
Phenotypic Plasticity of Cancer Cells Based on Remodeling of the Actin Cytoskeleton and Adhesive Structures



Abstract
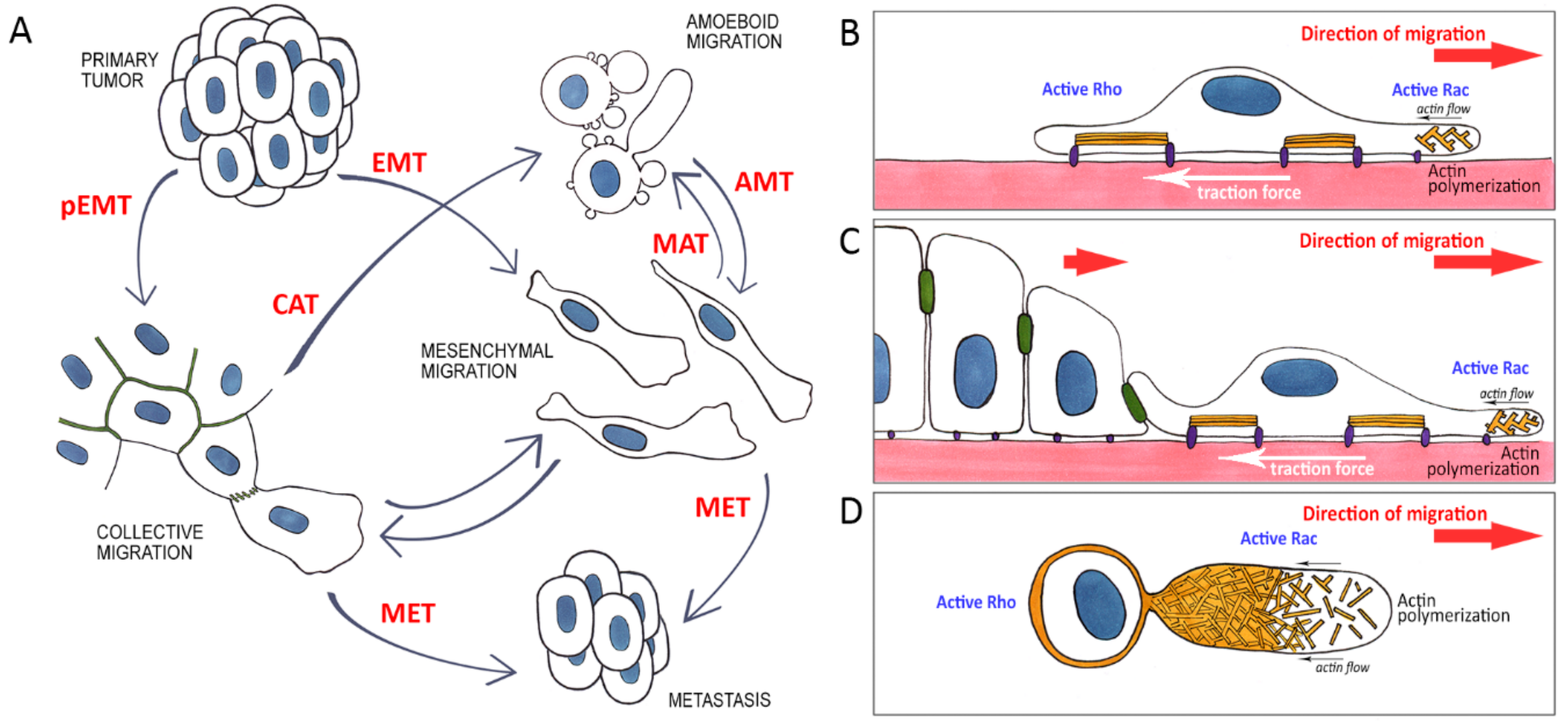
:1. Introduction
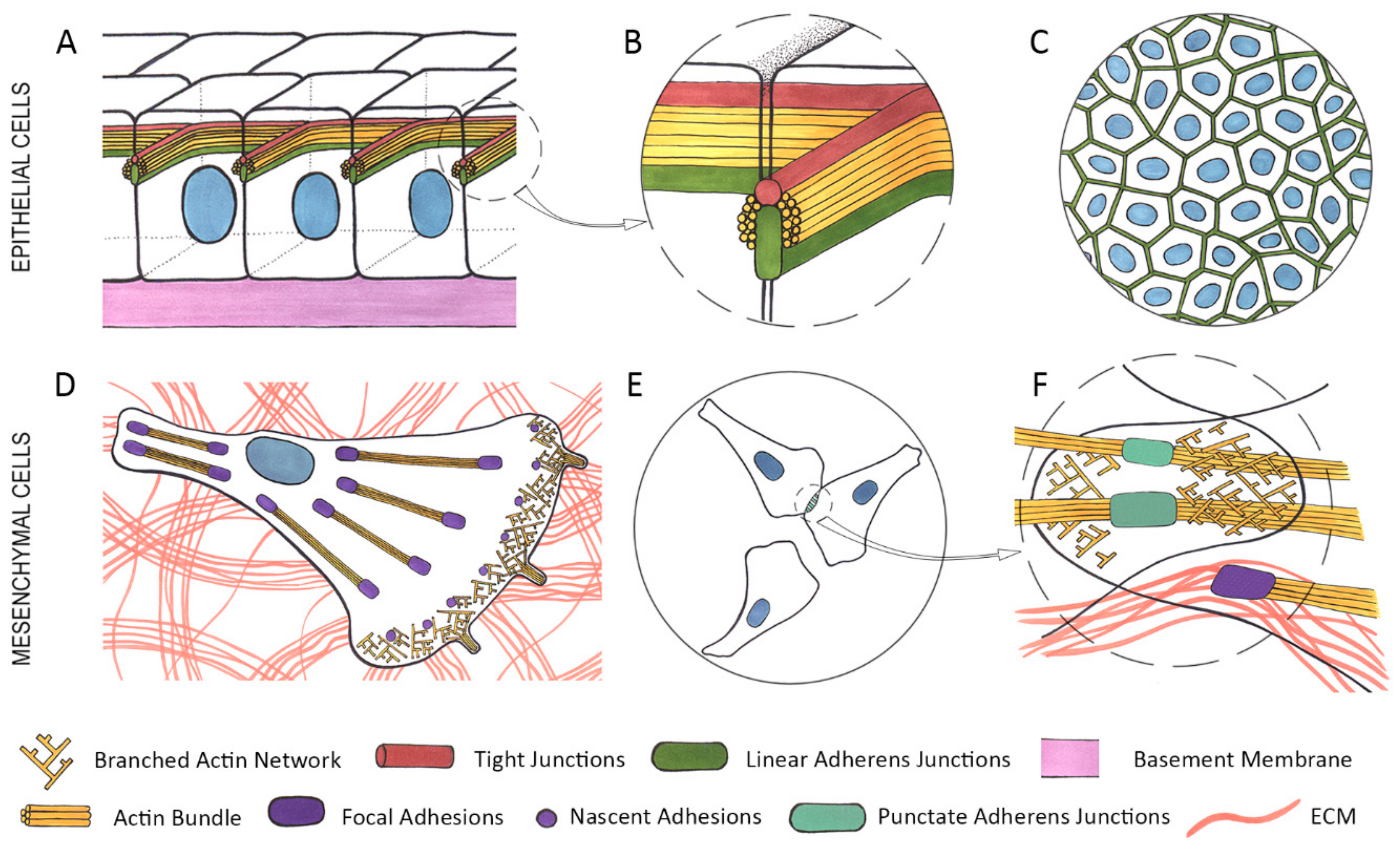
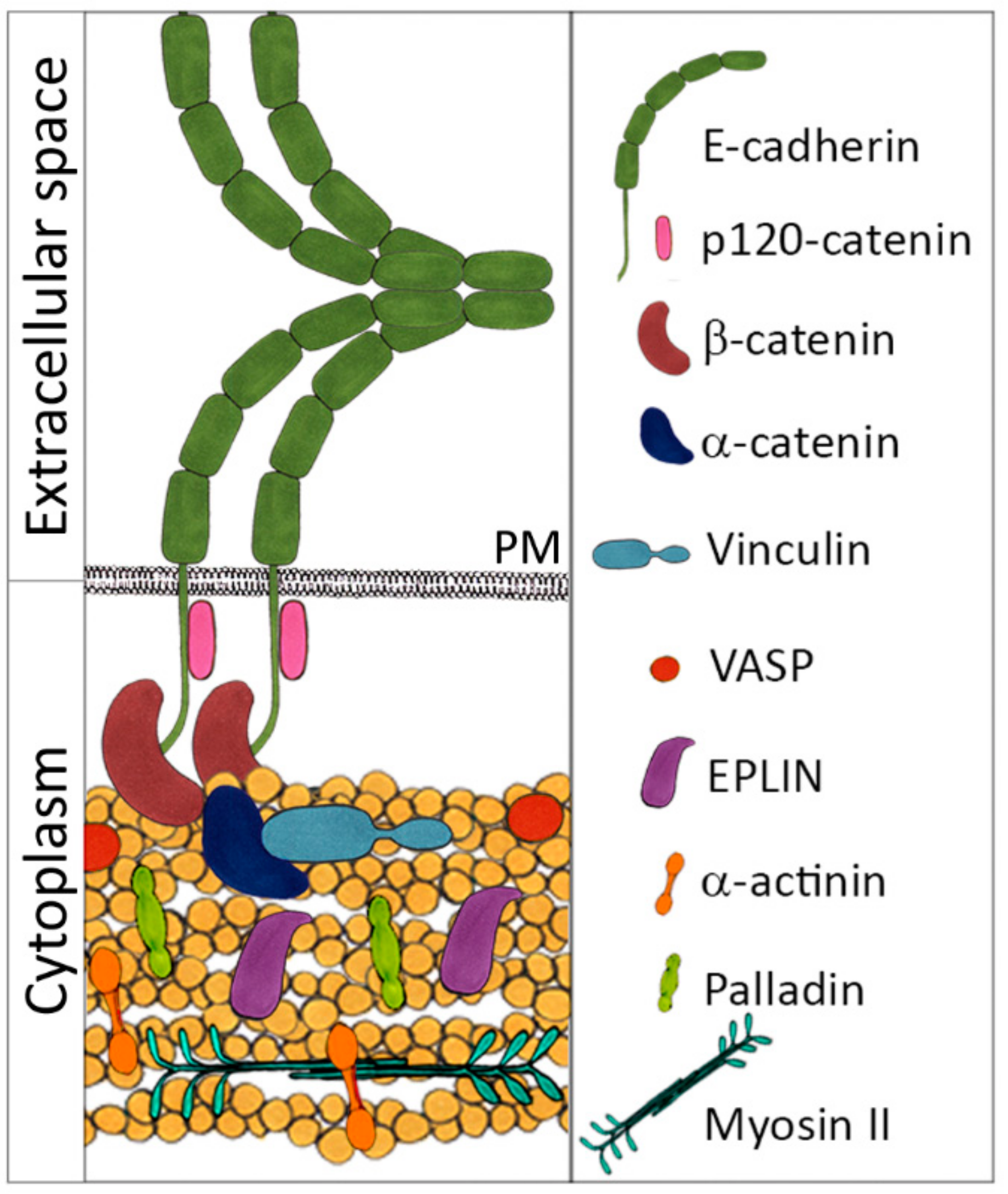
2. Epithelial Cells
3. Epithelial-Mesenchymal Transition (EMT)
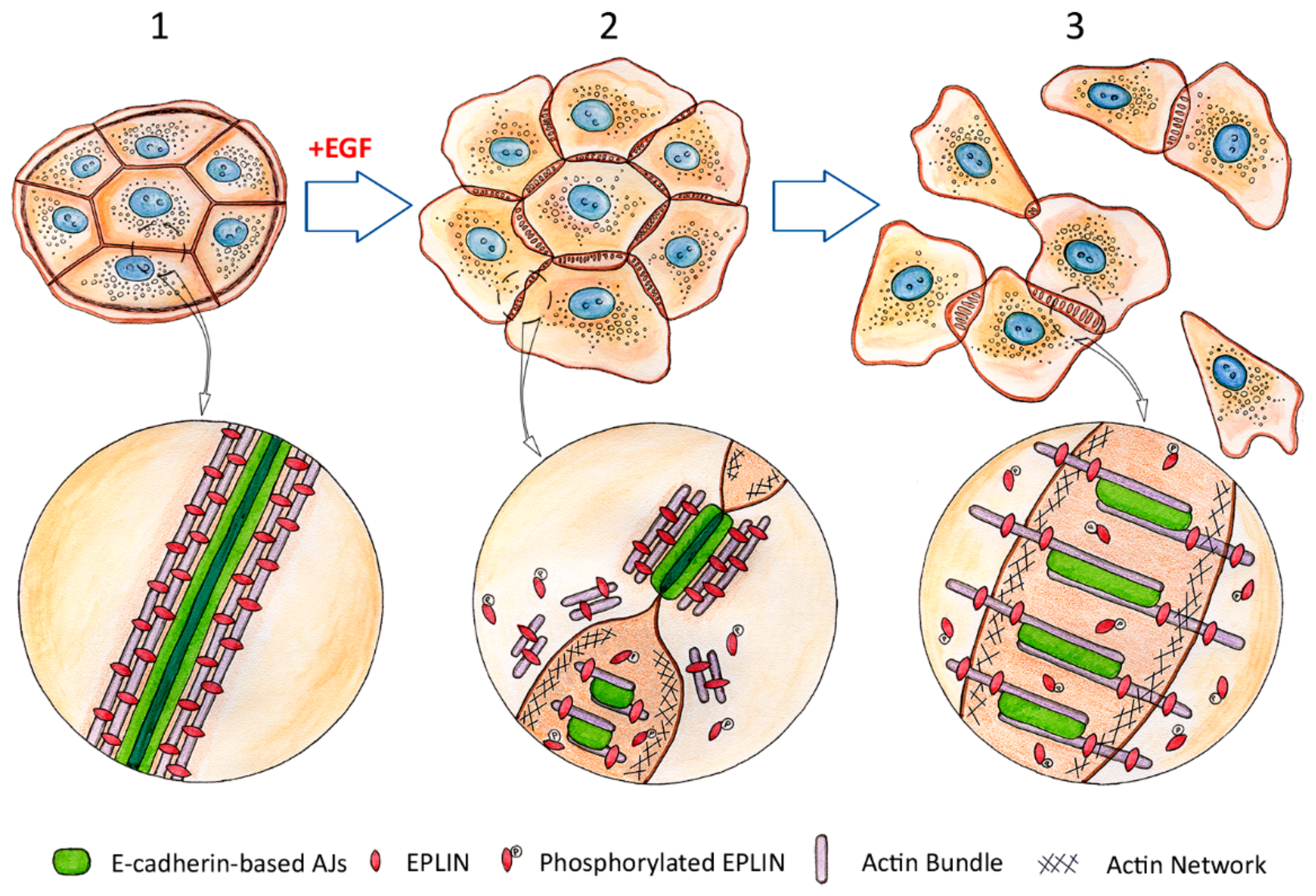
4. Hybrid Epithelial-Mesenchymal Phenotype
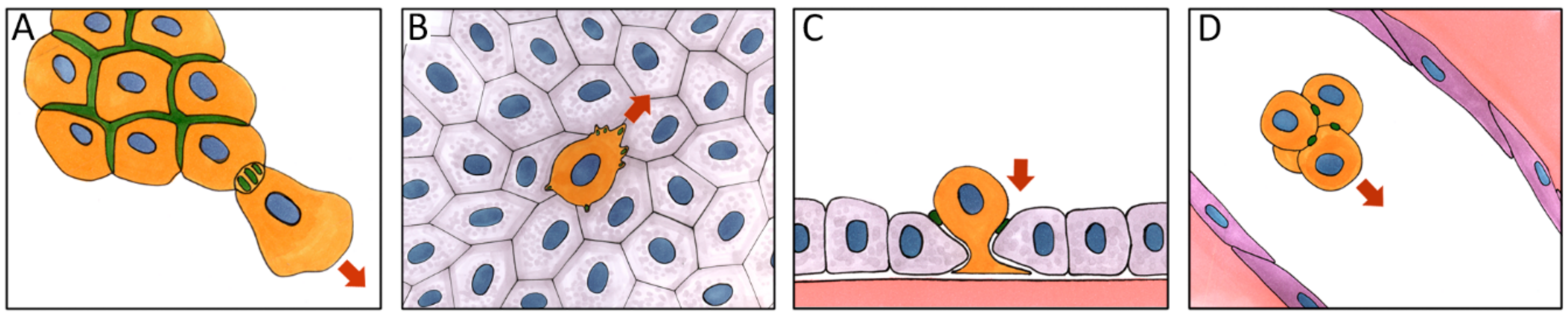
5. Role of E-Cadherin in Carcinoma Cell Dissemination
6. Rearrangement of the Cytoskeleton and Adhesive Structures in Cells Undergoing EMT
7. Mesenchymal Migration
8. Amoeboid Migration
9. Mesenchymal-Ameboid Transition (MAT)
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takeichi, M. Dynamic contacts: Rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell. Biol. 2014, 15, 397–410. [Google Scholar] [CrossRef]
- Vasileva, E.; Citi, S. The role of microtubules in the regulation of epithelial junctions. Tissue Barriers 2018, 6, 1539596. [Google Scholar] [CrossRef] [Green Version]
- Riga, A.; Castiglioni, V.G.; Boxem, M. New insights into apical-basal polarization in epithelia. Curr. Opin. Cell Biol. 2020, 62, 1–8. [Google Scholar] [CrossRef]
- Takeichi, M. Self-organization of animal tissues: Cadherin-mediated processes. Dev. Cell 2011, 21, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.A.; Ireton, R.C.; Reynolds, A.B. A core function for p120-catenin in cadherin turnover. J. Cell Biol. 2003, 163, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Yonemura, S.; Wada, Y.; Watanabe, T.; Nagafuchi, A.; Shibata, M. Alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Ishiyama, N.; Tanaka, N.; Abe, K.; Yang, Y.J.; Abbas, Y.M.; Umitsu, M.; Nagar, B.; Bueler, S.A.; Rubinstein, J.L.; Takeichi, M.; et al. An autoinhibited structure of α-catenin and its implications for vinculin recruitment to adherens junctions. J. Biol. Chem. 2013, 288, 15913–15925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, N.; Sarpal, R.; Wood, M.N.; Barrick, S.K.; Nishikawa, T.; Hayashi, H.; Kobb, A.B.; Flozak, A.S.; Yemelyanov, A.; Fernandez-Gonzalez, R.; et al. Force-dependent allostery of the α-catenin actin-binding domain controls adherens junction dynamics and functions. Nat. Commun. 2018, 9, 5121. [Google Scholar] [CrossRef]
- Maki, K.; Han, S.W.; Hirano, Y.; Yonemura, S.; Hakoshima, T.; Adachi, T. Mechano-adaptive sensory mechanism of α-catenin under tension. Sci. Rep. 2016, 6, 24878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, V. Spatial integration of E-cadherin adhesion, signalling and the epithelial cytoskeleton. Curr. Opin. Cell Biol. 2016, 42, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mège, R.M.; Ishiyama, N. Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harb. Perspect Biol. 2017, 9, a028738. [Google Scholar] [CrossRef] [Green Version]
- Bertocchi, C.; Wang, Y.; Ravasio, A.; Hara, Y.; Wu, Y.; Sailov, T.; Baird, M.A.; Davidson, M.W.; Zaidel-Bar, R.; Toyama, Y. Nanoscale architecture of cadherin-based cell adhesions. Nat. Cell Biol. 2017, 19, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maul, R.S.; Song, Y.; Amann, K.J.; Gerbin, S.C.; Pollard, T.D.; Chang, D.D. EPLIN regulates actin dynamics by cross-linking and stabilizing filaments. J. Cell Biol. 2003, 160, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Takeichi, M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc. Natl. Acad. Sci. USA 2008, 105, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, M. Nonmuscle myosin IIA is involved in recruitment of apical junction components through activation of α-catenin. Biol. Open 2018, 7, bio031369. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.I.; Hunt, D.; Utech, M.; Nusrat, A.; Parkos, C.A. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junctional complex. Mol. Biol. Cell 2005, 16, 2636–2650. [Google Scholar] [CrossRef] [Green Version]
- Smutny, M.; Cox, H.L.; Leerberg, J.M.; Kovacs, E.M.; Conti, M.A.; Ferguson, C.; Hamilton, N.A.; Parton, R.G.; Adelstein, R.S.; Yap, A.S. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat. Cell Biol. 2010, 12, 696–702. [Google Scholar] [CrossRef]
- Carramusa, L.; Ballestrem, C.; Zilberman, Y.; Bershadsky, A.D. Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J. Cell Sci. 2007, 120, 3870–3882. [Google Scholar] [CrossRef] [Green Version]
- Grikscheit, K.; Frank, T.; Wang, Y.; Grosse, R. Junctional actin assembly is mediated by Formin-like 2 downstream of Rac1. J. Cell Biol. 2015, 209, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloushankova, N.A.; Alieva, N.A.; Krendel, M.F.; Bonder, E.M.; Feder, H.H.; Vasiliev, J.M.; Gelfand, I.M. Cell-cell contact changes the dynamics of lamellar activity in nontransformed epitheliocytes but not in their ras-transformed descendants. Proc. Natl. Acad. Sci. USA 1997, 94, 879–883. [Google Scholar] [CrossRef] [Green Version]
- Gloushankova, N.A.; Krendel, M.F.; Alieva, N.O.; Bonder, E.M.; Feder, H.H.; Vasiliev, J.M.; Gelfand, I.M. Dynamics of contacts between lamellae of fibroblasts: Essential role of the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 1998, 95, 4362–4367. [Google Scholar] [CrossRef] [Green Version]
- Ayollo, D.V.; Zhitnyak, I.Y.; Vasiliev, J.M.; Gloushankova, N.A. Rearrangements of the actin cytoskeleton and E-cadherin-based adherens junctions caused by neoplasic transformation change cell-cell interactions. PLoS ONE 2009, 4, e8027. [Google Scholar] [CrossRef] [Green Version]
- Stramer, B.; Mayor, R. Mechanisms and in vivo functions of contact inhibition of locomotion. Nat. Rev. Mol. Cell Biol. 2017, 18, 43–55. [Google Scholar] [CrossRef]
- Krendel, M.F.; Bonder, E.M. Analysis of actin filament bundle dynamics during contact formation in live epithelial cells. Cell Motil. Cytoskeleton 1999, 43, 296–309. [Google Scholar] [CrossRef]
- Yamada, S.; Nelson, W.J. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J. Cell Biol. 2007, 178, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.W.; Nagaraj, S.H. Transition states that allow cancer to spread. Nature 2018, 556, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Nikolaou, S.; Machesky, L.M. The stressful tumour environment drives plasticity of cell migration programmes, contributing to metastasis. J. Pathol. 2020, 250, 612–623. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Rosivatz, E.; Becker, I.; Specht, K.; Fricke, E.; Luber, B.; Busch, R.; Höfler, H.; Becker, K.F. Differential expression of the epithelial-mesenchymal transition regulators Snail, SIP1, and Twist in gastric cancer. Am. J. Pathol. 2002, 161, 1881–1891. [Google Scholar] [CrossRef]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Spaderna, S.; Schmalhofer, O.; Wahlbuhl, M.; Dimmler, A.; Bauer, K.; Sultan, A.; Hlubek, F.; Jung, A.; Strand, D.; Eger, A.; et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008, 68, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, C.; Lahes, S.; Radhakrishnan, P.; Dutta, S.; Mogler, C.; Herpel, E.; Brand, K.; Steinert, G.; Schneider, M.; Mollenhauer, M.; et al. Overexpression of ZEB2 at the invasion front of colorectal cancer is an independent prognostic marker and regulates tumor invasion in vitro. Clin. Cancer Res. 2011, 17, 7654–7663. [Google Scholar] [CrossRef] [Green Version]
- Shioiri, M.; Shida, T.; Koda, K.; Oda, K.; Seike, K.; Nishimura, M.; Takano, S.; Miyazaki, M. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br. J. Cancer 2006, 94, 1816–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial– mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Hazan, R.B.; Phillips, G.R.; Qiao, R.F.; Norton, L.; Aaronson, S.A. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 2000, 148, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Seetharaman, S.; Etienne-Manneville, S. Cytoskeletal Crosstalk in Cell Migration. Trends Cell Biol. 2020, 30, 720–735. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, C.; Kim, E.J.; Jang, J.; Kim, S.-J.; Kang, S.M.; Kim, M.G.; Jung, H.; Park, D.; Kim, C. Vimentin filaments regulate integrin-ligand interactions by binding to the cytoplasmic tail of integrin β3. J. Cell Sci. 2016, 129, 2030–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- De Craene, B.; Gilbert, B.; Stove, C.; Bruyneel, E.; van Roy, F.; Berx, G. The transcription factor snail induces tumor cell invasion through modulation of the epithelial cell differentiation program. Cancer Res. 2005, 65, 6237–6244. [Google Scholar] [CrossRef] [Green Version]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajita, M.; McClinic, K.N.; Wade, P.A. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol. Cell Biol. 2004, 24, 7559–7566. [Google Scholar] [CrossRef] [Green Version]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraguchi, M.; Okubo, T.; Miyashita, Y.; Miyamoto, Y.; Hayashi, M.; Crotti, T.N.; McHugh, K.P.; Ozawa, M. Snail Regulates Cell-Matrix Adhesion by Regulation of the Expression of Integrins and Basement Membrane Proteins. J. Biol. Chem. 2008, 283, 23514–23523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, M.A.; Krantz, S.B.; Bentrem, D.J.; Dangi-Garimella, S.; Munsh, H.G. Interplay between β1-Integrin and Rho Signaling Regulates Differential Scattering and Motility of Pancreatic Cancer Cells by Snail and Slug Proteins. J. Biol. Chem. 2012, 287, 6218–6229. [Google Scholar] [CrossRef] [Green Version]
- Whiteman, E.L.; Liu, C.-J.; Fearon, E.R.; Margolis, B. The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene 2008, 27, 3875–3879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, K.; Kamata, N.; Fujimoto, R.; Tsutsumi, S.; Tomonari, M.; Taki, M.; Hosokawa, H.; Nagayama, M. Increased invasion and matrix metalloproteinase-2 expression by Snail-induced mesenchymal transition in squamous cell carcinomas. Int. J. Oncol. 2003, 22, 891–898. [Google Scholar] [CrossRef]
- Miyoshi, A.; Kitajima, Y.; Sumi, K.; Sato, K.; Hagiwara, A.; Koga, Y.; Miyazaki, K. Snail and SIP1 increase cancer invasion by upregulating MMP family in hepatocellular carcinoma cells. Br. J. Cancer 2004, 90, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-F.; Chen, J.-Y.; Ho, Y.-H.; Hsu, W.-H.; Wu, L.-C.; Lan, H.-Y.; Hsu, D.S.-S.; Tai, S.-K.; Chang, Y.-C.; Yang, M.-H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef]
- Taki, M.; Verschueren, K.; Yokoyama, K.; Nagayama, M.; Kamata, N. Involvement of Ets-1 transcription factor in inducing matrix metalloproteinase-2 expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Int. J. Oncol. 2006, 28, 487–496. [Google Scholar] [CrossRef]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar]
- Bolos, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wade, P.; Mandell, K.J.; Akyildiz, A.; Parkos, C.A.; Mrsny, R.J.; Nusrat, A. Raf 1 represses expression of the tight junction protein occludin via activation of the zinc-finger transcription factor slug. Oncogene 2007, 26, 1222–1230. [Google Scholar] [CrossRef] [Green Version]
- Kwok, W.K.; Ling, M.-T.; Lee, T.-W.; Lau, T.C.M.; Zhou, C.; Zhang, X.; Chua, C.W.; Chan, K.W.; Chan, F.L.; Glackin, C.; et al. Up-Regulation of TWIST in Prostate Cancer and Its Implication as a Therapeutic Target. Cancer Res. 2005, 65, 5153–5162. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, N.; Shiraha, H.; Fujikawa, T.; Takaoka, N.; Ueda, N.; Tanaka, S.; Nishina, S.; Nakanishi, Y.; Uemura, M.; Takaki, A.; et al. Twist expression promotes migration and invasion in hepatocellular carcinoma. BMC Cancer 2009, 9, 240. [Google Scholar] [CrossRef] [Green Version]
- Alexander, N.R.; Tran, N.L.; Rekapally, H.; Summers, C.E.; Glackin, C.; Heimark, R.L. N-cadherin gene expression in prostate carcinoma is modulated by integrin dependent nuclear translocation of Twist1. Cancer Res. 2006, 66, 3365–3369. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Chen, S.; Han, J.-X.; Qian, B.; Wang, X.-R.; Zhong, W.-L.; Qin, Y.; Zhang, H.; Gao, W.-F.; Lei, Y.-Y.; et al. Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4150–4162. [Google Scholar] [CrossRef] [Green Version]
- Nam, E.-H.; Lee, Y.; Moon, B.; Lee, J.W.; Kim, S. Twist1 and AP-1 cooperatively upregulate integrin α5 expression to induce invasion and the epithelial-mesenchymal transition. Carcinogenesis 2015, 36, 327–337. [Google Scholar] [CrossRef]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.M.; Strohbehn, G.; Bair, T.B.; Moreland, J.G.; Henry, M.D. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol. Biol. Cell. 2009, 20, 2207–2217. [Google Scholar] [CrossRef] [Green Version]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef] [Green Version]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like Growth Factor-I–Dependent Up-regulation of ZEB1 Drives Epithelial-to-Mesenchymal Transition in Human Prostate Cancer Cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Tilló, E.; Lázaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewalle, C.; Comijn, J.; De Craene, B.; Vermassen, P.; Bruyneel, E.; Andersen, H.; Tulchinsky, E.; Van Roy, F.; Berx, G. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005, 33, 6566–6578. [Google Scholar] [CrossRef] [PubMed]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noël, A.; van Roy, F.; Berx, G.; Foidart, J.M.; Gilles, C. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osorio, L.A.; Farfán, N.M.; Castellón, E.A.; Contreras, H.R. SNAIL transcription factor increases the motility and invasive capacity of prostate cancer cells. Mol. Med. Rep. 2016, 13, 778–786. [Google Scholar] [CrossRef] [Green Version]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell. Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visciano, C.; Liotti, F.; Prevete, N.; Cali’, G.; Franco, R.; Collina, F.; de Paulis, A.; Marone, G.; Santoro, M.; Melillo, R.M. Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene 2015, 34, 5175–5186. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell. Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Zhang, Q.; Zhang, Q.; Sun, P.; Xiang, R.; Ren, G.; Yang, S. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. 2018, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, P.G.; Moreno-Bueno, G.; Portillo, F.; Cano, A. EMT: Present and future in clinical oncology. Mol. Oncol. 2017, 11, 718–738. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schäfer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Bajor, D.L.; Norgard, R.J.; Sahmoud, A.; Bhagwat, N.; Pham, M.N.; Cornish, T.C.; Iacobuzio-Donahue, C.A.; Vonderheide, R.H.; Stanger, B.Z. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 2016, 7, 12819. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.-Y.; et al. Single-cell analysis reveals a stem cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhu, X.; Cui, K.; Mancuso, J.; Federley, R.; Fischer, K.; Teng, G.; Mittal, V.; Gao, D.; Zhao, H.; et al. In Vivo Visualization and Characterization of Epithelial-Mesenchymal Transition in Breast Tumors. Cancer Res. 2016, 76, 2094–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Fuyuhiro, Y.; Yashiro, M.; Noda, S.; Matsuoka, J.; Hasegawa, T.; Kato, Y.; Sawada, T.; Hirakawa, K. Cancer-associated orthotopic myofibroblasts stimulates the motility of gastric carcinoma cells. Cancer Sci. 2012, 103, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kandl, C.; D’Arcy Hamilton, C.; Shnayder, Y.; Tsue, T.; Kakarala, K.; Ledgerwood, L.; Sun, X.S.; Huang, H.; Girod, D.; et al. Mitigation of tumor-associated fibroblast-facilitated head and neck cancer progression with anti–hepatocyte growth factor antibody ficlatuzumab. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 1133–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Vanesa Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.-H.; Sun, R.; Zhou, X.-M.; Zhang, M.-Y.; Lu, J.-B.; Yang, Y.; Zeng, L.-S.; Yang, X.-Z.; Shi, L.; Xiao, R.-W.; et al. Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 2018, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell 2017, 20, 191–204.e5. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.; Herold, C.I.; Marcom, P.K.; George, D.; Garcia-Blanco, M.A. Circulating Tumor Cells from Patients with Advanced Prostate and Breast Cancer Display Both Epithelial and Mesenchymal Markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Liu, S.; Liu, Z.; Huang, J.; Pu, X.; Li, J.; Yang, D.; Deng, H.; Yang, N.; Xu, J. Classification of circulating tumor cells by epithelial-mesenchymal transition markers. PLoS ONE 2015, 10, e0123976. [Google Scholar] [CrossRef]
- De Wit, S.; Manicone, M.; Rossi, E.; Lampignano, R.; Yang, L.; Zill, B.; Rengel-Puertas, A.; Ouhlen, M.; Crespo, M.; Berghuis, A.M.S.; et al. EpCAM high and EpCAM low circulating tumor cells in metastatic prostate and breast cancer patients. Oncotarget 2018, 9, 35705–35716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadaki, M.A.; Stoupis, G.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol. Cancer Ther. 2019, 18, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Z.; Wang, C.; Ye, Z.; Austin, L.; Civan, J.; Hyslop, T.; Palazzo, J.P.; Jaslow, R.; Li, B.; Myers, R.E.; et al. Prospective assessment of the prognostic value of circulating tumor cells and their clusters in patients with advanced-stage breast cancer. Breast Cancer Res. Treat. 2015, 154, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Kulasinghe, A.; Schmidt, H.; Perry, C.; Whitfield, B.; Kenny, L.; Nelson, C.; Warkiani, M.E.; Punyadeera, C. A Collective Route to Head and Neck Cancer Metastasis. Sci. Rep. 2018, 8, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murlidhar, V.; Reddy, R.M.; Fouladdel, S.; Zhao, L.; Ishikawa, M.K.; Grabauskiene, S.; Zhang, Z.; Lin, J.; Chang, A.C.; Carrott, P.; et al. Poor Prognosis Indicated by Venous Circulating Tumor Cell Clusters in Early-Stage Lung Cancers. Cancer Res. 2017, 77, 5194–5206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.M.; Krebs, M.; Ward, T.; Sloane, R.; Priest, L.; Hughes, A.; Clack, G.; Ranson, M.; Blackhall, F.; Dive, C. Circulating tumor cells as a window on metastasis biology in lung cancer. Am. J. Pathol. 2011, 178, 989–996. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Au, S.H.; Storey, B.D.; Moore, J.C.; Tang, Q.; Chen, Y.L.; Javaid, S.; Sarioglu, A.F.; Sullivan, R.; Madden, M.W.; O’Keefe, R.; et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. USA 2016, 113, 4947–4952. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Berx, G.; van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef]
- Ling, Z.Q.; Li, P.; Ge, M.H.; Zhao, X.; Hu, F.J.; Fang, X.H.; Dong, Z.M.; Mao, W.M. Hypermethylation-modulated down-regulation of CDH1 expression contributes to the progression of esophageal cancer. Int. J. Mol. Med. 2011, 27, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Johnson, J.P.; Leitl, F.; Jauch, K.W.; Heiss, M.M.; Schildberg, F.W.; Birchmeier, W.; Funke, I. E-cadherin expression in primary and metastatic gastric cancer: Down-regulation correlates with cellular dedifferentiation and glandular disintegration. Cancer Res. 1993, 53, 1690–1695. [Google Scholar]
- Umbas, R.; Schalken, J.A.; Aalders, T.W.; Carter, B.S.; Karthaus, H.F.; Schaafsma, H.E.; Debruyne, F.M.; Isaacs, W.B. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992, 52, 5104–5109. [Google Scholar] [PubMed]
- Schuhmacher, C.; Becker, I.; Oswald, S.; Atkinson, M.J.; Nekarda, H.; Becker, K.F.; Mueller, J.; Siewert, J.R.; Höfler, H. Loss of immunohistochemical E-cadherin expression in colon cancer is not due to structural gene alterations. Virchows Arch. 1999, 434, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Yan, H.X.; Liu, S.Q.; Chen, L.; Wu, M.C.; Wang, H.Y. Reduced expression of E-cadherin/catenin complex in hepatocellular carcinomas. World J. Gastroenterol. 2008, 14, 5665–5673. [Google Scholar] [CrossRef]
- Jeschke, U.; Mylonas, I.; Shabani, N.; Kunert-Keil, C.; Schindlbeck, C.; Gerber, B.; Friese, K. Expression of sialyllewis X, sialyl Lewis A, E-cadherin and cathepsin-D in human breast cancer: Immunohistochemical analysis in mammary carcinoma in situ, invasive carcinomas and their lymph node metastasis. Anticancer Res. 2005, 25, 1615–1622. [Google Scholar]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Karpova, T.; Sheppard, A.M.; McNally, J.; Lowy, D.R. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004, 23, 1739–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrais, M.; Chen, X.; Perez-Moreno, M.; Gumbiner, B.M. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell 2007, 18, 2013–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtidis, A.; Necela, B.; Lin, W.H.; Lu, R.; Feathers, R.W.; Asmann, Y.W.; Thompson, E.A.; Anastasiadis, P.Z. Cadherin complexes recruit mRNAs and RISC to regulate epithelial cell signaling. J. Cell Biol. 2017, 216, 3073–3085. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Koizumi, H.; Ihara, A.; Ohta, T.; Uchikoshi, T. Expression of beta-catenin in normal breast tissue and breast carcinoma: A comparative study with epithelial cadherin and alpha-catenin. Histopathology 1996, 29, 139–146. [Google Scholar] [CrossRef]
- Kartenbeck, J.; Haselmann, U.; Gassler, N. Synthesis of junctional proteins in metastasizing colon cancer cells. Eur. J. Cell Biol. 2005, 84, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef]
- Whittle, M.C.; Hingorani, S.R. Disconnect between EMT and metastasis in pancreas cancer. Oncotarget 2015, 6, 30445–30446. [Google Scholar] [CrossRef]
- Rubin, M.A.; Mucci, N.R.; Figurski, J.; Fecko, A.; Pienta, K.J.; Day, M.L. E-cadherin expression in prostate cancer: A broad survey using high-density tissue microarray technology. Hum. Pathol. 2001, 32, 690–697. [Google Scholar] [CrossRef]
- Vered, M.; Allon, I.; Buchner, A.; Dayan, D. E-cadherin in oral SCC: An analysis of the confusing literature and new insights related to its immunohistochemical expression. Histol. Histopathol. 2012, 27, 141–150. [Google Scholar] [CrossRef]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Gavert, N.; Vivanti, A.; Hazin, J.; Brabletz, T.; Ben-Ze’ev, A. L1-mediated colon cancer cell metastasis does not require changes in EMT and cancer stem cell markers. Mol. Cancer Res. 2011, 9, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Koelzer, V.H.; Zlobec, I.; Lugli, A. Tumor budding in colorectal cancer - ready for diagnostic practice? Hum. Pathol. 2016, 47, 4–19. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Kowalski, P.J.; Rubin, M.A.; Kleer, C.G. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003, 5, R217–R222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocaña, O.H.; Córcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massagué, J.; Benezra, R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [Green Version]
- Del Pozo Martin, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Chakraborty, G.; Zhang, Z.; Akalay, I.; Gadiya, M.; Gao, Y.; Sinha, S.; Hu, J.; Jiang, C.; Akram, M.; et al. Multi-organ Site Metastatic Reactivation Mediated by Non-canonical Discoidin Domain Receptor 1 Signaling. Cell 2016, 166, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitnyak, I.Y.; Gloushankova, N.A. Morphology, cell-cell interactions, and migratory activity of IAR-2 epithelial cells transformed with the RAS oncogene: Contribution of cell adhesion protein E-cadherin. Russ. J. Dev. Biol. 2011, 42, 402–411. [Google Scholar] [CrossRef]
- Rubtsova, S.N.; Zhitnyak, I.Y.; Gloushankova, N.A. A Novel Role of E-Cadherin-Based Adherens Junctions in Neoplastic Cell Dissemination. PLoS ONE 2015, 10, e0133578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indra, I.; Choi, J.; Chen, C.S.; Troyanovsky, R.B.; Shapiro, L.; Honig, B.; Troyanovsky, S.M. Spatial and temporal organization of cadherin in punctate adherens junctions. Proc. Natl. Acad. Sci. USA 2018, 115, E4406–E4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indra, I.; Troyanovsky, R.B.; Shapiro, L.; Honig, B.; Troyanovsky, S.M. Sensing Actin Dynamics through Adherens Junctions. Cell Rep. 2020, 30, 2820–2833. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Nevolo, M.; Lehmann, K.; Downward, J.; Beug, H.; Grieco, M. Raf plus TGFβ-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene 2006, 25, 7117–7130. [Google Scholar] [CrossRef] [Green Version]
- Zhitnyak, I.Y.; Rubtsova, S.N.; Litovka, N.I.; Gloushankova, N.A. Early Events in Actin Cytoskeleton Dynamics and E-Cadherin-Mediated Cell-Cell Adhesion during Epithelial-Mesenchymal Transition. Cells 2020, 9, 578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, X.; Iqbal, S.; Wang, Y.; Osunkoya, A.O.; Chen, Z.; Chen, Z.; Shin, D.M.; Yuan, H.; Wang, Y.A.; et al. Epidermal growth factor promotes protein degradation of epithelial protein lost in neoplasm (EPLIN), a putative metastasis suppressor, during epithelial-mesenchymal transition. J. Biol. Chem. 2013, 288, 1469–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.W.; Collins, S.R.; Meyer, T. Locally excitable Cdc42 signals steer cells during chemotaxis. Nat. Cell Biol. 2016, 18, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Manneville, J.-B.; Nicholls, S.; Ferenczi, M.A.; Hall, A. Cdc42 and Par6–PKC regulate the spatially localized association of Dlg1 and APC to control cell polarization. J. Cell Biol. 2005, 170, 895–901. [Google Scholar] [CrossRef]
- Burute, M.; Prioux, M.; Blin, G.; Truchet, S.; Letort, G.; Tseng, Q.; Bessy, T.; Lowell, S.; Young, J.; Filhol, O.; et al. Polarity Reversal by Centrosome Repositioning Primes Cell Scattering during Epithelial-to-Mesenchymal Transition. Dev. Cell 2017, 40, 168–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiring, J.C.M.; Shneyer, B.I.; Akhmanova, A. Generation and regulation of microtubule network asymmetry to drive cell polarity. Curr. Opin. Cell Biol. 2020, 62, 86–95. [Google Scholar] [CrossRef]
- Mardakheh, F.K.; Paul, A.; Kümper, S.; Sadok, A.; Paterson, H.; Mccarthy, A.; Yuan, Y.; Marshall, C.J. Global Analysis of mRNA, Translation, and Protein Localization: Local Translation Is a Key Regulator of Cell Protrusions. Dev. Cell 2015, 35, 344–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henty-Ridilla, J.L.; Rankova, A.; Eskin, J.A.; Kenny, K.; Goode, B.L. Accelerated actin filament polymerization from microtubule plus ends. Science 2016, 352, 1004–1009. [Google Scholar] [CrossRef] [Green Version]
- De Rooij, J.; Kerstens, A.; Danuser, G.; Schwartz, M.A.; Waterman-Storer, C.M. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J. Cell Biol. 2005, 171, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolega, J. Asymmetric Distribution of Myosin IIB in Migrating Endothelial Cells Is Regulated by a rho-dependent Kinase and Contributes to Tail Retraction. Mol. Biol. Cell 2003, 14, 4745–4757. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Manzanares, M.; Koach, M.A.; Whitmore, L.; Lamers, M.L.; Horwitz, A.F. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 2008, 183, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, A.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef] [Green Version]
- Rottner, K.; Schaks, M. Assembling actin filaments for protrusion. Curr. Opin. Cell Biol. 2019, 56, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campellone, K.G.; Welch, M.D. A Nucleator Arms Race: Cellular Control of Actin Assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burianek, L.E.; Soderling, S.H. Under lock and key: Spatiotemporal regulation of WASP family proteins coordinates separate dynamic cellular processes. Semin. Cell Dev. Biol. 2013, 24, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, H.; Suetsugu, S.; Takenawa, T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998, 17, 6932–6941. [Google Scholar] [CrossRef] [Green Version]
- Barzik, M.; Kotova, T.I.; Higgs, H.N.; Hazelwood, L.; Hanein, D.; Gertler, F.B.; Schafer, D.A. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J. Biol. Chem. 2005, 280, 28653–28662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Urbanke, C.; Resch, G.P.; Small, J.V.; Faix, J. Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J. 2008, 27, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Romero, S.; Le Clainche, C.; Didry, D.; Egile, C.; Pantaloni, D.; Carlier, M.-F. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell 2004, 119, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Law, A.L.; Vehlow, A.; Kotini, M.; Dodgson, L.; Soong, D.; Theveneau, E.; Bodo, C.; Taylor, E.; Navarro, C.; Perera, U.; et al. Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J. Cell Biol. 2013, 203, 673–689. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Leslie, J.S.; Stewart, M.; Lafuente, E.M.; Valderrama, F.; Jagannathan, R.; Strasser, G.A.; Rubinson, D.A.; Liu, H.; Way, M.; et al. Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev. Cell 2004, 7, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Dimchev, G.; Amiri, B.; Humphries, A.C.; Schaks, M.; Dimchev, V.; Stradal, T.E.B.; Faix, J.; Krause, M.; Way, M.; Falcke, M.; et al. Lamellipodin tunes cell migration by stabilizing protrusions and promoting adhesion formation. J. Cell Sci. 2020, 133, jcs239020. [Google Scholar] [CrossRef] [PubMed]
- Plastino, J.; Blanchoin, L. Dynamic stability of the actin ecosystem. J. Cell Sci. 2019, 132, jcs219832. [Google Scholar] [CrossRef] [Green Version]
- Molinie, N.; Gautreau, A. The Arp2/3 Regulatory System and Its Deregulation in Cancer. Physiol. Rev. 2018, 98, 215–238. [Google Scholar] [CrossRef] [PubMed]
- Carmona, G.; Perera, U.; Gillett, C.; Naba, A.; Law, A.-L.; Sharma, V.P.; Wang, J.; Wyckoff, J.; Balsamo, M.; Mosis, F.; et al. Lamellipodin promotes invasive 3D cancer cell migration via regulated interactions with Ena/VASP and SCAR/WAVE. Oncogene 2016, 35, 5155–5169. [Google Scholar] [CrossRef]
- Di Modugno, F.; Mottolese, M.; Di Benedetto, A.; Conidi, A.; Novelli, F.; Perracchio, L.; Venturo, I.; Botti, C.; Jager, E.; Santoni, A.; et al. The cytoskeleton regulatory protein hMena (ENAH) is overexpressed in human benign breast lesions with high risk of transformation and human epidermal growth factor receptor-2-positive/hormonal receptor-negative tumors. Clin. Cancer Res. 2006, 12, 1470–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurzu, S.; Jung, I.; Prantner, I.; Ember, I.; Pavai, Z.; Mezei, T. The expression of cytoskeleton regulatory protein Mena in colorectal lesions. Rom. J. Morphol. Embryol. 2008, 49, 345–349. [Google Scholar]
- Pino, M.S.; Balsamo, M.; Di Modugno, F.; Mottolese, M.; Alessio, M.; Melucci, E.; Milella, M.; McConkey, D.J.; Philippar, U.; Gertler, F.B.; et al. Human Mena+11a isoform serves as a marker of epithelial phenotype and sensitivity to epidermal growth factor receptor inhibition in human pancreatic cancer cell lines. Clin. Cancer Res. 2008, 14, 4943–4950. [Google Scholar] [CrossRef] [Green Version]
- Gurzu, S.; Jung, I.; Prantner, I.; Chira, L.; Ember, I. The immunohistochemical aspects of protein Mena in cervical lesions. Rom. J. Morphol. Embryol. 2009, 50, 213–216. [Google Scholar] [PubMed]
- Roussos, E.T.; Balsamo, M.; Alford, S.K.; Wyckoff, J.B.; Gligorijevic, B.; Wang, Y.; Pozzuto, M.; Stobezki, R.; Goswami, S.; Segall, J.E.; et al. Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J. Cell Sci. 2011, 124, 2120–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidel-Bar, R.; Geiger, G. The switchable integrin adhesome. J. Cell Sci. 2010, 123, 1385–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca-Cusachs, P.; del Rio, A.; Puklin-Faucher, E.; Gauthier, N.C.; Biais, N.; Sheetz, M.P. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. USA 2013, 110, E1361–E1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golding, A.E.; Visco, I.; Bieling, P.; Bement, W.M. Extraction of active RhoGTPases by RhoGDI regulates spatiotemporal patterning of RhoGTPases. Elife 2019, 8, e50471. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.M.; Rademacher, J.; Bagshaw, R.D.; Wortmann, C.; Barth, C.; van Unen, J.; Alp, K.M.; Giudice, G.; Eccles, R.L.; Heinrich, L.E.; et al. Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions. Nat. Cell Biol. 2020, 22, 498–511. [Google Scholar] [CrossRef]
- Papalazarou, V.; Salmeron-Sanchez, M.; Machesky, L.M. Tissue engineering the cancer microenvironment - challenges and opportunities. Biophys. Rev. 2018, 10, 1695–1711. [Google Scholar] [CrossRef] [Green Version]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.H.; Mui, K.L.; Hsu, B.Y.; Liu, S.-L.; Cretu, A.; Razinia, Z.; Xu, T.; Puré, E.; Assoian, R.K. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 2014, 7, ra57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. Beta 4 Integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariotti, A.; Kedeshian, P.A.; Dans, M.; Curatola, A.M.; Gagnoux-Palacios, L.; Giancotti, F.G. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: Role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 2001, 155, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 2001, 107, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.; Giancotti, F.G. Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Cruz da Silva, E.; Dontenwill, M.; Choulier, L.; Lehmann, M. Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer. Cancers 2019, 11, 692. [Google Scholar] [CrossRef] [Green Version]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen Type I Induces Disruption of E-Cadherin–Mediated Cell-Cell Contacts and Promotes Proliferation of Pancreatic Carcinoma Cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef] [Green Version]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 2015, 17, 1412–1421. [Google Scholar] [CrossRef] [Green Version]
- Te Boekhorst, V.; Friedl, P. Plasticity of Cancer Cell Invasion - Mechanisms and Implications for Therapy. Adv. Cancer Res. 2016, 132, 209–264. [Google Scholar] [CrossRef]
- Friedl, P.; Mayor, R. Tuning Collective Cell Migration by Cell-Cell Junction Regulation. Cold Spring Harb. Perspect. Biol. 2017, 9, a029199. [Google Scholar] [CrossRef] [Green Version]
- Bazellières, E.; Conte, V.; Elosegui-Artola, A.; Serra-Picamal, X.; Bintanel-Morcillo, M.; Roca-Cusachs, P.; Muñoz, J.J.; Sales-Pardo, M.; Guimerà, R.; Trepat, X. Control of cell-cell forces and collective cell dynamics by the intercellular adhesome. Nat. Cell Biol. 2015, 17, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuana, L.; Boström, A.; Etienne-Manneville, S. Multicellular scale front-to-rear polarity in collective migration. Curr. Opin. Cell Biol. 2020, 62, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, M.; Di Russo, J.; Probst, D.; Schwarz, U.S.; Das, T.; Spatz, J.P. Mechanical interactions among followers determine the emergence of leaders in migrating epithelial cell collectives. Nat. Commun. 2018, 9, 3469. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, V.; Wieser, S.; Callan-Jones, A.; Smutny, M.; Morita, H.; Sako, K.; Barone, V.; Ritsch-Marte, V.; Sixt, M.; Voituriez, R.; et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 2015, 160, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chugh, P.; Paluch, E.K. The actin cortex at a glance. J. Cell Sci. 2018, 131, jcs186254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol. Oncol. 2017, 11, 5–27. [Google Scholar] [CrossRef] [Green Version]
- Paluch, E.K.; Aspalter, I.M.; Sixt, M. Focal adhesion-independent cell migration. Annu. Rev. Cell Dev. Biol. 2016, 32, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.T.; Hu, C.-K.; Coughlin, M.; Mitchison, T. J Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Chikina, A.S.; Svitkina, T.M.; Alexandrova, A.Y. Time-resolved ultrastructure of the cortical actin cytoskeleton in dynamic membrane blebs. J. Cell Biol. 2019, 218, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Logue, J.S.; Cartagena-Rivera, A.X.; Baird, M.A.; Davidson, M.W.; Chadwick, R.S.; Waterman, C.M. Erk regulation of actin capping and bundling by Eps8 promotes cortex tension and leader bleb-based migration. eLife 2015, 4, e08314. [Google Scholar] [CrossRef] [PubMed]
- Poincloux, R.; Collin, O.; Lizárraga, F.; Romao, M.; Debray, M.; Piel, M.; Chavrier, M. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc. Natl. Acad. Sci. USA 2011, 108, 1943–1948. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.R.; Castillo-Badillo, J.A.; Meshik, X.; Kalyanaraman, V.; Melgarejo, K.; Gautam, N. Membrane Flow Drives an Adhesion-Independent Amoeboid Cell Migration Mode. Dev. Cell 2018, 46, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Welf, E.S.; Driscoll, M.K.; Sapoznik, E.; Murali, V.S.; Weems, A.; Roh-Johnson, M.; Dean, K.M.; Fiolka, R.; Danuser, G. Worrying drives cell migration in mechanically unrestrained environments. bioRxiv 2020, 372912. [Google Scholar] [CrossRef]
- Bergert, M.; Chandradoss, S.D.; Desai, R.A.; Paluch, E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc. Natl. Acad. Sci. USA 2012, 109, 14434–14439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrova, A.Y.; Chikina, A.S.; Svitkina, T.M. Actin cytoskeleton in mesenchymal-to-amoeboid transition of cancer cells. Int. Rev. Cell Mol. Biol. 2020, 356, 197–256. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Le Berre, M.; Lautenschlaeger, F.; Maiuri, P.; Callan-Jones, A.; Heuzé, M.; Takaki, T.; Voituriez, R.; Piel, M. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 2015, 160, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holle, A.W.; Devi, N.G.K.; Clar, K.; Fan, A.; Saif, T.; Kemkemer, R.; Spatz, J.P. Cancer Cells Invade Confined Microchannels via a Self-Directed Mesenchymal-to-Amoeboid Transition. Nano Lett. 2019, 19, 2280–2290. [Google Scholar] [CrossRef]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef] [Green Version]
- Laser-Azogui, A.; Diamant-Levi, T.; Israeli, S.; Roytman, Y.; Tsarfaty, I. Met-induced membrane blebbing leads to amoeboid cell motility and invasion. Oncogene 2014, 33, 1788–1798. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- López-Luque, J.; Bertran, E.; Crosas-Molist, E.; Maiques, O.; Malfettone, A.; Caja, L.; Serrano, T.; Ramos, E.; Sanz-Moreno, V.; Fabregat, I. Downregulation of Epidermal Growth Factor Receptor in hepatocellular carcinoma facilitates Transforming Growth Factor-β-induced epithelial to amoeboid transition. Cancer Lett. 2019, 464, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Geiger, F.; Rüdiger, D.; Zahler, S.; Engelke, H. Fiber stiffness, pore size and adhesion control migratory phenotype of MDA-MB-231 cells in collagen gels. PLoS ONE 2019, 14, e0225215. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; te Boekhorst, V.; Odenthal, J.; Bianchi, R.; van Helvert, S.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia Induces a HIF-1-Dependent Transition from Collective-to-Amoeboid Dissemination in Epithelial Cancer Cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EMT-TF | Down-Regulated Expression | References | Up-Regulated Expression | References |
|---|---|---|---|---|
| SNAIL | E-cadherin Plakophilin-2 Claudin-4 Cytokeratins 17, 18, 19, 20 Gelsolin Occludin Integrins α3, α6, β4 Crumbs3 | [46,47] [48] [48] [48] [48] [48,49,50,51] [52,53] [54] | Vimentin Claudin-11 MMP1 MMP2 MMP7 MT1-MMP Fibronectin Integrins α2, β1, β3 | [55,56] [57] [56] [55,56,58] [56] [56] [56] [31,41,53] |
| SLUG | E-cadherin Occludin | [59,60] [50,61] | Vimentin Fibronectin | [60] [60] |
| TWIST1 | E-cadherin α-catenin γ-catenin | [62,63,64] [62,63] [63] | N-cadherin Vimentin Smooth muscle actin Fibronectin Integrin α5 | [64,65] [63,64,66] [62,63] [62,63] [67] |
| ZEB1 | E-cadherin Occludin Crumbs3 | [68,69] [70] [70] | N-cadherin Fibronectin Vimentin | [71] [71] [72] |
| ZEB2 | E-cadherin α-catenin | [56,73] [73] | N-cadherin Vimentin MMP1 MMP2 MT1-MMP Fibronectin | [73] [56,74] [56] [56] [56] [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubtsova, S.N.; Zhitnyak, I.Y.; Gloushankova, N.A. Phenotypic Plasticity of Cancer Cells Based on Remodeling of the Actin Cytoskeleton and Adhesive Structures. Int. J. Mol. Sci. 2021, 22, 1821. https://doi.org/10.3390/ijms22041821
Rubtsova SN, Zhitnyak IY, Gloushankova NA. Phenotypic Plasticity of Cancer Cells Based on Remodeling of the Actin Cytoskeleton and Adhesive Structures. International Journal of Molecular Sciences. 2021; 22(4):1821. https://doi.org/10.3390/ijms22041821
Chicago/Turabian StyleRubtsova, Svetlana N., Irina Y. Zhitnyak, and Natalya A. Gloushankova. 2021. "Phenotypic Plasticity of Cancer Cells Based on Remodeling of the Actin Cytoskeleton and Adhesive Structures" International Journal of Molecular Sciences 22, no. 4: 1821. https://doi.org/10.3390/ijms22041821