Transcriptomic Analysis Reveals Candidate Genes Responsive to Sclerotinia scleroterum and Cloning of the Ss-Inducible Chitinase Genes in Morus laevigata


Abstract
:1. Introduction
2. Results
2.1. RNA-seq and de Novo Transcriptome Assembly
2.2. Functional Annotation of the Unigenes
2.3. Analysis of Differentially Expressed Genes
2.4. Candidate Genes Screening
2.5. Organ-Specificity of Transription Patterns of the Candidate Genes
2.6. Stock-Diferred Expression and Ss-Inducibility Dynamics of the Candidate Genes
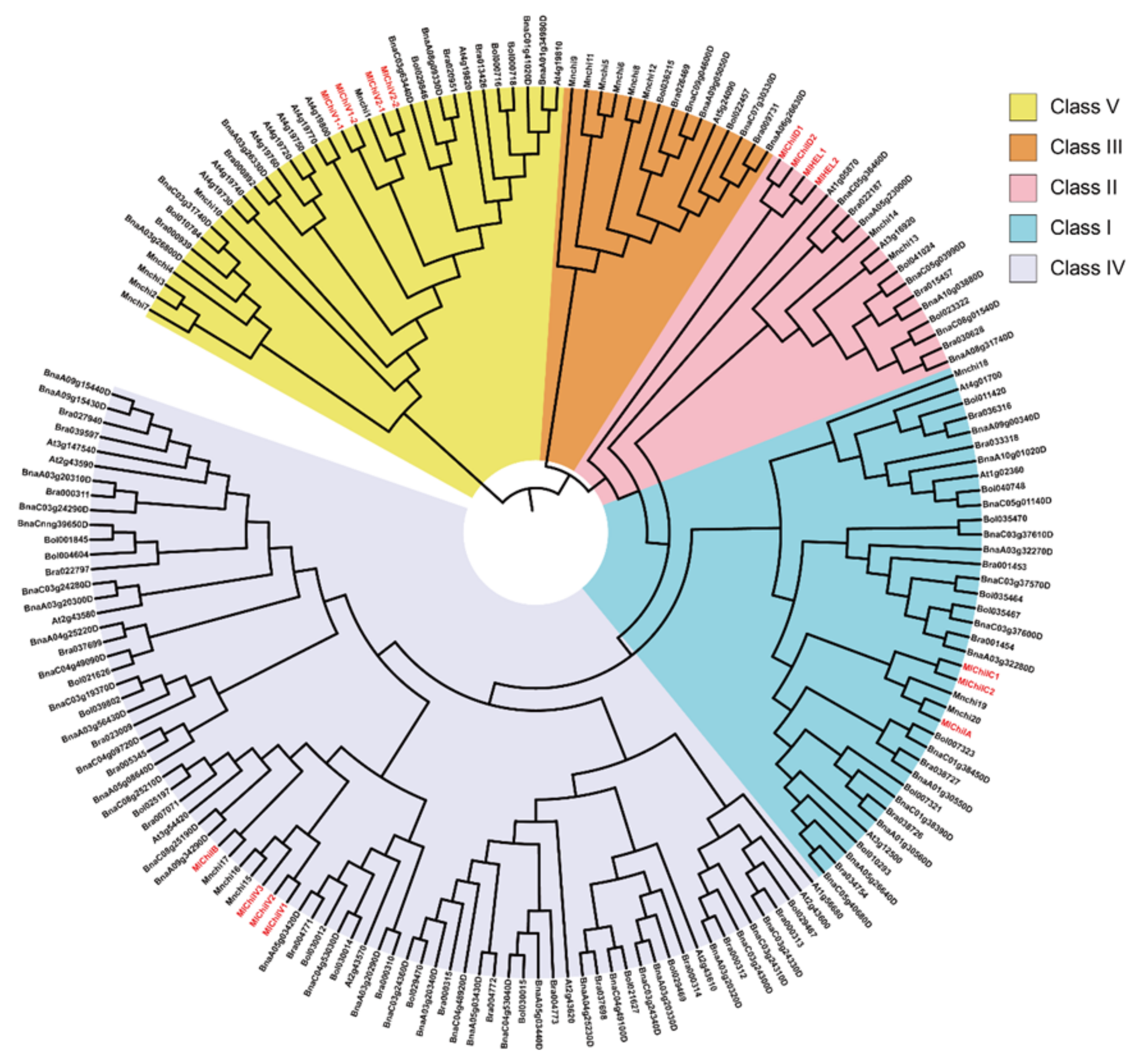
2.7. Cloning and Molecular Features of the Ss-Inducible Chitinase Genes from M. Laevigata
2.8. Prokaryotic Expression of the Ss-Inducible Chitinase Genes from M. Laevigata
3. Discussions
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. RNA Extraction, and cDNA Library Preparation and Sequencing
4.3. Data Processing, and Transcriptome Assembly and Annotation
4.4. Differential Gene Expression, GO, and KEGG Enrichment Analysis
4.5. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.6. Cloning of cDNA and gDNA Sequences of Ss-Responsive Chitinase Genes from M. Laevigata
4.7. Sequence Characteristics, and Phylogenetic and Interaction Networks Analysis
4.8. Prokaryotic Expression
4.9. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SsSSVP1 | A small secreted virulence-related protein 1 in Sclerotinia scleroterum |
| QCR8 | Cytochrome b-c1 complex, subunit 8 protein |
| SsCP1 | Cerato-platanin protein 1 in Sclerotinia scleroterum |
| PR1 | Pathogenesis-related protein 1 |
| MaMLX-Q1 | Latex chitinase protein 1 isolated from Mulberry variety Qiangsang 1 (Q1) |
| BLAST | Basic Local Alignment Search Tool |
| IPTG | Isopropyl beta-D-thiogalactopyranoside |
References
- Hegedus, D.D.; Rimmer, S.R. Sclerotinia sclerotiorum: When “to be or not to be” a pathogen? Fems Microbiol. Lett. 2005, 251, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhao, Q.; Yang, Q.; Liu, H.; Li, Q.; Yi, X.; Cheng, Y.; Guo, L.; Fan, C.; Zhou, Y. Comparative transcriptomic analysis uncovers the complex genetic network for resistance to Sclerotinia sclerotiorum in Brassica napus. Sci. Rep. 2016, 6, 19007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, M.D.; Thomma, B.P.H.J.; Nelson, B.D. Sclerotinia sclerotiorum (Lib.) de Bary: Biology and molecular traits of a cosmopolitan pathogen. Mol. Plant Pathol. 2006, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, N.; Babai-Ahary, A.; Assadi, Y.; Valizadeh, M.; Passebaneslam, B. The Effects of Sclerotinia Stem Rot of Oilseed Rape on the Production and Quality of Extracted Oil. J. Sci. Technol. Agric. Nat. Resour. 2006, 3, 485–495. [Google Scholar]
- Boland, G.J.; Hall, R. Index of plant hosts of Sclerotinia sclerotiorum. Can. J. Plant Pathol. 1994, 16, 93–108. [Google Scholar] [CrossRef]
- Cao, J.Y.; Xu, Y.P.; Zhao, L.; Li, S.S.; Cai, X.Z. Tight regulation of the interaction between Brassica napus and Sclerotinia sclerotiorumat the microRNA level. Plant Mol. Biol. 2016, 92, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Ding, Y.; Li, Y.; Tong, C.; Du, H.; Yu, Y.; Wan, H.; Xiong, Q.; Yu, J.; Liu, S. Transcriptomic comparison between Brassica oleracea and rice (Oryza sativa) reveals diverse modulations on cell death in response to Sclerotinia sclerotiorum. Sci Rep. 2016, 6, 33706. [Google Scholar] [CrossRef]
- Seifbarghi, S.; Borhan, M.H.; Wei, Y.; Coutu, C.; Robinson, S.J.; Hegedus, D.D. Changes in the Sclerotinia sclerotiorum transcriptome during infection of Brassica napus. BMC Genom. 2017, 18, 1–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monazzah, M.; Tahmasebi Enferadi, S.; Rabiei, Z. Enzymatic activities and pathogenesis-related genes expression in sunflower inbred lines affected by Sclerotinia sclerotiorum culture filtrate. J. Appl. Microbiol. 2018, 125, 227–242. [Google Scholar] [CrossRef]
- Ranjan, A.; Westrick, N.M.; Jain, S.; Piotrowski, J.S.; Ranjan, M.; Kessens, R.; Stiegman, L.; Grau, C.R.; Conley, S.P.; Smith, D.L. Resistance against Sclerotinia sclerotiorum in soybean involves a reprogramming of the phenylpropanoid pathway and upregulation of anti-fungal activity targeting ergosterol biosynthesis. Plant Biotechnol. J. 2019, 17, 1567–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, N.; Zhang, C.; Qi, X.; Zhao, S.; Tao, Y.; Yang, G.; Lee, T.; Wang, X.; Cai, Q.; Li, D. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 2013, 4, 2445. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.Y.; Li, K.T.; Yang, W.J.; Chung, M.C.; Chang, J.C.; Chang, M.W. Ploidy level and their relationship with vegetative traits of mulberry (Morus spp.) species in Taiwan. Sci. Hortic. 2018, 235, 78–85. [Google Scholar] [CrossRef]
- Liu, Y.; Ji, D.; Chen, J.; Lin, T.; Wei, J.; Zhu, Y.; Lv, Z. Overexpression of the mulberry latex gene MaMLX-Q1 enhances defense against Plutella xylostella in Arabidopsis thaliana. Arch. Biol. Sci. 2017, 69, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Yuan-Zhang, K.; Fu-An, W.U. A Review on Pathogens of Mulberry Fruit Sclerotiniosis and its Control Technology. Sci. Seric. 2012, 38, 1099–1104. [Google Scholar]
- Bao, L.; Gao, H.; Zheng, Z.; Zhao, X.; Qian, Y. Integrated Transcriptomic and Un-Targeted Metabolomics Analysis Reveals Mulberry Fruit (Morus atropurpurea) in Response to Sclerotiniose Pathogen Ciboria shiraiana Infection. Int. J. Mol. Ences. 2020, 21, 1789. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.K.; Wan, G.K.; Sung, G.B.; Nam, S.H. Identification and distribution of two fungal species causing sclerotial disease on mulberry fruits in Korea. Mycobiology 2007, 35, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Sultana, R.; Kim, K. Bacillus thuringiensis C25 suppresses popcorn disease caused by Ciboria shiraianain mulberry (Morus australis L.). Biocontrol Sci. Technol. 2016, 26, 145–162. [Google Scholar] [CrossRef]
- Lu, R.; Jin, X.; Zhao, A.; Ji, J.; Liu, C.; Li, J.; Pu, L.; Lu, C.; Yu, M. Cross Infection, Biological Characteristics and Genetic Relationship between Pathogens of Hypertrophy Sorosis Sclerotenisis from Mulberry and Sclerotinia Stem Rot from Oilseed Rape. Acta Agron. Sin. 2015, 41, 42–48. [Google Scholar] [CrossRef]
- Zh, Z.; Ya, Y.; Zh, M.; Re, J.; Re, X. Field Resistance Evaluation of 21 Fruit Mulberry Germplasm Resources against Mulberry Fruit Hypertrophic Sclerote Disease. Sci. Seric. 2015, 6, 1011–1016. [Google Scholar]
- Tikader, A.; Kamble, C.K. Mulberry wild species in India and their use in crop improvement—A review. Aust. J. Crop Sci. 2008, 2, 64–72. [Google Scholar]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Sharma, N.; Sharma, K.P.; Gaur, R.K.; Gupta, V.K. Role of Chitinase in Plant Defense. Asian J. Biochem. 2011, 6, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, G.; Jiang, D.; Chen, W. Sclerotinia sclerotiorum: An Evaluation of Virulence Theories. Annu. Rev. Phytopathol. 2018, 56, 311–338. [Google Scholar] [CrossRef]
- Lyu, X.; Shen, C.; Fu, Y.; Xie, J.; Jiang, D.; Li, G.; Cheng, J. A Small Secreted Virulence-Related Protein is Essential for the Necrotrophic Interactions of Sclerotinia sclerotiorum with its Host Plants. Plos Pathog. 2016, 12, 1005435. [Google Scholar] [CrossRef]
- Yang, G.; Tang, L.; Gong, Y.; Xie, J.; Fu, Y. A cerato-platanin protein SsCP1 targets plant PR1 and contributes to virulence of Sclerotinia sclerotiorum. New Phytol. 2017, 217, 739–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, R.; Khan, M.A.; Ahmad, M.; Ahmad, M.M.; Abdin, M.Z.; Musarrat, J.; Javed, S. Chitinases: An update. J. Pharm. Bioallied Sci. 2013, 5, 21–29. [Google Scholar]
- Schlumbaum, A.; Mauch, F.; Vögeli, U.; Boller, T. Plant chitinases are important inhibitors of fungal growth. Nature 1986, 324, 365–367. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Fang, H.; Chen, Y.; Chen, K.; Li, G.; Gu, S.; Tan, X. Overexpression of BnWRKY33 in oilseed rape enhances resistance to Sclerotinia sclerotiorum. Mol. Plant Pathol. 2014, 15, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, Y.; Tang, Y.; Chen, J.; Ding, W. Overexpression of NtWRKY50 Increases Resistance to Ralstonia solanacearum and Alters Salicylic Acid and Jasmonic Acid Production in Tobacco. Front. Plant Sci. 2017, 8, 1710. [Google Scholar] [CrossRef] [Green Version]
- Zarinpanjeh, N.; Motallebi, M.; Zamani, M.R.; Ziaei, M. Enhanced resistance to Sclerotinia sclerotiorumin Brassica napus by co-expression of defensin and chimeric chitinase genes. J. Appl. Genet. 2016, 57, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, M.; Motallebi, M.; Zamani, M.R.; Panjeh, N.Z. Co-expression of chimeric chitinase and a polygalacturonase-inhibiting protein in transgenic canola (Brassica napus) confers enhanced resistance to Sclerotinia sclerotiorum. Biotechnol. Lett. 2016, 38, 1021–1032. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; He, N.; Zeng, Q.; Xiang, Z. Identification and expression analyses of chitinase genes in mulberry (Morus L.) plants. Plant Omics 2015, 8, 183–189. [Google Scholar]
- Passarinho, P.A.; de Vries, S.C. Arabidopsis chitinases: A genomic survey. Arab. Book. 2002, 1, e0023. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.K.; Megha, S.; Rahman, M.H.; Basu, U.; Kav, N.N.V. A global study of transcriptome dynamics in canola (Brassica napus L.) responsive to Sclerotinia sclerotiorum infection using RNA-Seq. Gene 2016, 590, 57–67. [Google Scholar] [CrossRef]
- Dai, F.; Wang, Z.; Li, Z.; Luo, G.; Wang, Y.; Tang, C. Transcriptomic and proteomic analyses of mulberry (Morus atropurpurea) fruit response to Ciboria carunculoides. J. Proteom. 2019, 193, 142–153. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, J.; An, L.; Doerge, R.W.; Chen, Z.J.; Grau, C.R.; Osborn, M.T.C. Analysis of gene expression profiles in response to Sclerotinia sclerotiorum in Brassica napus. Planta 2007, 227, 13–24. [Google Scholar] [CrossRef]
- Zhao, J.; Buchwaldt, L.; Rimmer, S.R.; Sharpe, A.; Mcgregor, L.; Bekkaoui, D.; Hegedus, D. Patterns of differential gene expression in Brassica napus cultivars infected with Sclerotinia sclerotiorum. Mol. Plant Pathol. 2009, 10, 635–649. [Google Scholar] [CrossRef]
- Arfaoui, A.; Hadrami, A.E.; Daayf, F. Pre-treatment of soybean plants with calcium stimulates ROS responses and mitigates infection by Sclerotinia sclerotiorum. Plant Physiol. Biochem. 2017, 122, 121–128. [Google Scholar] [CrossRef]
- Li, J.; Brader, G.; Palva, E.T. The WRKY70 Transcription Factor: A Node of Convergence for Jasmonate-Mediated and Salicylate-Mediated Signals in Plant Defense. Plant Cell 2004, 16, 319–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishikaboshi, M.; Seo, S.; Takahashi, A.; Hirochika, H. The MAMP-Responsive MYB Transcription Factors MYB30, MYB55 and MYB110 Activate the HCAA Synthesis Pathway and Enhance Immunity in Rice. Plant Cell Physiol. 2018, 59, 903–915. [Google Scholar] [CrossRef]
- Navarro-González, S.S.; Ramírez-Trujillo, J.A.; Peña-Chora, G.; Gaytán, P.; Suárez-Rodríguez, R. Enhanced Tolerance against a Fungal Pathogen and Insect Resistance in Transgenic Tobacco Plants Overexpressing an Endochitinase Gene from Serratia marcescens. Int. J. Mol. Sci. 2019, 20, 3482. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Li, C.; Wang, X.; Zhang, F.; Ruan, X. Overexpression of a Chitinase Gene from Trichoderma asperellum Increases Disease Resistance in Transgenic Soybean. Appl. Biochem. Biotechnol. 2016, 180, 1542–1558. [Google Scholar]
- Karmakar, S.; Molla, K.A.; Chanda, P.K.; Sarkar, S.N.; Datta, S.K.; Datta, K. Green tissue-specific co-expression ofchitinaseandoxalateoxidase 4genes in rice for enhanced resistance against sheath blight. Planta 2016, 243, 115–130. [Google Scholar] [CrossRef]
- Singh, H.R.; Deka, M.; Das, S. Enhanced resistance to blister blight in transgenic tea (Camellia sinensis [L.] O. Kuntze) by overexpression of class I chitinase gene from potato (Solanum tuberosum). Funct. Integr. Genom. 2015, 15, 461–480. [Google Scholar] [CrossRef]
- Amselem, J.; Cuomo, C.A.; Van Kan, J.A.L.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; De Vries, R.P.; Dyer, P.S.; Fillinger, S. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. Plos Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Piao, Y.; Liu, Y.; Li, X.; Piao, Z. Genome-wide identification and expression analysis of chitinase gene family in Brassica rapa reveals its role in clubroot resistance. Plant Sci. 2018, 270, 257–267. [Google Scholar] [CrossRef]
- Gerhardt, L.B.D.A.; Sachettomartins, G.; Contarini, M.G.; Sandroni, M.; Ferreira, R.P.; Lima, V.M.; Cordeiro, M.C.R.; De Oliveira, D.E.; Margispinheiro, M. Arabidopsis thaliana class IV chitinase is early induced during the interaction with Xanthomonas campestris. Febs Lett. 1997, 419, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Faris, J.D.; Muthukrishnan, S.; Liu, D.; Chen, P.; Gill, B.S. Isolation and characterization of novel cDNA clones of acidic chitinases and β-1,3-glucanases from wheat spikes infected by Fusarium graminearum. Theor. Appl. Genet. 2001, 102, 353–362. [Google Scholar] [CrossRef]
- Moghadam, M.; Ganji, A.; Varasteh, A.; Falak, R.; Sankian, M. Refolding process of cysteine-rich proteins: Chitinase as a model. Rep. Biochem. Mol. Biol. 2015, 4, 19. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Chao, Q.; Gao, Z.F.; Zhang, D.; Zhao, B.G.; Wang, B.C. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnol. J. 2018, 17, 206–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Song, F.; Zhu, J.; Zhang, S.; Yang, Y.; Chen, T.; Tang, B. GSA: Genome Sequence Archive. Genom. Proteom. Bioinform. 2017, 15, 14–18. [Google Scholar] [CrossRef]
- National Genomics Data Center Members and Partners. Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 2020, 48, D24–D33. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Databases | Annotated Unigenes | Percentage (%) |
|---|---|---|
| Nr | 27,991 | 45.15 |
| Nt | 14,341 | 23.13 |
| KEGG | 7258 | 11.71 |
| Swiss-Prot | 18,506 | 29.85 |
| Pfam | 18,463 | 29.78 |
| GO | 19,030 | 30.70 |
| KOG | 9779 | 15.77 |
| Annotated in all Databases | 3804 | 6.13 |
| Annotated in at least one Database | 30,334 | 48.93 |
| Total Unigenes | 61,985 | 100.00 |
| Seq_ID | log2 (FC) | Gene Annotation | BP Description |
|---|---|---|---|
| c49683_g1 | 7.25 | Protein SRG1 [Morus notabilis] | oxidation |
| c30732_g1 | 5.04 | Myb-related protein 305 [Morus notabilis] | reduction |
| c40595_g2 | 3.34 | Cytochrome P450 82A3 [Morus notabilis] | process |
| c46262_g2 | 3.33 | NAC domain-containing protein 8 [Morus notabilis] | |
| c38775_g1 | 3.18 | Hyoscyamine 6-dioxygenase [Morus notabilis] | |
| c40192_g1 | 3.09 | putative inactive poly [ADP-ribose] polymerase SRO5 [Morus notabilis] | |
| c35789_g1 | 2.15 | putative linoleate 9S-lipoxygenase 5 [Morus notabilis] | |
| c38165_g1 | 2.04 | Extracellular ribonuclease LE [Morus notabilis] | |
| c36175_g1 | 2.01 | Myb-related protein 305 [Morus notabilis] | |
| c47127_g2 | 3.98 | Calcium-dependent protein kinase 3 [Morus notabilis] | protein |
| c23623_g1 | 3.94 | type II proteinase inhibitor [Potato] | phosphorylation |
| c40703_g1 | 3.65 | indole-3-acetic acid amido synthetase [Morus alba var. multicaulis] | and proteolysis |
| c31769_g1 | 3.54 | Salutaridinol 7-O-acetyltransferase [Morus notabilis] | |
| c43119_g1 | 3.17 | Protein BONZAI 3 [Morus notabilis] | |
| c40199_g1 | 2.87 | Protein kinase byr2 [Morus notabilis] | |
| c42162_g2 | 2.12 | xyloglucanase inhibitor 3 [Humulus lupulus] | |
| c48054_g1 | 1.78 | G-type lectin S-receptor-like serine/threonine-protein kinase RKS1 [Morus notabilis] | |
| c47511_g1 | 1.69 | L-type lectin-domain containing receptor kinase VII.1 [Morus notabilis] | |
| c31798_g1 | 1.69 | Cysteine proteinase inhibitor 5 [Morus notabilis] | |
| c31985_g1 | 1.64 | Proteinase inhibitor [Morus notabilis] | |
| c36313_g1 | 3.87 | Transcription factor MYB86 [Arabidopsis thaliana] | transcription |
| c37744_g1 | 2.87 | Ethylene-responsive transcription factor [Morus notabilis] | factor |
| c42874_g1 | 2.85 | Ethylene-responsive transcription factor [Morus notabilis] | |
| c39290_g1 | 2.12 | Pathogenesis-related protein transcriptional activator PTI5 [Morus notabilis] | |
| c43863_g1 | 2.09 | Transcription factor TGA1 [Morus notabilis] | |
| c45307_g1 | 1.83 | putative WRKY transcription factor 33 [Morus notabilis] | |
| c30412_g1 | 1.76 | Transcription factor MYB44 [Arabidopsis thaliana] | |
| c44653_g2 | 1.58 | putative WRKY transcription factor 33 [Morus notabilis] | |
| c38466_g2 | 6.02 | Zeatin O-glucosyltransferase [Morus notabilis] | metabolic |
| c47334_g1 | 5.39 | putative cysteine desulfurase [Morus notabilis] | process |
| c35494_g1 | 5.38 | Flavonol reductase [Morus notabilis] | |
| c41475_g1 | 5.34 | Galactose oxidase [Morus notabilis] | |
| c47088_g3 | 5.26 | Carbonic anhydrase 2 [Morus notabilis] | |
| c41666_g1 | 1.75 | Glucan endo-1,3-beta-glucosidase, basic vacuolar isoform [Morus notabilis] | |
| c43476_g1 | 1.62 | GDSL esterase/lipase 1 [Morus notabilis] | |
| c36895_g1 | 3.47 | Endochitinase [Morus notabilis] | chitin metabolic process |
| c42297_g2 | 2.09 | Pathogenesis-related protein P2 [Morus notabilis] | |
| c46798_g2 | 2.05 | class V chitinase [Morus alba] | |
| c46612_g1 | 2 | Endochitinase 1 [Morus notabilis] | |
| c34352_g1 | 1.26 | Endochitinase [Morus notabilis] | |
| c36015_g1 | 1.24 | class I chitinase, putative [Ricinus communis] | |
| c42529_g1 | 1.23 | class I pathogenesis-related protein 4 [Ficus pumila] |
| Gene name | Seq_ID | gDNA | cDNA | ORF | AA | SP | MW (kDa) | pI | GRAVY | Domain | Subcellular localization |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (bp) | (bp) | (bp) | |||||||||
| MlChiIA | c46612 | 1997 | 1265 | 972 | 323 | Y | 34.95 | 6.84 | −0.37 | Chitin_bind_1 and Glyco_hydro_19 | vacuole |
| MlChiIB | c34352 | 2167 | 1176 | 840 | 279 | Y | 30.39 | 4.63 | −0.32 | Chitin_bind_1 and Glyco_hydro_19 | vacuole |
| MlChiIC1 | c36015 | N | 1052 | 750 | 249 | Y | 27.36 | 5.57 | −0.36 | Glyco_hydro_19 | vacuole |
| MlChiIC2 | c36015 | 1548 | 1039 | 750 | 249 | Y | 27.73 | 5.10 | −0.39 | Glyco_hydro_19 | vacuole |
| MlChiID1 | c42529 | 1664 | 1052 | 633 | 210 | Y | 22.34 | 6.76 | −0.31 | Chitin_bind_1 and Barwin | vacuole |
| MlChiID2 | c42529 | 1666 | 1054 | 636 | 211 | Y | 22.41 | 6.76 | −0.29 | Chitin_bind_1 and Barwin | vacuole |
| MlHEL1 | c42297 | 1629 | 753 | 435 | 144 | Y | 15.78 | 6.79 | −0.20 | Barwin | cell wall |
| MlHEL2 | c42297 | 1284 | 753 | 435 | 144 | Y | 15.75 | 6.04 | −0.21 | Barwin | cell wall |
| MlChiV1-1 | c46798 | 3600 | 1307 | 1140 | 379 | Y | 42.15 | 4.96 | −0.22 | Glyco_hydro_18 | cell wall |
| MlChiV1-2 | c46798 | 3603 | 1307 | 1140 | 379 | Y | 41.98 | 4.88 | −0.21 | Glyco_hydro_18 | cell wall |
| MlChiV2-1 | c46798 | N | 1308 | 1173 | 390 | Y | 43.51 | 5.89 | −0.23 | Glyco_hydro_18 | cell wall |
| MlChiV2-2 | c46798 | N | 1308 | 1173 | 390 | Y | 43.57 | 6.14 | −0.27 | Glyco_hydro_18 | cell wall |
| MlChiIV1 | c36895 | 1847 | 1329 | 822 | 273 | Y | 29.48 | 4.66 | −0.42 | Chitin_bind_1 and Glyco_hydro_19 | vacuole |
| MlChiIV2 | c36895 | 1849 | 1332 | 822 | 273 | Y | 29.48 | 4.62 | −-0.42 | Chitin_bind_1 and Glyco_hydro_19 | vacuole |
| MlChiIV3 | c36895 | 1848 | 1331 | 822 | 273 | Y | 29.39 | 4.61 | −0.38 | Chitin_bind_1 and Glyco_hydro_19 | vacuole |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Jin, X.; Shi, X.; Xue, Y.; Jiang, J.; Yuan, C.; Du, Y.; Liu, X.; Xie, R.; Liu, X.; et al. Transcriptomic Analysis Reveals Candidate Genes Responsive to Sclerotinia scleroterum and Cloning of the Ss-Inducible Chitinase Genes in Morus laevigata. Int. J. Mol. Sci. 2020, 21, 8358. https://doi.org/10.3390/ijms21218358
Jiang H, Jin X, Shi X, Xue Y, Jiang J, Yuan C, Du Y, Liu X, Xie R, Liu X, et al. Transcriptomic Analysis Reveals Candidate Genes Responsive to Sclerotinia scleroterum and Cloning of the Ss-Inducible Chitinase Genes in Morus laevigata. International Journal of Molecular Sciences. 2020; 21(21):8358. https://doi.org/10.3390/ijms21218358
Chicago/Turabian StyleJiang, Huanhuan, Xiaoyun Jin, Xiaofeng Shi, Yufei Xue, Jiayi Jiang, Chenglong Yuan, Youjie Du, Xiaodan Liu, Ruifang Xie, Xuemei Liu, and et al. 2020. "Transcriptomic Analysis Reveals Candidate Genes Responsive to Sclerotinia scleroterum and Cloning of the Ss-Inducible Chitinase Genes in Morus laevigata" International Journal of Molecular Sciences 21, no. 21: 8358. https://doi.org/10.3390/ijms21218358