
Identification of Overexpressed Genes in Malignant Pleural Mesothelioma

 , , and
, , and
Abstract
:1. Introduction
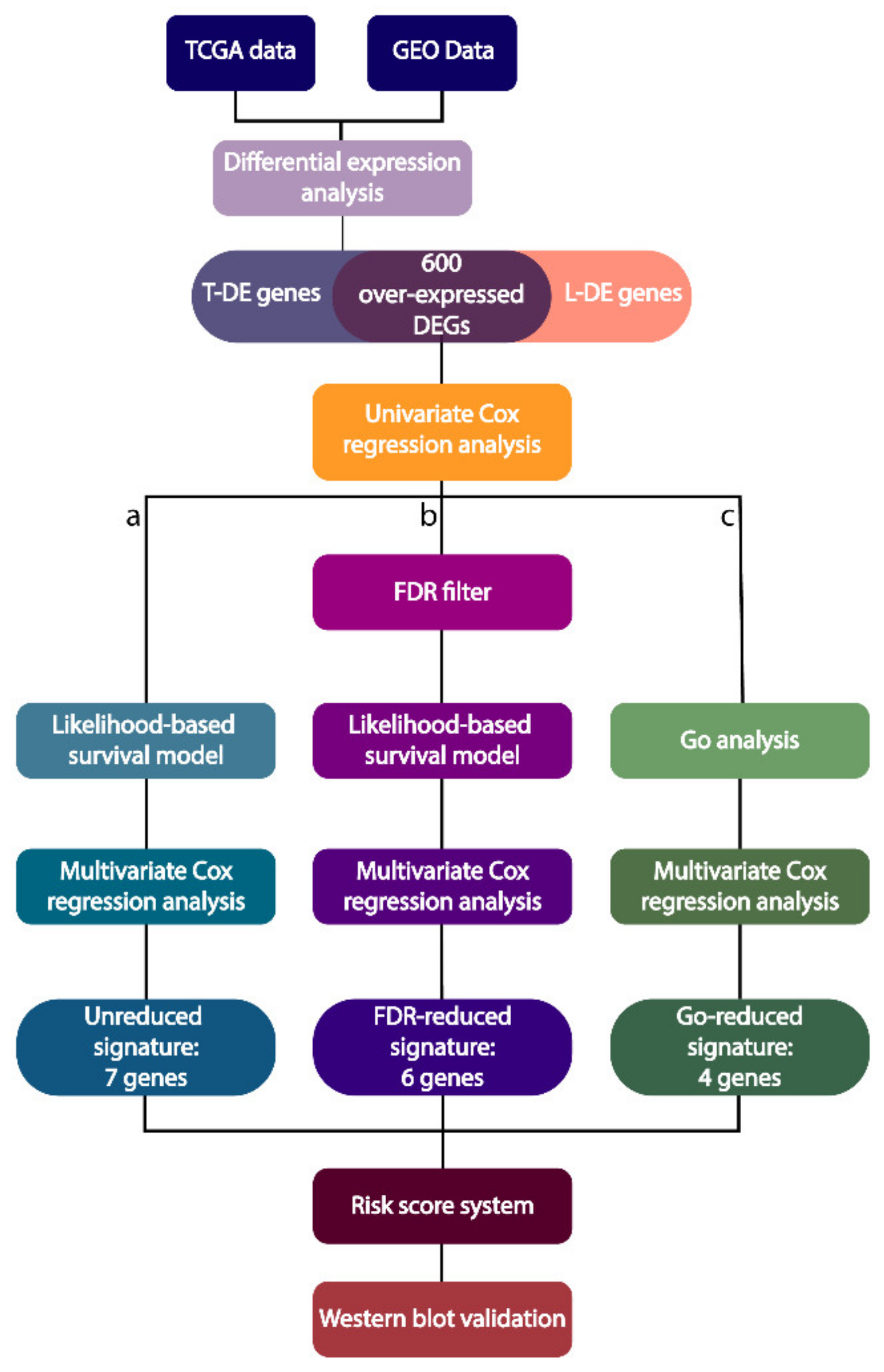
2. Results
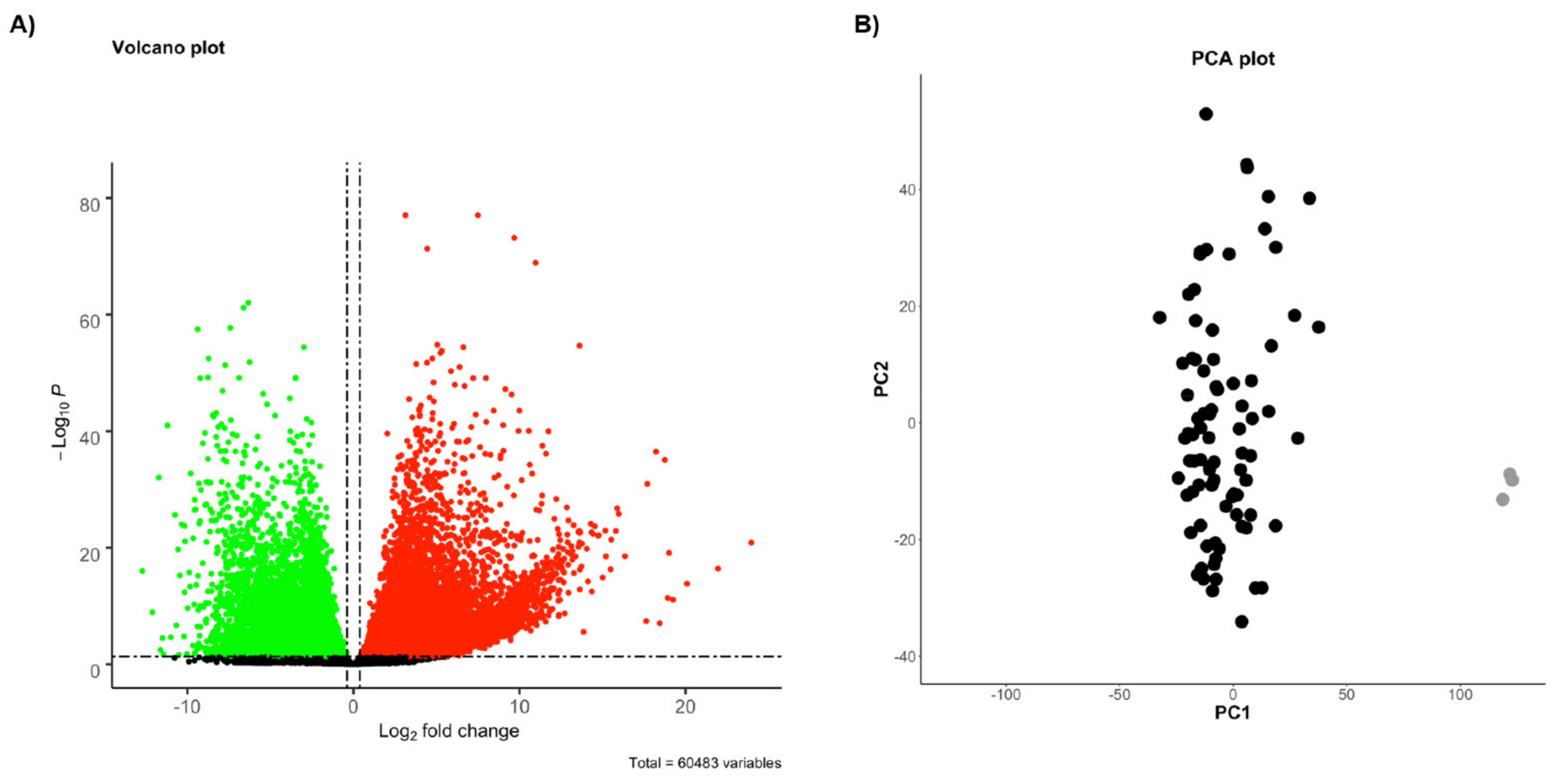
2.1. Identification of Differentially Expressed Genes (DEGs)
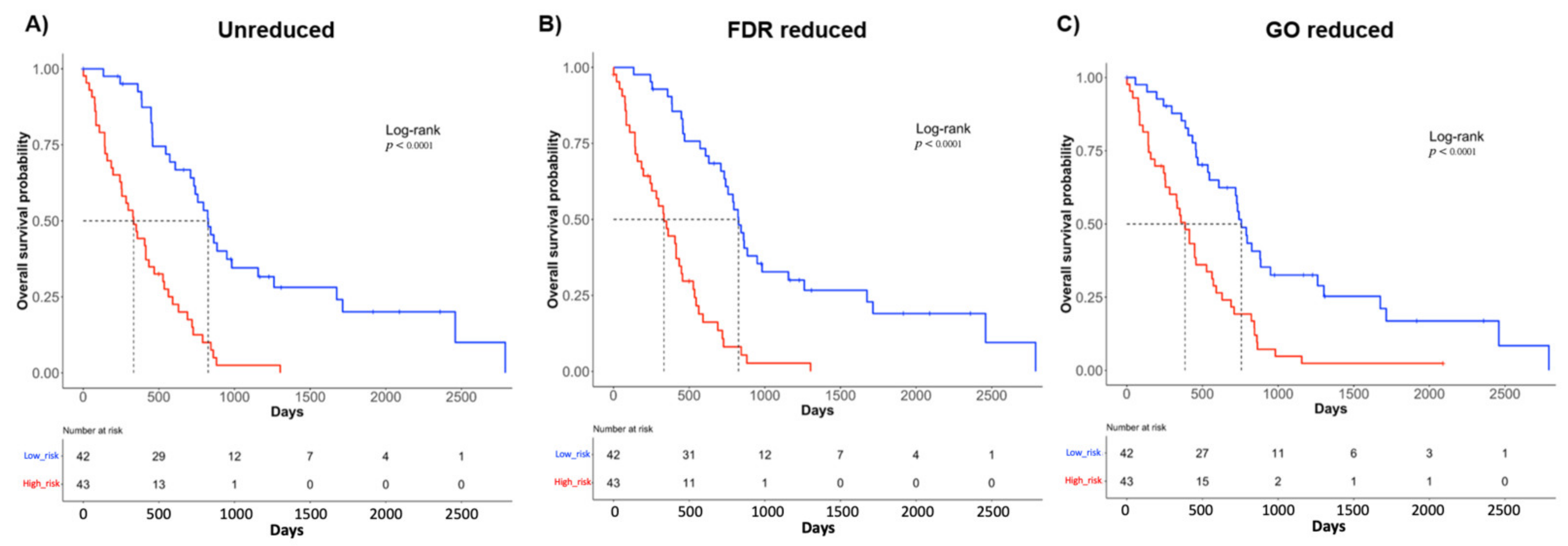
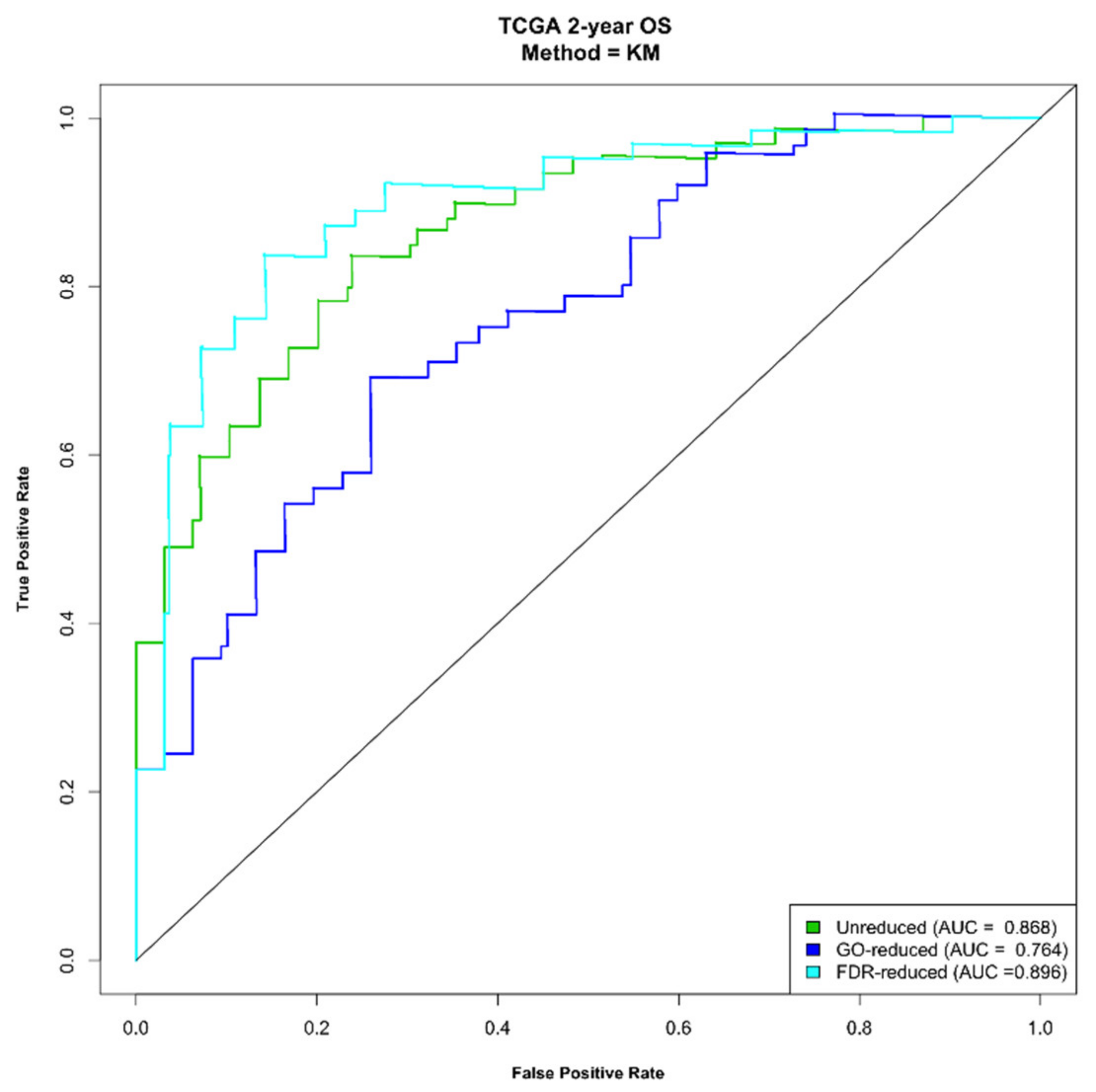
2.2. Molecular Signatures Associated with the OS
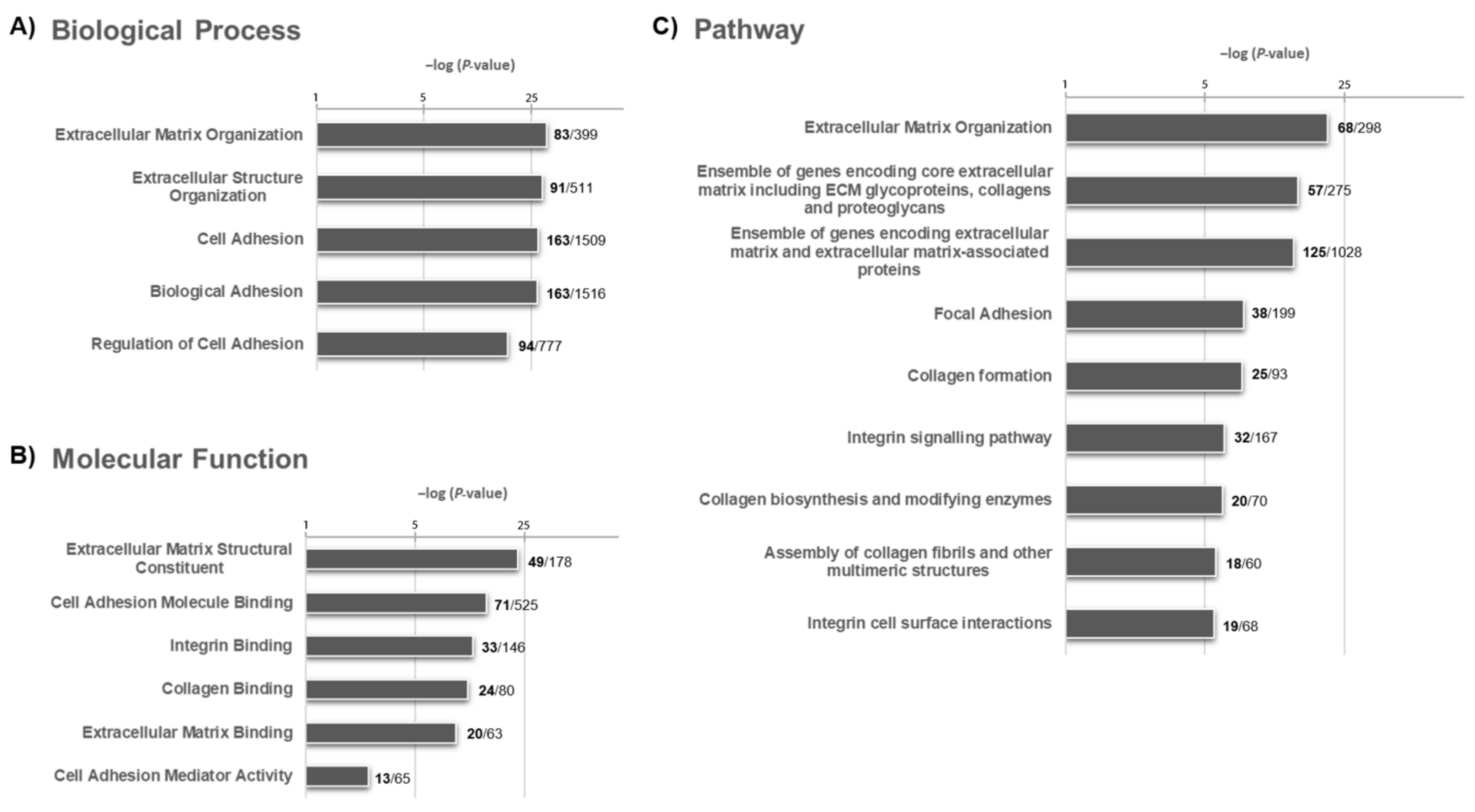
2.3. Molecular Signature Following a Functional Enrichment Analysis and Associated with the OS
2.4. Construction of a Prognostic Risk Scoring System Using the Three Molecular Signatures
2.5. The Signatures Are Independent Prognostic Factors
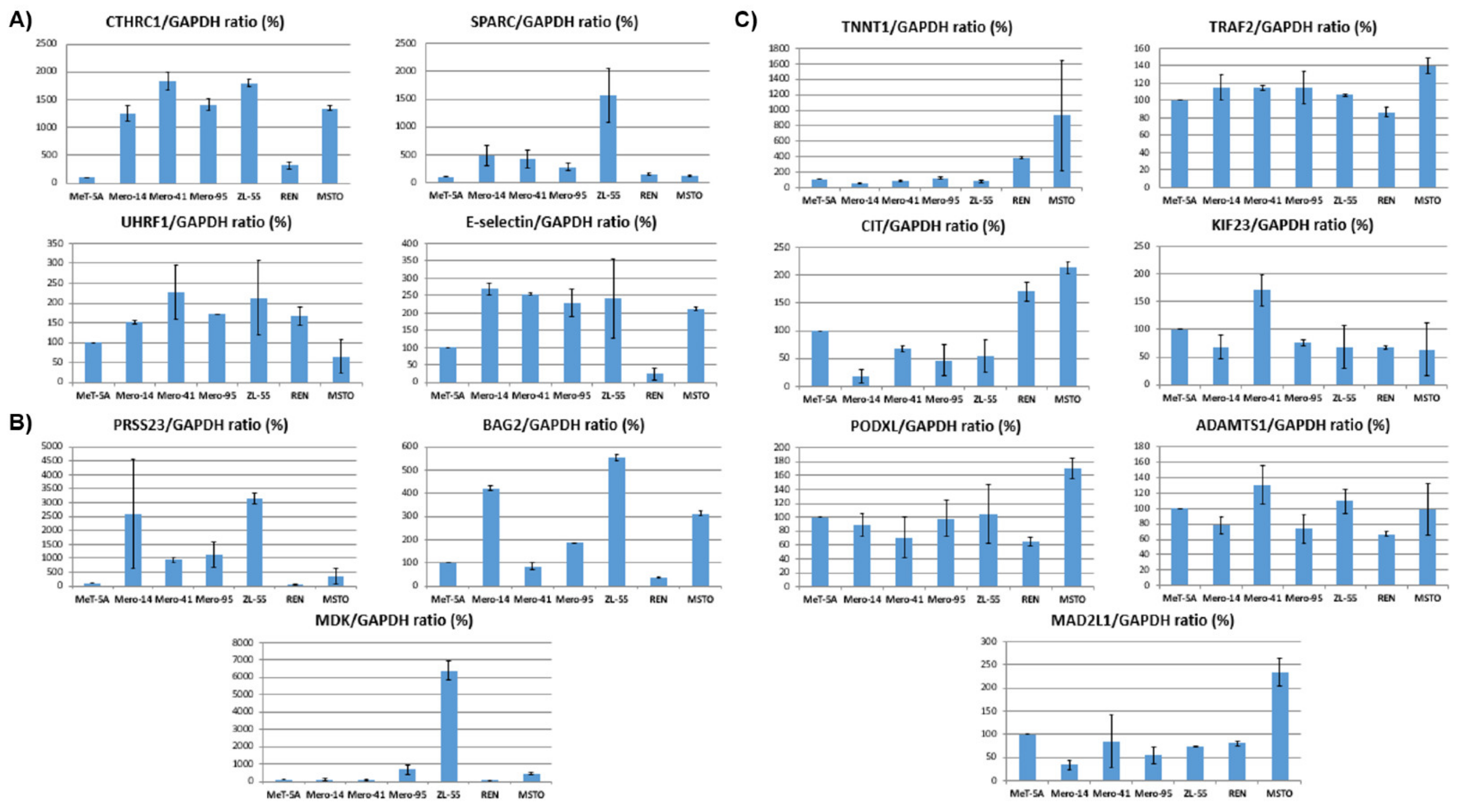
2.6. In Vitro Validation of the Prognostic Signatures by Protein Analysis of MPM Cell Lines
3. Discussion
4. Materials and Methods
4.1. Public Databases
4.2. Data Processing and Computational Analysis
4.3. Differential Expression Analysis
4.4. Literature Search Strategy
4.5. Functional Enrichment Analysis
4.6. Gene Signature Identification
4.7. Survival Analysis
4.8. Cell Culture
4.9. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCambridge, A.J.; Napolitano, A.; Mansfield, A.S.; Fennell, D.A.; Sekido, Y.; Nowak, A.K.; Reungwetwattana, T.; Mao, W.; Pass, H.I.; Carbone, M.; et al. Progress in the Management of Malignant Pleural Mesothelioma in 2017. J. Thorac. Oncol. 2018, 13, 606–623. [Google Scholar] [CrossRef] [Green Version]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin Versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [Green Version]
- Rouka, E.; Beltsios, E.; Goundaroulis, D.; Vavougios, G.D.; Solenov, E.I.; Hatzoglou, C.; Gourgoulianis, K.I.; Zarogiannis, S.G. In Silico Transcriptomic Analysis of Wound-Healing-Associated Genes in Malignant Pleural Mesothelioma. Medicina 2019, 55, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.-G.; Zhong, H.; Zhang, J.; Jin, S.-H.; Roudi, R.; Ma, H. Development and Validation of a Prognostic Signature for Malignant Pleural Mesothelioma. Front. Oncol. 2019, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Quackenbush, J. Microarray Analysis and Tumor Classification. N. Engl. J. Med. 2006, 354, 2463–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon Gavin, J.; Jensen Roderick, V.; Hsiao, L.-L.; Gullans Steven, R.; Blumenstock, J.E.; Ramaswamy, S.; Richards, W.G.; Sugarbaker David, J.B.R. Translation of microarray data into clinically relevant cancer diagnostic tests using gene expression ratios in lung cancer and mesothelioma. Cancer Res. 2002, 62, 4963–4967. [Google Scholar]
- Gordon, G.J.; Rockwell, G.N.; Jensen, R.V.; Rheinwald, J.G.; Glickman, J.N.; Aronson, J.P.; Pottorf, B.J.; Nitz, M.D.; Richards, W.G.; Sugarbaker, D.J.; et al. Identification of Novel Candidate Oncogenes and Tumor Suppressors in Malignant Pleural Mesothelioma Using Large-Scale Transcriptional Profiling. Am. J. Pathol. 2005, 166, 1827–1840. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, X.; Hou, J.; Geng, L.; Liang, X.; Ruan, Z.; Guo, H.; Nan, K.; Jiang, L. Identification of a Five-Gene Signature for Predicting Survival in Malignant Pleural Mesothelioma Patients. Front. Genet. 2020, 11, 899. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Lee, D.; Wu, L.; Patel, P.; Young, A.J.; Wada, H.; Hu, H.-P.; Ujiie, H.; Kaji, M.; Kano, S.; et al. Kinesin family members KIF11 and KIF23 as potential therapeutic targets in malignant pleural mesothelioma. Int. J. Oncol. 2016, 49, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Suraokar, M.B.; Nunez, M.I.; Diao, L.; Chow, C.W.; Kim, D.; Behrens, C.; Lin, H.; Lee, S.; Raso, G.; Moran, C.; et al. Expression profiling stratifies mesothelioma tumors and signifies deregulation of spindle checkpoint pathway and microtubule network with therapeutic implications. Ann. Oncol. 2014, 25, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Ak, G.; Tada, Y.; Shimada, H.; Metintas, S.; Ito, M.; Hiroshima, K.; Tagawa, M.; Metintas, M. Midkine is a potential novel marker for malignant mesothelioma with different prognostic and diagnostic values from mesothelin. BMC Cancer 2017, 17, 212. [Google Scholar] [CrossRef] [Green Version]
- Reardon, E.S.; Shukla, V.; Xi, S.; Gara, S.K.; Liu, Y.; Straughan, D.; Zhang, M.; Hong, J.A.; Payabyab, E.C.; Kumari, A.; et al. UHRF1 Is a Novel Druggable Epigenetic Target in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2020, 16, 89–103. [Google Scholar] [CrossRef]
- Kao, S.C.; Kirschner, M.B.; Cooper, W.A.; Tran, T.; Burgers, S.; Wright, C.; Korse, T.; van den Broek, D.; Edelman, J.; Vallely, M.; et al. A proteomics-based approach identifies secreted protein acidic and rich in cysteine as a prognostic biomarker in malignant pleural mesothelioma. Br. J. Cancer 2016, 114, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, D.; Zhu, Y.; Zhang, L.; Wei, W. The Role of CTHRC1 in Regulation of Multiple Signaling and Tumor Progression and Metastasis. Mediat. Inflamm. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Jubeli, E.; Moine, L.; Vergnaud-Gauduchon, J.; Barratt, G. E-selectin as a target for drug delivery and molecular imaging. J. Control. Release 2012, 158, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, S.; Visweswaran, G.R.R.; Storm, G.; Hennink, W.E.; Kamps, J.A.A.M.; Kok, R.J. E-selectin targeted immunoliposomes for rapamycin delivery to activated endothelial cells. Int. J. Pharm. 2018, 548, 759–770. [Google Scholar] [CrossRef]
- Xu, X.-L.; Zhu, M.-L.; Liu, D.; Shu, G.-F.; Qi, J.; Lu, Y.; Wang, F.; Ying, X.-Y.; Chen, J.; Du, Y.-Z. Highly Integrated Nanoplatform Based on an E-Selectin-Targeting Strategy for Metastatic Breast Cancer Treatment. Mol. Pharm. 2019, 16, 3694–3702. [Google Scholar] [CrossRef]
- Filippou, P.S.; Karagiannis, G.S.; Constantinidou, A. Midkine (MDK) growth factor: A key player in cancer progression and a promising therapeutic target. Oncogene 2020, 39, 2040–2054. [Google Scholar] [CrossRef] [PubMed]
- Camacho, D.; Jesus, J.P.; Palma, A.M.; Martins, S.A.; Afonso, A.; Peixoto, M.L.; Pelham, C.J.; Moreno, E.; Gogna, R. SPARC-p53: The Double Agents of Cancer. Adv. Cancer Res. 148, 171–199. [CrossRef]
- Glotzer, M. The Molecular Requirements for Cytokinesis. Science (80-) 2005, 307, 1735–1739. [Google Scholar] [CrossRef] [Green Version]
- Neef, R.; Klein, U.R.; Kopajtich, R.; Barr, F.A. Cooperation between Mitotic Kinesins Controls the Late Stages of Cytokinesis. Curr. Biol. 2006, 16, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Fusaki, N.; Ohta, S.; Iwahori, Y.; Iizuka, Y.; Inagawa, K.; Kawakami, Y.; Yoshida, K.; Toda, M. Downregulation of KIF23 suppresses glioma proliferation. J. Neurooncol. 2012, 106, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.X.; Duan, Z.; Wang, J.; Sokolov, A.; Xu, J.; Chen, C.Z.; Li, J.J.; Chen, H.-W. Kinesin Family Deregulation Coordinated by Bromodomain Protein ANCCA and Histone Methyltransferase MLL for Breast Cancer Cell Growth, Survival, and Tamoxifen Resistance. Mol. Cancer Res. 2014, 12, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, H.; Ito, S.; Tanaka, H.; Kondo, E.; Kodera, Y.; Nakanishi, H. Establishment of new intraperitoneal paclitaxel-resistant gastric cancer cell lines and comprehensive gene expression analysis. Anticancer Res. 2013, 33, 4299–4308. [Google Scholar]
- Pulverer, B. Spindle checkpoint protein links Rb pathway to aneuploidy. Nat. Cell Biol. 2004, 6, 806. [Google Scholar] [CrossRef]
- Wang, Z.; Katsaros, D.; Shen, Y.; Fu, Y.; Canuto, E.M.; Benedetto, C.; Lu, L.; Chu, W.-M.; Risch, H.A.; Yu, H. Biological and Clinical Significance of MAD2L1 and BUB1, Genes Frequently Appearing in Expression Signatures for Breast Cancer Prognosis. PLoS ONE 2015, 10, e0136246. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Yang, M.; Miao, X.; Shi, Y.; Yao, J.; Tan, W.; Sun, T.; Zhao, D.; Yu, D.; et al. Functional evaluation of missense variations in the human MAD1L1 and MAD2L1 genes and their impact on susceptibility to lung cancer. J. Med. Genet. 2010, 47, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Benezra, R. Identification of a Human Mitotic Checkpoint Gene: hsMAD2. Science (80-) 1996, 274, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.; Alhosin, M.; Hamiche, A.; Mousli, M. Coordinated Dialogue between UHRF1 and DNMT1 to Ensure Faithful Inheritance of Methylated DNA Patterns. Genes 2019, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, H.; Capalash, N. UHRF1: The key regulator of epigenetics and molecular target for cancer therapeutics. Tumor Biol. 2017, 39, 101042831769220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, H.; Chen, Y.; Yang, X.; Wang, P.; Liu, T.; Deng, M.; Qin, B.; Correia, C.; Lee, S.; et al. A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat. Commun. 2016, 7, 10201. [Google Scholar] [CrossRef] [PubMed]
- Mousli, M.; Hopfner, R.; Abbady, A.-Q.; Monté, D.; Jeanblanc, M.; Oudet, P.; Louis, B.; Bronner, C. ICBP90 belongs to a new family of proteins with an expression that is deregulated in cancer cells. Br. J. Cancer 2003, 89, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Chen, J.; Xie, W.; Brown, S.M.; Cai, Y.; Wu, K.; Fan, D.; Nie, Y.; Yegnasubramanian, S.; Tiedemann, R.L.; et al. Defining UHRF1 Domains that Support Maintenance of Human Colon Cancer DNA Methylation and Oncogenic Properties. Cancer Cell 2019, 35, 633–648.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Guo, J.; Zheng, Q.; Zhang, H. BAG2 structure, function and involvement in disease. Cell. Mol. Biol. Lett. 2016, 21, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-S.; Wei, B. Over-expression of Bcl2-associated athanogene 2 in oral cancer promotes cellular proliferation and is associated with poor prognosis. Arch. Oral Biol. 2019, 102, 164–170. [Google Scholar] [CrossRef]
- Sun, L.; Chen, G.; Sun, A.; Wang, Z.; Huang, H.; Gao, Z.; Liang, W.; Liu, C.; Li, K. BAG2 Promotes Proliferation and Metastasis of Gastric Cancer via ERK1/2 Signaling and Partially Regulated by miR186. Front. Oncol. 2020, 10, 31. [Google Scholar] [CrossRef]
- Yang, K.-M.; Bae, E.; Ahn, S.G.; Pang, K.; Park, Y.; Park, J.; Lee, J.; Ooshima, A.; Park, B.; Kim, J.; et al. Co-chaperone BAG2 Determines the Pro-oncogenic Role of Cathepsin B in Triple-Negative Breast Cancer Cells. Cell Rep. 2017, 21, 2952–2964. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Zhao, Y.; Liu, J.; Zhang, C.; Yu, H.; Wang, J.; Zheng, T.; Liu, L.; Li, J.; Feng, Z.; et al. BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. eLife 2015, 4, e08401. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, K.; Murphy, M.J.; Yeoman, R.R.; Mitra, S.; Dubay, C.J.; Hennebold, J.D. The Identification of Novel Ovarian Proteases Through the Use of Genomic and Bioinformatic Methodologies1. Biol. Reprod. 2006, 75, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Bayoumi, A.S.; Teoh, J.-P.; Aonuma, T.; Yuan, Z.; Ruan, X.; Tang, Y.; Su, H.; Weintraub, N.L.; Kim, I.-M. MicroRNA-532 protects the heart in acute myocardial infarction, and represses prss23, a positive regulator of endothelial-to-mesenchymal transition. Cardiovasc. Res. 2017, 113, 1603–1614. [Google Scholar] [CrossRef]
- Chan, H.-S.; Chang, S.-J.; Wang, T.-Y.; Ko, H.-J.; Lin, Y.-C.; Lin, K.-T.; Chang, K.-M.; Chuang, Y.-J. Serine Protease PRSS23 Is Upregulated by Estrogen Receptor α and Associated with Proliferation of Breast Cancer Cells. PLoS ONE 2012, 7, e30397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Yang, Y.; Chen, J.; He, X.; Lv, N.; Yan, R. PRSS23 knockdown inhibits gastric tumorigenesis through EIF2 signaling. Pharmacol. Res. 2019, 142, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Okada, R.; Hozaka, Y.; Wada, M.; Moriya, S.; Satake, S.; Idichi, T.; Kurahara, H.; Ohtsuka, T.; Seki, N. Molecular Pathogenesis of Pancreatic Ductal Adenocarcinoma: Impact of miR-30c-5p and miR-30c-2-3p Regulation on Oncogenic Genes. Cancers 2020, 12, 2731. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: https://qubeshub.org/resources/fastqc (accessed on 10 January 2021).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data, version 0.14.4; Bioinformatics: Oxford, UK, 2015. [Google Scholar]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Rihn, B.H.; Mohr, S.; McDowell, S.A.; Binet, S.; Loubinoux, J.; Galateau, F.; Keith, G.; Leikauf, G.D. Differential gene expression in mesothelioma. FEBS Lett. 2000, 480, 95–100. [Google Scholar] [CrossRef] [Green Version]
- De Rienzo, A.; Richards, W.G.; Yeap, B.Y.; Coleman, M.H.; Sugarbaker, P.E.; Chirieac, L.R.; Wang, Y.E.; Quackenbush, J.; Jensen, R.V.; Bueno, R. Sequential Binary Gene Ratio Tests Define a Novel Molecular Diagnostic Strategy for Malignant Pleural Mesothelioma. Clin. Cancer Res. 2013, 19, 2493–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuraoka, M.; Amatya, V.J.; Kushitani, K.; Mawas, A.S.; Miyata, Y.; Okada, M.; Kishimoto, T.; Inai, K.; Nishisaka, T.; Sueda, T.; et al. Identification of DAB2 and Intelectin-1 as Novel Positive Immunohistochemical Markers of Epithelioid Mesothelioma by Transcriptome Microarray Analysis for Its Differentiation From Pulmonary Adenocarcinoma. Am. J. Surg. Pathol. 2017, 41, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Gueugnon, F.; Leclercq, S.; Blanquart, C.; Sagan, C.; Cellerin, L.; Padieu, M.; Perigaud, C.; Scherpereel, A.; Gregoire, M. Identification of Novel Markers for the Diagnosis of Malignant Pleural Mesothelioma. Am. J. Pathol. 2011, 178, 1033–1042. [Google Scholar] [CrossRef]
- Kettunen, E.; Nissn, A.M.; Ollikainen, T.; Taavitsainen, M.; Tapper, J.; Mattson, K.; Linnainmaa, K.; Knuutila, S.; El-Rifai, W. Gene expression profiling of malignant mesothelioma cell lines: cDNA array study. Int. J. Cancer 2001, 91, 492–496. [Google Scholar] [CrossRef]
- Singhal, S.; Wiewrodt, R.; Malden, L.D.; Amin, K.M.; Matzie, K.; Friedberg, J.; Kucharczuk, J.C.; Litzky, L.A.; Johnson, S.W.; Kaiser, L.R.; et al. Gene expression profiling of malignant mesothelioma. Clin. Cancer Res. 2003, 9, 3080–3097. [Google Scholar] [PubMed]
- Hoang, C.D.; D’Cunha, J.; Kratzke, M.G.; Casmey, C.E.; Frizelle, S.P.; Maddaus, M.A.; Kratzke, R.A. Gene Expression Profiling Identifies Matriptase Overexpression in Malignant Mesothelioma. Chest 2004, 125, 1843–1852. [Google Scholar] [CrossRef]
- Mohr, S.; Keith, G.; Galateau-Salle, F.; Icard, P.; Rihn, B.H. Cell protection, resistance and invasiveness of two malignant mesotheliomas as assessed by 10K-microarray. Biochim. Biophys. Acta. Mol. Basis Dis. 2004, 1688, 43–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Nymoen, D.A.; Stavnes, H.T.; Rosnes, A.K.; Bjørang, O.; Wu, C.; Nesland, J.M.; Davidson, B. Tenascin-X is a Novel Diagnostic Marker of Malignant Mesothelioma. Am. J. Surg. Pathol. 2009, 33, 1673–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Jensen, R.V.; De Rienzo, A.; Gordon, G.J.; Xu, Y.; Sugarbaker, D.J.; Bueno, R. Differentially expressed alternatively spliced genes in Malignant Pleural Mesothelioma identified using massively parallel transcriptome sequencing. BMC Med. Genet. 2009, 10, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Røe, O.D.; Anderssen, E.; Helge, E.; Pettersen, C.H.; Olsen, K.S.; Sandeck, H.; Haaverstad, R.; Lundgren, S.; Larsson, E. Genome-Wide Profile of Pleural Mesothelioma versus Parietal and Visceral Pleura: The Emerging Gene Portrait of the Mesothelioma Phenotype. PLoS ONE 2009, 4, e6554. [Google Scholar] [CrossRef] [Green Version]
- Røe, O.D.; Anderssen, E.; Sandeck, H.; Christensen, T.; Larsson, E.; Lundgren, S. Malignant pleural mesothelioma: Genome-wide expression patterns reflecting general resistance mechanisms and a proposal of novel targets. Lung Cancer 2010, 67, 57–68. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Yu, A.; Kim, S.; Kang, J.H.S. Robust likelihood-based survival modeling with microarray data. J. Stat. Softw. 2009, 29, 1–16. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in R, version 3.2-7; 2020. Available online: https://github.com/therneau/survival (accessed on 10 January 2021).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||||||||||
| Multivariate Cox Results | |||||||||||
| Symbol | Ensembl ID | Coef | HR | Se(Coef) | Z | p-Value | |||||
| SPARC | ENSG00000113140.9 | 1.40 | 4.05 | 0.53 | 2.63 | 8.44 × 10−3 | |||||
| CIT | ENSG00000122966.12 | 2.03 | 7.64 | 0.65 | 3.11 | 1.9 × 10−3 | |||||
| TRAF2 | ENSG00000127191.16 | 3.26 | 26.13 | 0.97 | 3.36 | 7.86 × 10−4 | |||||
| PODXL | ENSG00000128567.15 | 0.74 | 2.10 | 0.32 | 2.29 | 2.2 × 10−2 | |||||
| KIF23 | ENSG00000137807.12 | 3.94 | 51.57 | 1.19 | 3.31 | 9.21 × 10−4 | |||||
| PRSS23 | ENSG00000150687.10 | 1.20 | 3.31 | 0.35 | 3.44 | 5.86 × 10−4 | |||||
| UHRF1 | ENSG00000276043.3 | 3.83 | 46.05 | 0.99 | 3.88 | 1.04 × 10−4 | |||||
| (B) | |||||||||||
| Multivariate Cox Results | |||||||||||
| Symbol | Ensembl ID | Coef | HR | Se(Coef) | Z | p-Value | |||||
| MAD2L1 | ENSG00000164109.12 | 1.85 | 6.34 | 0.40 | 4.68 | 2.93 × 10−6 | |||||
| KIF23 | ENSG00000137807.12 | 1.39 | 4.02 | 0.67 | 2.08 | 3.75 × 10−2 | |||||
| UHRF1 | ENSG00000276043.3 | 1.35 | 3.87 | 0.52 | 2.62 | 8.86 × 10−3 | |||||
| ADAMTS1 | ENSG00000154734.13 | 0.68 | 1.98 | 0.24 | 2.89 | 3.86 × 10−3 | |||||
| TNNT1 | ENSG00000105048.15 | 0.63 | 1.87 | 0.23 | 2.78 | 5.43 × 10−3 | |||||
| BAG2 | ENSG00000112208.11 | 0.40 | 1.49 | 0.17 | 2.39 | 1.71 × 10−2 | |||||
| (C) | |||||||||||
| Multivariate Cox Results | |||||||||||
| Symbol | Ensembl ID | Coef | HR | Se(Coef) | Z | p-Value | |||||
| MDK | ENSG00000110492.14 | 1.46 | 4.29 | 0.40 | 3.60 | 3.17 × 10−4 | |||||
| SELE | ENSG00000007908.14 | 1.09 | 2.99 | 0.33 | 3.29 | 9.88 × 10−4 | |||||
| CTHRC1 | ENSG00000164932.11 | 1.69 | 5.40 | 0.70 | 2.40 | 1.66 × 10−2 | |||||
| DSC3 | ENSG00000134762.15 | 0.86 | 2.37 | 0.39 | 2.23 | 2.57 × 10−2 | |||||
| Variable | Beta | HR (95% CI for HR) | Wald Test | p-Value |
|---|---|---|---|---|
| Age at index median | 0.29 | 1.3 (0.83–2.1) | 1.5 | 0.23 |
| Primary diagnosis: epithelioid vs. other | −0.64 | 0.53 (0.32–0.86) | 6.4 | 0.01 |
| Primary diagnosis: biphasic vs. other | 0.63 | 1.9 (1.1–3.2) | 5.3 | 0.02 |
| Gender: male vs. female | −0.008 | 0.99 (0.54–1.8) | 0 | 0.98 |
| Tumor stage | −0.036 | 0.96 (0.58–1.6) | 0.02 | 0.89 |
| RS: unreduced median | 1.4 | 4 (2.4–6.7) | 28 | 1.2 × 10−7 |
| RS: FDR-reduced median | 1.5 | 4.3 (2.6–7.2) | 31 | 2.6 × 10−8 |
| RS: GO-reduced median | 0.98 | 2.7 (1.6–4.3) | 16 | 7.6 × 10−5 |
| Variable | Beta | HR | Se(Coef) | Z | p-Value |
|---|---|---|---|---|---|
| Age at index median | 0.192 | 12.122 | 0.268 | 0.718 | 0.472 |
| Primary diagnosis: epithelioid vs. other | −0.625 | 0.535 | 0.463 | −1.352 | 0.176 |
| Primary diagnosis: biphasic vs. other | −0.280 | 0.756 | 0.512 | −0.547 | 0.584 |
| Gender: male vs. female | −0.480 | 0.619 | 0.342 | −1.405 | 0.160 |
| Tumor stage | −0.317 | 0.728 | 0.282 | −1.124 | 0.261 |
| RS: unreduced median | 0.683 | 19.800 | 0.591 | 1.156 | 0.248 |
| RS: FDR-reduced median | 12.228 | 33.967 | 0.469 | 2.607 | 0.009 |
| RS: GO-reduced median | 0.980 | 26.655 | 0.279 | 3.520 | 4 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morani, F.; Bisceglia, L.; Rosini, G.; Mutti, L.; Melaiu, O.; Landi, S.; Gemignani, F. Identification of Overexpressed Genes in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2021, 22, 2738. https://doi.org/10.3390/ijms22052738
Morani F, Bisceglia L, Rosini G, Mutti L, Melaiu O, Landi S, Gemignani F. Identification of Overexpressed Genes in Malignant Pleural Mesothelioma. International Journal of Molecular Sciences. 2021; 22(5):2738. https://doi.org/10.3390/ijms22052738
Chicago/Turabian StyleMorani, Federica, Luisa Bisceglia, Giulia Rosini, Luciano Mutti, Ombretta Melaiu, Stefano Landi, and Federica Gemignani. 2021. "Identification of Overexpressed Genes in Malignant Pleural Mesothelioma" International Journal of Molecular Sciences 22, no. 5: 2738. https://doi.org/10.3390/ijms22052738