
4.4. Alizarin Red Staining of Urine Crystals
4.5. Isolation and Primary Culture of PT Cells
4.6. Time-Lapse [Ca2+]i Fluorescence Measurements
4.7. Whole-Cell Patch Clamp Experiments
4.8. RNA Extraction, cDNA Synthesis and Polymerase Chain Reaction (PCR)
4.9. Protein Analysis and Western Blotting
4.10. Histochemistry of Kidney Sections
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodríguez Soriano, J. Renal tubular acidosis: The clinical entity. J. Am. Soc. Nephrol. 2002, 13, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Smulders, Y.M.; Frissen, P.H.; Slaats, E.H.; Silberbusch, J. Renal tubular acidosis: Pathophysiology and diagnosis. Arch. Intern. Med. 1996, 156, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.; Tiselius, H.G. The effect of pH on the risk of calcium oxalate crystallization in urine. Eur. Urol. 1986, 12, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Mateos Antón, F.; García Puig, J.; Gaspar, G.; Martínez, M.E.; Ramos, T.; Martínez Piñeiro, J.A. Renal tubular acidosis in recurrent renal stone formers. Eur. Urol. 1984, 10, 55–59. [Google Scholar] [CrossRef]
- Hamm, L.; Nakhoul, N. Renal Acidification. In Brenner and Rector’s the Kidney, 8th ed.; Brenner, B.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 248–267. ISBN 978-1416031055. [Google Scholar]
- Pak, C.Y.; Koenig, K.; Khan, R.; Haynes, S.; Padalino, P. Physicochemical action of potassium-magnesium citrate in nephrolithiasis. J. Bone Miner. Res. 1992, 7, 281–285. [Google Scholar] [CrossRef]
- Goldfarb, D.S. A woman with recurrent calcium phosphate kidney stones. Clin. J. Am. Soc. Nephrol. 2012, 7, 1172–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiselius, H.G. A hypothesis of calcium stone formation: An interpretation of stone research during the past decades. Urol. Res. 2011, 39, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Ahlstrand, C.; Tiselius, H.G. Urine composition and stone formation during treatment with acetazolamide. Scand. J. Urol. Nephrol. 1987, 21, 225–228. [Google Scholar] [CrossRef]
- Krishnan, D.; Liu, L.; Wiebe, S.A.; Casey, J.R.; Cordat, E.; Alexander, R.T. Carbonic anhydrase II binds to and increases the activity of the epithelial sodium-proton exchanger, NHE3. Am. J. Physiol. Renal Physiol. 2015, 309, F383–F392. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, D.; Pan, W.; Beggs, M.R.; Trepiccione, F.; Chambrey, R.; Eladari, D. Deficiency of Carbonic Anhydrase II Results in a Urinary Concentrating Defect. Front. Physiol. 2017, 8, 1108. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; An, Y.; Gao, J.; Han, J.; Pan, X.; Pan, Y. Aquaporin-1 translocation and degradation mediates the water transportation mechanism of acetazolamide. PLoS ONE 2012, 7, e45976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evan, A.P. Physiopathology and etiology of stone formation in the kidney and the urinary tract. Pediatr. Nephrol. 2010, 25, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Thorleifsson, G.; Holm, H.; Edvardsson, V.; Walters, G.B.; Styrkarsdottir, U.; Gudbjartsson, D.F. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat. Genet. 2009, 41, 926–930. [Google Scholar] [CrossRef]
- Boadi, E.A.; Deems, N.J.; Raub, C.B.; Bandyopadhyay, B.C. Matting Calcium Crystals by Melamine Improves Stabilization and Prevents Dissolution. Cryst. Growth Des. 2019, 19, 6636–6648. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.M.; Assimos, D.G. Hypocitraturia: Pathophysiology and medical management. Rev. Urol. 2009, 11, 134–144. [Google Scholar] [PubMed]
- Nouvenne, A.; Meschi, T.; Prati, B.; Guerra, A.; Allegri, F.; Vezzoli, G. Effects of a low-salt diet on idiopathic hypercalciuria in calcium-oxalate stone formers: A 3-mo randomized controlled trial. Am. J. Clin. Nutr. 2010, 91, 565–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibeh, C.L.; Yiu, A.J.; Kanaras, Y.L.; Paal, E.; Birnbaumer, L.; Jose, P.A. Evidence for a regulated Ca2+ entry in proximal tubular cells and its implication in calcium stone formation. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.R.; Pearle, M.S.; Robertson, W.G.; Gambaro, G.; Canales, B.K.; Doizi, S. Kidney stones. Nat. Rev. Dis. Primers 2016, 2, 16008. [Google Scholar] [CrossRef]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.E.; Armstrong, W.P.; Benham, C.D.; Hallam, T.J.; Jacob, R.; Jaxa-Chamiec, A. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 1990, 271, 515–522. [Google Scholar] [PubMed]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.F. The search for calcium receptor antagonists (calcilytics). J. Mol. Endocrinol. 2002, 29, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.J.; Gao, Y.D.; Geng, S.; Yang, J. Role of STIM1/Orai1-mediated store-operated Ca2⁺ entry in airway smooth muscle cell proliferation. J. Appl. Physiol. 2011, 110, 1256–1263. [Google Scholar] [CrossRef]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca(2+) Entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curthoys, N.P.; Moe, O.W. Proximal tubule function and response to acidosis. Clin. J. Am. Soc. Nephrol. 2014, 9, 1627–1638. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, A.; Vargas, L.A.; Aiello, E.A.; Álvarez, B.V. Elevated carbon dioxide upregulates NBCn1 Na+/HCO3(-) cotransporter in human embryonic kidney cells. Am. J. Physiol. Renal Physiol. 2013, 305, F1765–F1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-kappaB in renal inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef]
- Yamamoto, T.; Noble, N.A.; Miller, D.E.; Border, W.A. Sustained expression of TGF-beta 1 underlies development of progressive kidney fibrosis. Kidney Int. 1994, 45, 916–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M.; Hewitson, T.D.; Becker, G.J.; Jones, C.L. Ischemic acute renal failure: Long-term histology of cell and matrix changes in the rat. Kidney Int. 2000, 57, 2375–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, N.L.; Miguel, V.; Ding, W.; Singh, A.K.; Malik, S.; Rotllan, N.; Moshnikova, A.; Toczek, J.; Zeiss, C.; Sadeghi, M.M.; et al. Genetic deficiency or pharmacological inhibition of miR-33 protects from kidney fibrosis. JCI Insight 2019, 4, e131102. [Google Scholar] [CrossRef] [Green Version]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholze, A.; Jankowski, J.; Pedraza-Chaverri, J.; Evenepoel, P. Oxidative Stress in Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2016, 2016, 8375186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarng, D.C.; Wen Chen, T.; Huang, T.P.; Chen, C.L.; Liu, T.Y.; Wei, Y.H. Increased oxidative damage to peripheral blood leukocyte DNA in chronic peritoneal dialysis patients. J. Am. Soc. Nephrol. 2002, 13, 1321–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Esteve, J.M.; Mompo, J.; Garcia de la Asuncion, J.; Sastre, J.; Asensi, M.; Boix, J. Oxidative damage to mitochondrial DNA and glutathione oxidation in apoptosis: Studies in vivo and in vitro. FASEB J. 1999, 13, 1055–1064. [Google Scholar] [CrossRef]
- Sanz, A.B.; Santamaría, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Mechanisms of renal apoptosis in health and disease. J. Am. Soc. Nephrol. 2008, 19, 1634–1642. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, J.; Katayama, T.; Taniguchi, M.; Honda, A.; Imaizumi, K.; Tohyama, M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci. Lett. 2004, 357, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, H.; Okamoto, H.; Yoshimura, A.; Yoshida, H. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J. Cell Sci. 2006, 119, 3958–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Sano, F.; Ferraro, E.; Tufi, R.; Achsel, T.; Piacentini, M.; Cecconi, F. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J. Biol. Chem. 2006, 281, 2693–2700. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.R. Reactive oxygen species, inflammation and calcium oxalate nephrolithiasis. Transl. Androl. Urol. 2014, 3, 256–276. [Google Scholar]
- Leaf, D.E.; Goldfarb, D.S. Mechanisms of action of acetazolamide in the prophylaxis and treatment of acute mountain sickness. J. Appl. Physiol. 2007, 102, 1313–1322. [Google Scholar] [CrossRef]
- Matlaga, B.R.; Shah, O.D.; Assimos, D.G. Drug-induced urinary calculi. Rev. Urol. 2003, 5, 227–231. [Google Scholar] [PubMed]
- Nensén, O.; Hansell, P.; Palm, F. Role of carbonic anhydrase in acute recovery following renal ischemia reperfusion injury. PLoS ONE 2019, 14, e0220185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterrett, S.P.; Penniston, K.L.; Wolf, J.S., Jr.; Nakada, S.Y. Acetazolamide is an effective adjunct for urinary alkalization in patients with uric acid and cystine stone formation recalcitrant to potassium citrate. Urology 2008, 72, 278–281. [Google Scholar] [CrossRef]
- Doroszewicz, J.; Waldegger, P.; Jeck, N.; Seyberth, H.; Waldegger, S. pH dependence of extracellular calcium sensing receptor activity determined by a novel technique. Kidney Int. 2005, 67, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Malnic, G.; Giebisch, G. Symposium on acid-base homeostasis. Mechanism of renal hydrogenion secretion. Kidney Int. 1972, 1, 280–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, S.K.; Ariceta, G.; Batlle, D. Proximal renal tubular acidosis: A not so rare disorder of multiple etiologies. Nephrol. Dial. Transplant. 2012, 27, 4273–4287. [Google Scholar] [CrossRef]
- Bomsztyk, K.; Swenson, E.R.; Calalb, M.B. HCO3 accumulation in proximal tubule: Roles of carbonic anhydrase, luminal buffers, and pH. Am. J. Physiol. 1987, 252, F501–F508. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.M.; Liu, Y.; Liu, M.; Wang, J.L.; Xie, Z.D.; Chen, K.J. Na+/HCO3-Cotransporter NBCn2 Mediates HCO3-Reclamation in the Apical Membrane of Renal Proximal Tubules. J. Am. Soc. Nephrol. 2017, 28, 2409–2419. [Google Scholar] [CrossRef] [Green Version]
- Clausen, T. Potassium and sodium transport and pH regulation. Can. J. Physiol. Pharmacol. 1992, 70, S219–S222. [Google Scholar] [CrossRef] [PubMed]
- Aronson, P.S.; Giebisch, G. Effects of pH on potassium: New explanations for old observations. J. Am. Soc. Nephrol. 2011, 22, 1981–1989. [Google Scholar] [CrossRef]
- Roy, A.; Al-bataineh, M.M.; Pastor-Soler, N.M. Collecting duct intercalated cell function and regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 305–324. [Google Scholar] [CrossRef]
- Wagner, C.A.; Mohebbi, N. Urinary pH and stone formation. J. Nephrol. 2010, 23, S165–S169. [Google Scholar]
- Hsu, Y.J.; Hoenderop, J.G.; Bindels, R.J. TRP channels in kidney disease. Biochim. Biophys. Acta 2007, 1772, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Gewin, L.S. Renal fibrosis: Primacy of the proximal tubule. Matrix Biol. 2018, 68–69, 248–262. [Google Scholar] [CrossRef]
- Humayun, Y.; Sanchez, P.; Norris, L.T.; Monga, D.; Lewin, J.; Fülöp, T. Kidney biopsy for renal tubular acidosis: When tissue diagnosis makes a difference. Clin. Nephrol. Case Stud. 2015, 3, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruana, R.J.; Buckalew, V.M. The syndrome of distal (type 1) renal tubular acidosis. Clinical and laboratory findings in 58 cases. Medicine 1988, 67, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Rustom, R.; Wang, B.; McArdle, F.; Shalamanova, L.; Alexander, J.; McArdle, A. Oxidative stress in a novel model of chronic acidosis in LLC-PK1 cells. Nephron. Exp. Nephrol. 2003, 95, e13–e23. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Yiu, A.J.; Ibeh, C.L.; Roy, S.K.; Bandyopadhyay, B.C. Melamine induces Ca2+-sensing receptor activation and elicits apoptosis in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2017, 313, C27–C41. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, J.F.; Magyar, C.E.; Sneddon, W.B.; Friedman, P.A. Impaired renal calcium absorption in mice lacking calcium channel beta 3 subunits. Can. J. Physiol. Pharmacol. 2009, 87, 522–530. [Google Scholar] [CrossRef]
- Gombedza, F.; Evans, S.; Shin, S.; Awuah Boadi, E.; Zhang, Q.; Nie, Z. Melamine promotes calcium crystal formation in three-dimensional microfluidic device. Sci. Rep. 2019, 9, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchý, P.; Straková, E.; Herzig, I.; Staňa, J.; Kalusová, R.; Pospíchalová, M. Toxicological risk of melamine and cyanuric acid in food and feed. Interdiscip. Toxicol. 2009, 2, 55–59. [Google Scholar] [CrossRef]
- Paul, H.; Reginato, A.J.; Schumacher, H.R. Alizarin red S staining as a screening test to detect calcium compounds in synovial fluid. Arthritis Rheum. 1983, 26, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Lau, I.; Potluri, A.; Ibeh, C.L.; Redman, R.S.; Paal, E.; Bandyopadhyay, B.C. Microcalcifications in stone-obstructed human submandibular gland are associated with apoptosis and cell proliferation. Arch. Oral Biol. 2017, 82, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.D.; Alavi, N.; Livingston, D.; Hiller, S.; Taub, M. Characterization of primary rabbit kidney cultures that express proximal tubule functions in a hormonally defined medium. J. Cell Biol. 1982, 95, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, B.C.; Swaim, W.D.; Sarkar, A.; Liu, X.; Ambudkar, I.S. Extracellular Ca(2+) sensing in salivary ductal cells. J. Biol. Chem. 2012, 287, 30305–30316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, B.C.; Pingle, S.C.; Ahern, G.P. Store-operated Ca2+ signaling in dendritic cells occurs independently of STIM1. J. Leukoc. Biol. 2011, 89, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, B.C.; Swaim, W.D.; Liu, X.; Redman, R.S.; Patterson, R.L.; Ambudkar, I.S. Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells. Role in apical Ca2+ influx. J. Biol. Chem. 2005, 280, 12908–12916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.E.; Lewin-Smith, M.R.; Kalasinsky, V.F.; Pizzolato, K.M.; Fleetwood, M.L.; McElhaney, M.R. Characterization of melamine-containing and calcium oxalate crystals in three dogs with suspected pet food-induced nephrotoxicosis. Vet. Pathol. 2008, 45, 417–426. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (Sense, Antisense) | Product Size (bp) |
|---|---|---|
| mGAPDH | 5′-ACTCCACTCACGGCAAATTC-3′ | 171 |
| 5′-TCTCCATGGTGGTGAAGACA-3′ | ||
| mNCX1 | 5′-CCTTGTGCATCTTAGCAATG-3′ | 309 |
| 5′-TCTCACTCATCTCCAGA-3′ | ||
| mPMCA1 | 5′TGGCAAACAACTCAGTTGCATATAGTGG3′ | 562 |
| 5′TCCTGTTCAATTCGACTCTGCAAGCCTCG3′ | ||
| mSMa | 5′-AGATTGTCCGTGACATCAAGG-3′ | 538 |
| 5′-TTGTGTGCTAGAGGCAGAGC-3 | ||
| mNBCe1 | 5′-CACTGAAAATGTGGAAGGGAA-3′ | 544 |
| 5′-TTATCACCCTTGTGCTTTGC-3′ | ||
| mNaPiIIa | 5′-AGACACAACAGAGGCTTC-3′ | 181 |
| 5′-CACAAGGAGGATAAGACAAG-3′ | ||
| mBMP2 | 5′-TGGAAGTGGCCCATTTAGAG-3′ | 166 |
| 5′-TGACGCTTTTCTCGTTTGTG-3′ | ||
| mBMP6 | 5′-CCCGCCCGGAGTAGTTTTAGC-3′ | 168 |
| 5′-AGTGCCCTTCTCCCCTCCATT-3′ | ||
| mOCL | 5′-CTGACAAAGCCTTCATGTCCAA-3′ | 59 |
| 5′-GCGCCGGAGTCTGTTCACTA-3′ | ||
| mOPN | 5′-GATGATGATGACGATGGAGACC-3′ | 148 |
| 5′-CGACTGTAGGGACGATTGGAG-3′ | ||
| mRUNX2 | 5′-CGGCCCTCCCTGAACTCT-3′ | 75 |
| 5′-TGCCTGCCTGGGATCTGTA-3′ | ||
| mTGFb | 5′-CTGAGTGGCTGTCTTTTG-3′ | 436 |
| 5′-TTGCTGTACTGTGTGTCC-3′ | ||
| mFN1 | 5′-TGCACGATGATATGGAGAGC-3′ | 93 |
| 5′-TGGGTGTCACCTGACTGA-3′ | ||
| mNLRP3 | 5′-AGAGCCTACAGTTGGGTGAAATG-3′ | 116 |
| 5′-CCACGCCTACCAGGAAATCTC-3′ | ||
| mIL-1b | 5′-TCCATGAGCTTTGTACAAGGA-3′ | 343 |
| 5′-AGCCCATACTTTAGGAAGACA-3′ | ||
| mIL-6 | 5′-TGGAGTCACAGAAGGAGTGGCTAA-3′ | 155 |
| 5′-TCTGACCACAGTGAGGAATGTCCA-3′ | ||
| mMCP1 | 5′-AGAGAGCCAGACGGGAGGAA-3′ | 520 |
| 5′-GTCACACTGGTCACTCCTAC-3′ | ||
| mNFkb | 5′-GTGGAGGCATGTTCGGTAGT-3′ | 367 |
| 5′-AGCTGCAGAGCCTTCTCAAG-3′ | ||
| mGPX3 | 5′-TGGCTTGGTCATTCTGGGC-3′ | 103 |
| 5′-CCCACCTGGTCGAACATACTT-3′ | ||
| mM18S | 5′-ACGGAAGGGCACCACCAGGA-3′ | 127 |
| 5′-CACCACCACCCACGGAATCG-3′ | ||
| mBAX | 5′-GAGACACCTGAGCTGACCTT-3′ | 244 |
| 5′-GCACCAGTTTGCTAGCAAAG-3′ | ||
| mBCL2 | 5′-CTCGTCGCTACCGTCGTGACTTCG-3′ | 242 |
| 5′-CAGAGTCCGGTTCAGGTACTCAGTC-3′ | ||
| mCaspase3 | 5′-AGAGAGCCAGACGGGAGGAA-3′ | 519 |
| 5′-GTCACACTGGTCACTCCTAC-3′ | ||
| mCaspase12 | 5′-GCTGGCCACATTGCCAATTCCC-3′ | 314 |
| 5′-GCCAGACGTGTTCGTCCCTCC-5′ | ||
| mGPX6 | 5′-GCCCAGAAGTTGTGGGGTTC-3′ | 129 |
| 5′-TCCATACTCATAGACGGTGCC-3′ | ||
| mSLC26a | 5′-AGATCTTCCTTGCGTCTGC-3′ | 149 |
| 5′-GCCTTTCCACATGGTAGTCTC-3′ |
| Antibody | Company Purchased/Ref. Number | Dilution Used |
|---|---|---|
| BAX1 | Santa Cruz Biotechnology; sc-20067 | 1:1000 |
| BCL2 | Santa Cruz Biotechnology; sc-23960 | 1:1000 |
| Caspase-3 | Santa Cruz Biotechnology; sc-7272 | 1:1000 |
| OPN | Santa Cruz Biotechnology; sc-10591 | 1:1000 |
| RUNX2 | Santa Cruz Biotechnology; sc-101145 | 1:1000 |
| BMP2 | Santa Cruz Biotechnology; sc-6895 | 1:1000 |
| BMP6 | Santa Cruz Biotechnology; sc-7406 | 1:1000 |
| OCL | Santa Cruz Biotechnology; sc-376726 | 1:1000 |
| Beta Actin | Santa Cruz Biotechnology; sc-47778 | 1:1000 |
| SLC4A4 (NBCe1) | Proteintech; 11885-1-AP | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awuah Boadi, E.; Shin, S.; Yeroushalmi, S.; Choi, B.-E.; Li, P.; Bandyopadhyay, B.C. Modulation of Tubular pH by Acetazolamide in a Ca2+ Transport Deficient Mice Facilitates Calcium Nephrolithiasis. Int. J. Mol. Sci. 2021, 22, 3050. https://doi.org/10.3390/ijms22063050
Awuah Boadi E, Shin S, Yeroushalmi S, Choi B-E, Li P, Bandyopadhyay BC. Modulation of Tubular pH by Acetazolamide in a Ca2+ Transport Deficient Mice Facilitates Calcium Nephrolithiasis. International Journal of Molecular Sciences. 2021; 22(6):3050. https://doi.org/10.3390/ijms22063050
Chicago/Turabian StyleAwuah Boadi, Eugenia, Samuel Shin, Samuel Yeroushalmi, Bok-Eum Choi, Peijun Li, and Bidhan C. Bandyopadhyay. 2021. "Modulation of Tubular pH by Acetazolamide in a Ca2+ Transport Deficient Mice Facilitates Calcium Nephrolithiasis" International Journal of Molecular Sciences 22, no. 6: 3050. https://doi.org/10.3390/ijms22063050