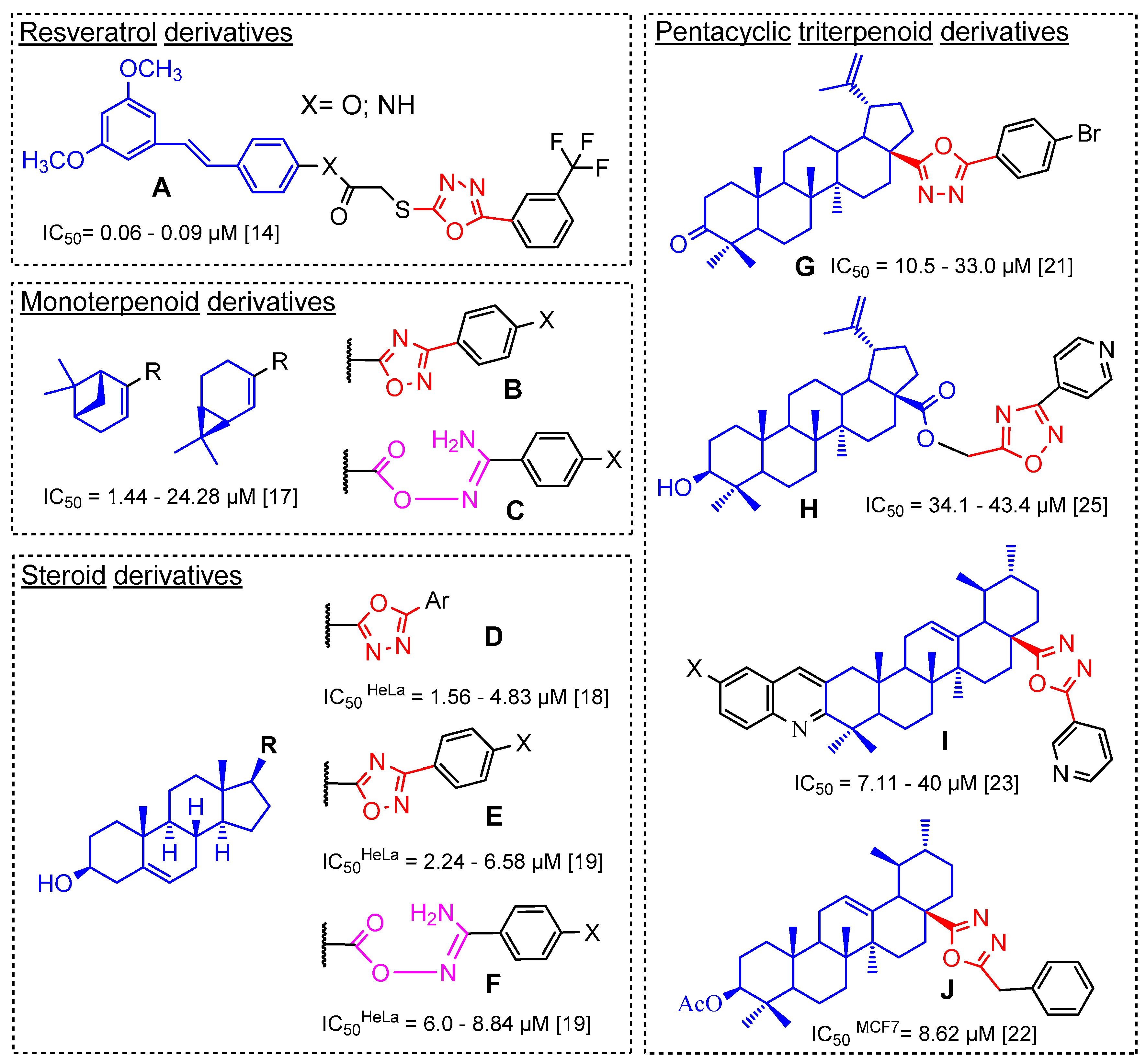
Novel 3′-Substituted-1′,2′,4′-Oxadiazole Derivatives of 18βH-Glycyrrhetinic Acid and Their O-Acylated Amidoximes: Synthesis and Evaluation of Antitumor and Anti-Inflammatory Potential In Vitro and In Vivo

 ,
,
Abstract
:1. Introduction
2. Results
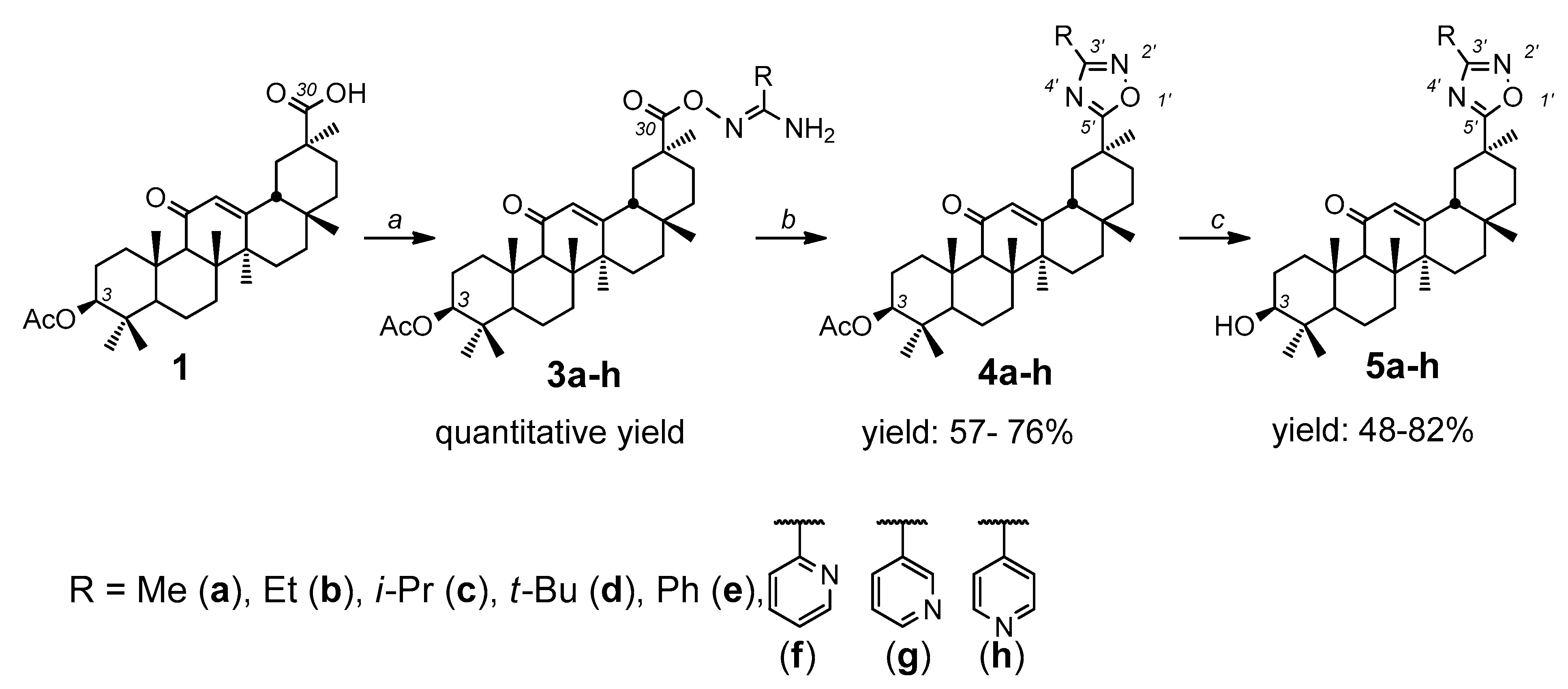
2.1. Synthesis of Novel Glycyrrhetinic Acid (GA) Derivatives
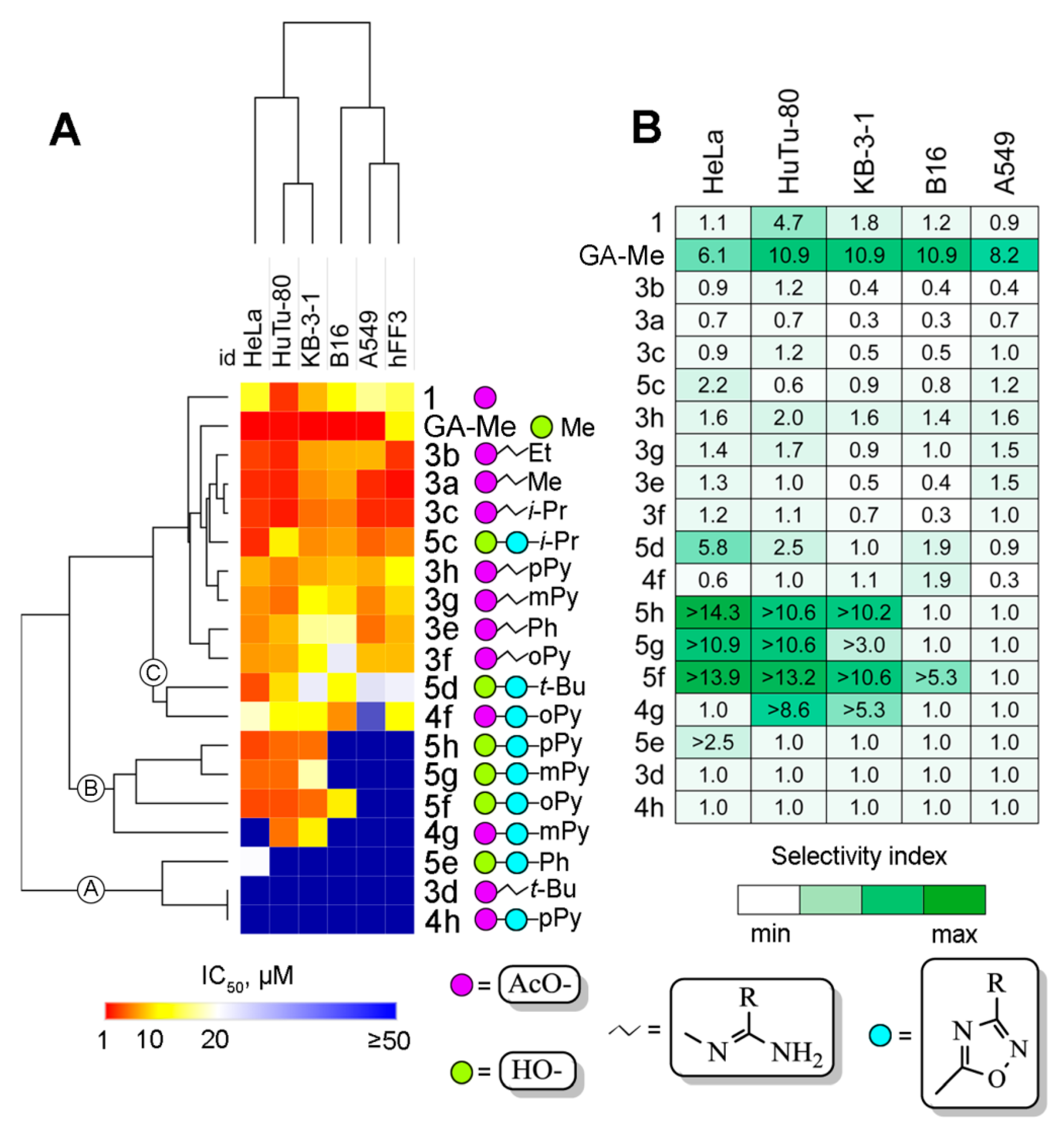
2.2. Cytotoxicity of Novel GA Derivatives
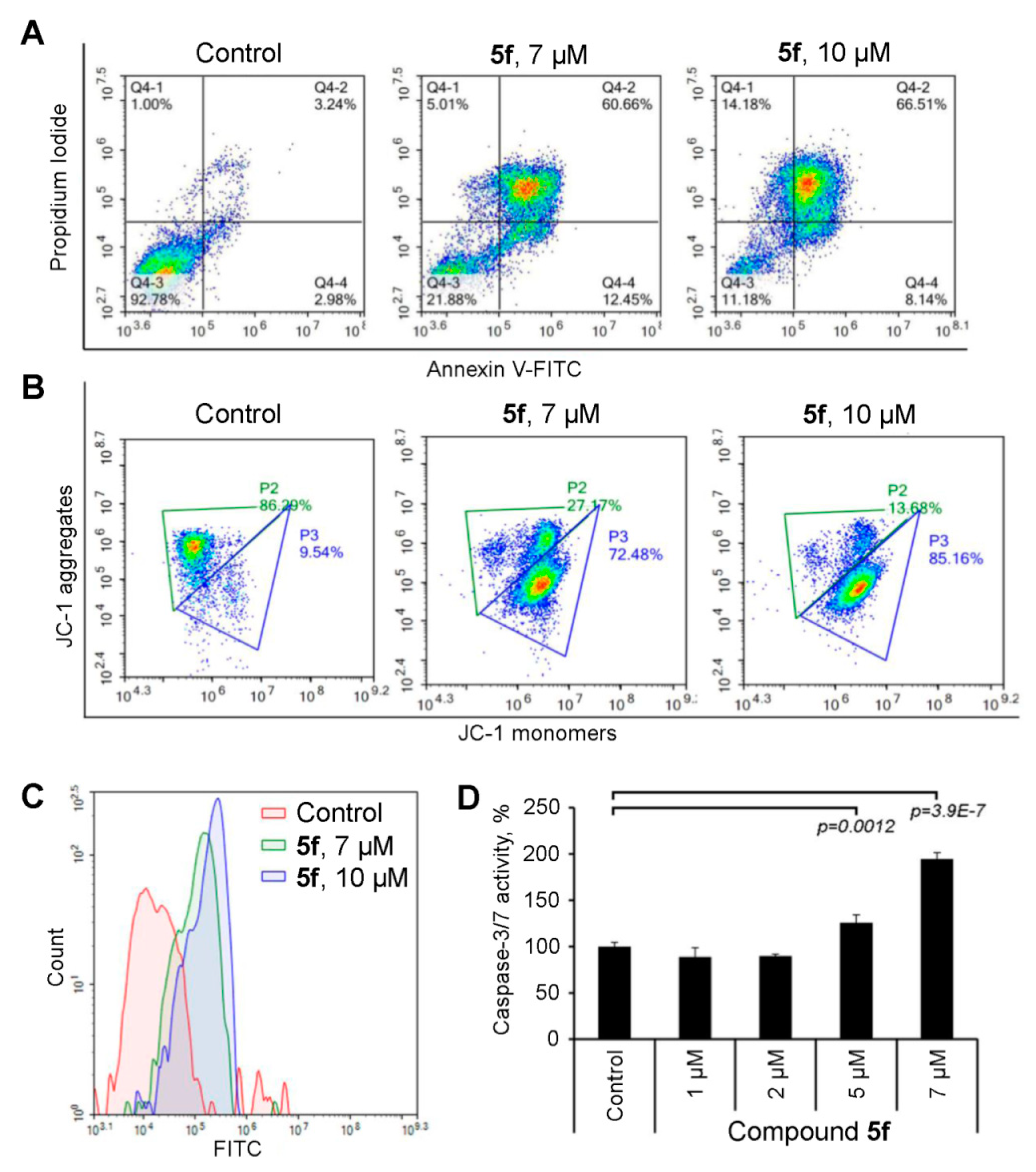
2.3. Compound 5f Triggers Mitochondrial Caspase-Dependent Apoptosis
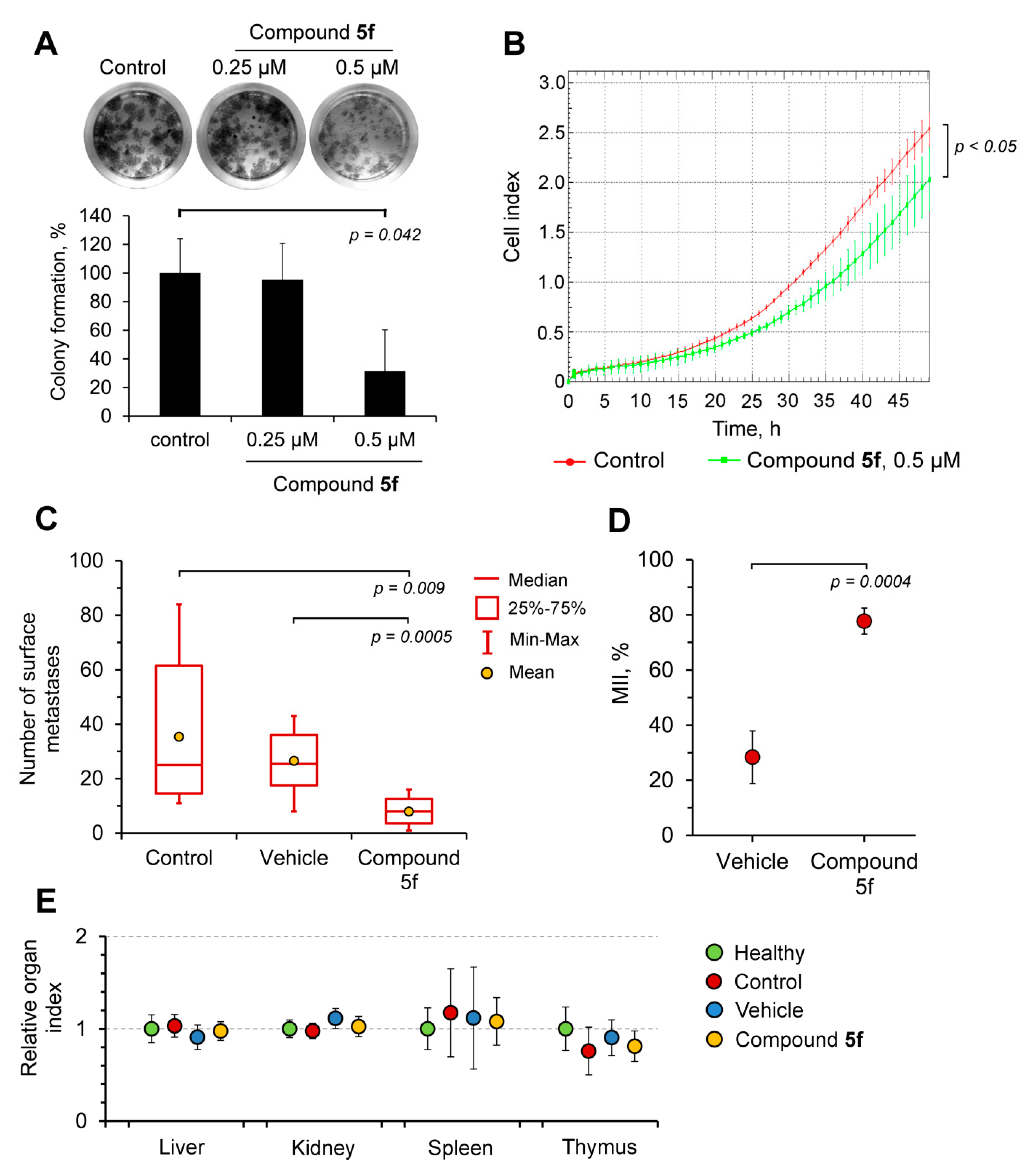
2.4. Compound 5f Inhibits the Metastatic Potential of HeLa Cells In Vitro and the Metastatic Growth of Melanoma B16 In Vivo
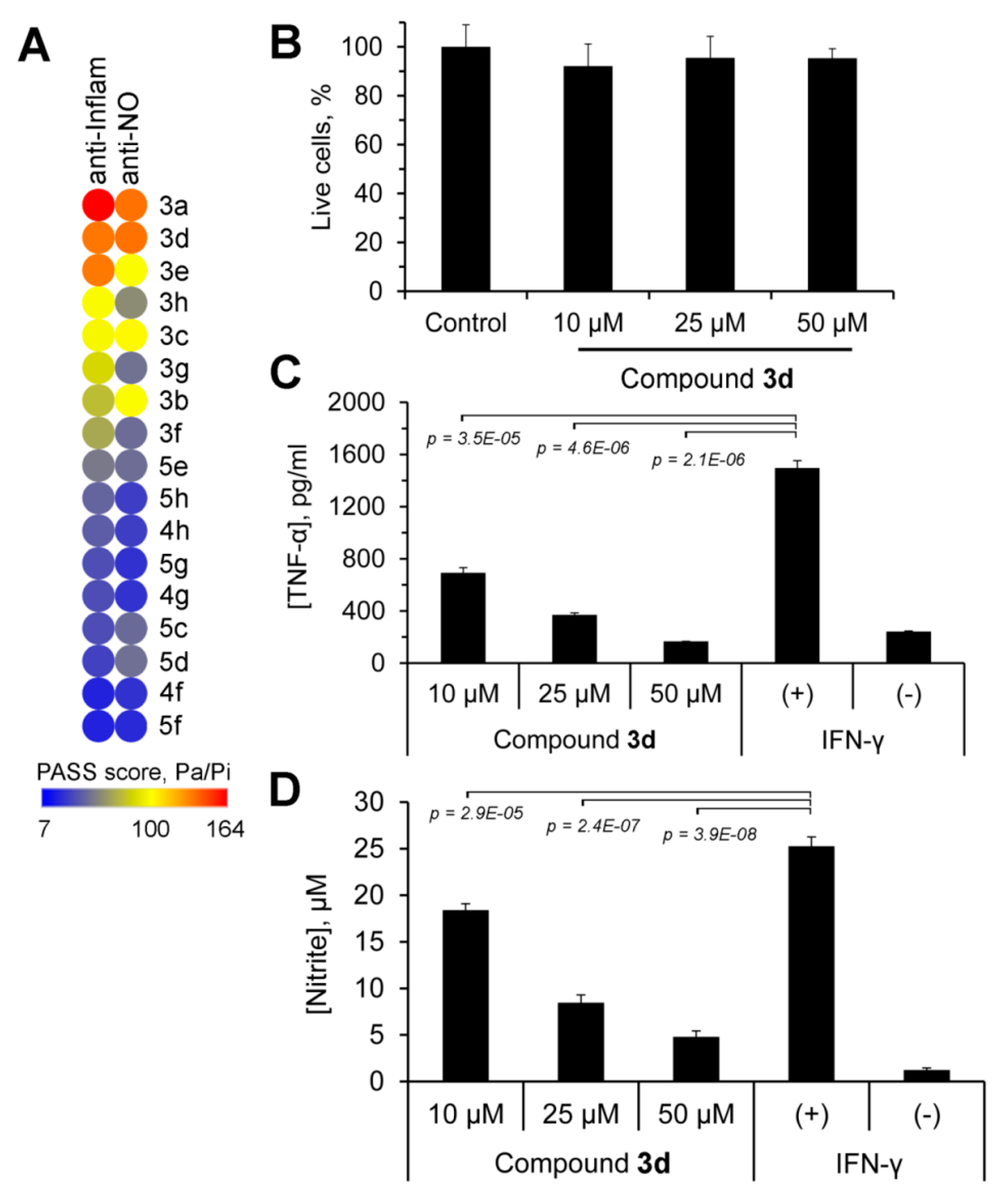
2.5. Anti-Inflammatory Potential of Novel GA Derivatives
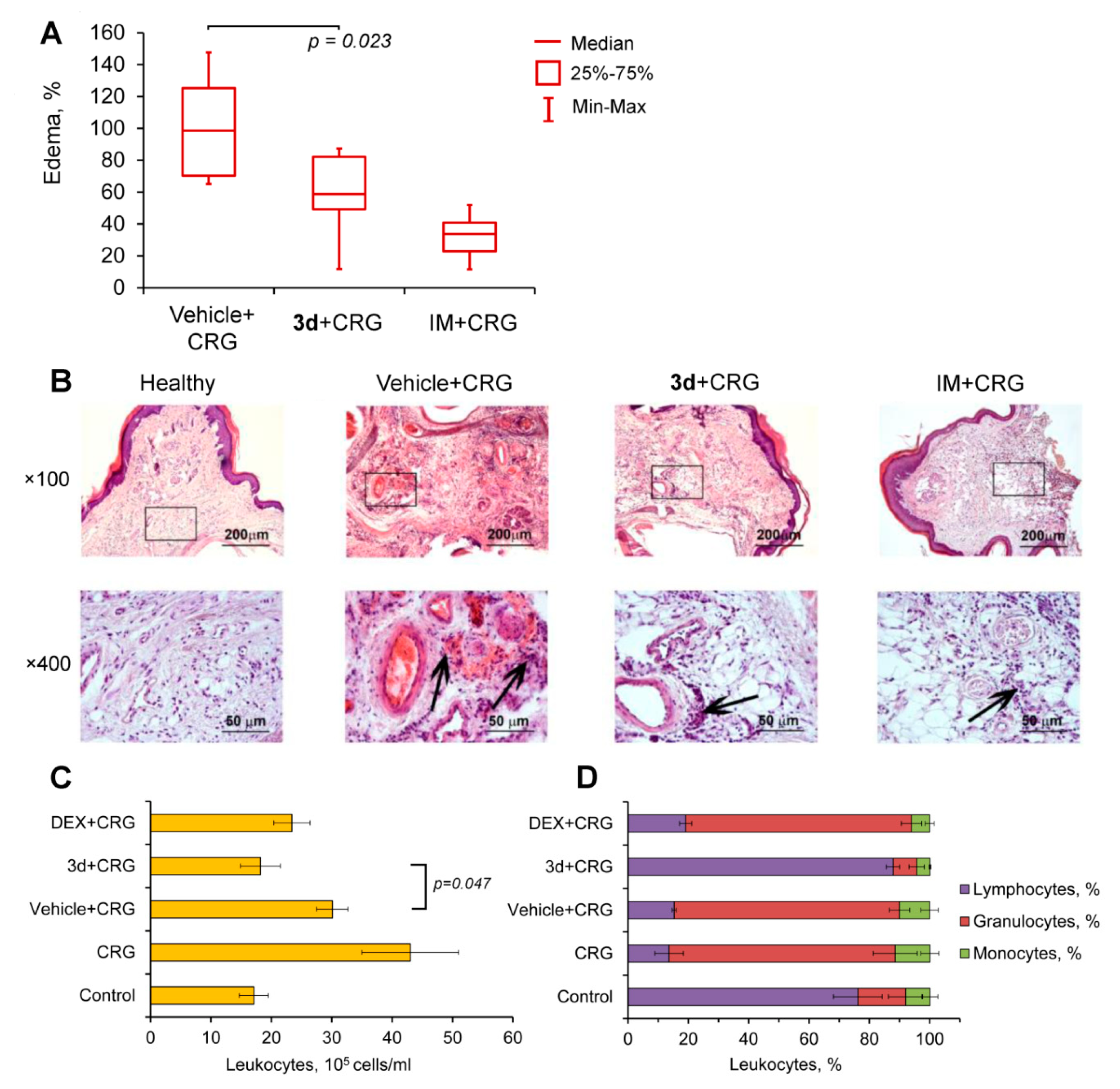
2.6. Compound 3d Effectively Inhibits Carrageenan-Induced Inflammation In Vivo
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Reagents
4.3. General Procedure A for 3β-acetoxy-11-oxo-18βH-olean-12-en-29-oic acid (1) Interaction with N′ Hydroxy(alkyl/aryl/hetaryl)Imidamides
4.4. General Procedure B for Cyclization of Compounds 3a–3h by n-Bu4NF in THF
4.5. General Procedure C for the Hydrolysis of Acetate Groups of Oxadiazole Derivatives 4a–4h
4.6. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)Acetimidamide (3a)
4.7. 30-nor-3β-acetoxy-11-oxo-20-(3′-methyl-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (4a)
4.8. 30-nor-3β-hydroxy-11-oxo-20-(3′-methyl-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5a)
4.9. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)propionimidamide (3b)
4.10. 30-nor-3β-acetoxy-11-oxo-20-(3′-ethyl-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (4b)
4.11. 30-nor-3β-hydroxy-11-oxo-20-(3′-ethyl-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5b)
4.12. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)isobutyrimidamide (3c)
4.13. 30-nor-3β-acetoxy-11-oxo-20-(3′-isopropyl -1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (4c)
4.14. 30-nor-3β-hydroxy-11-oxo-20-(3′-isopropyl-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5c)
4.15. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)pivalimidamide (3d)
4.16. 30-nor-3β-acetoxy-11-oxo-20-(3′-tert-butyl-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (4d)
4.17. 30-nor-3β-hydroxy-11-oxo-20-(3′-tert-butyl -1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5d)
4.18. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)benzimidamide (3e)
4.19. 30-nor-3β-Acetoxy-11-oxo-20-(3′-Phenyl-1′,2′,4′-Oxadiazol-5′-yl)-18βH-olean-12-en (4e)
4.20. 30-nor-3β-hydroxy-11-oxo-20-(3′-phenyl-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5e)
4.21. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)Picolinimidamide (3f)
4.22. 30-nor-3β-acetoxy-11-oxo-20-(3′-(pyridin-2′′-yl)-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (4f)
4.23. 30-nor-3β-hydroxy-11-oxo-20-(3′-(pyridin-2′′-yl)-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5f)
4.24. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)nicotinimidamide (3g)
4.25. 30-nor-3β-acetoxy-11-oxo-20-(3′-(pyridin-3′′-yl)-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (4g)
4.26. 30-nor-3β-hydroxy-11-oxo-20-(3′-(pyridin-3′′-yl)-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5g)
4.27. N′-(3β-Acetoxy-11-oxo-18βH-olean-12-en-29-oyl)isonicotinimidamide (3h)
4.28. 30-nor-3β-acetoxy-11-oxo-20-(3′-(pyridin-4″-yl)-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (4h)
4.29. 30-nor-3β-hydroxy-11-oxo-20-(3′-(pyridin-4′′-yl)-1′,2′,4′-oxadiazol-5′-yl)-18βH-olean-12-en (5h)
4.30. Cell Cultures and GA Derivatives
4.31. Mice
4.32. Analysis of Cytotoxicity of GA Derivatives by MTT Assay
4.33. Apoptosis Assay by Flow Cytometry
4.34. Analysis of Mitochondrial Membrane Potential
4.35. Metacaspases Activity Assay
4.36. Executioner Caspases-3/7 Activity Assay
4.37. Colony-Forming Assay
4.38. Measurement of Cell Motility Using xCelligence Technology
4.39. Tumor Transplantation and Design of Animal Experiments
4.40. Toxicity Assessment
4.41. Analysis of the Number of Metastases and Metastasis Inhibition Index
4.42. In Silico Prediction of the Anti-Inflammatory Potential of the Tested Compounds
4.43. The Measurement of TNF-α and NO Production by Inflamed Macrophages
4.44. Carrageenan-Induced Paw Edema
4.45. Histology
4.46. Carrageenan-Induced Peritonitis
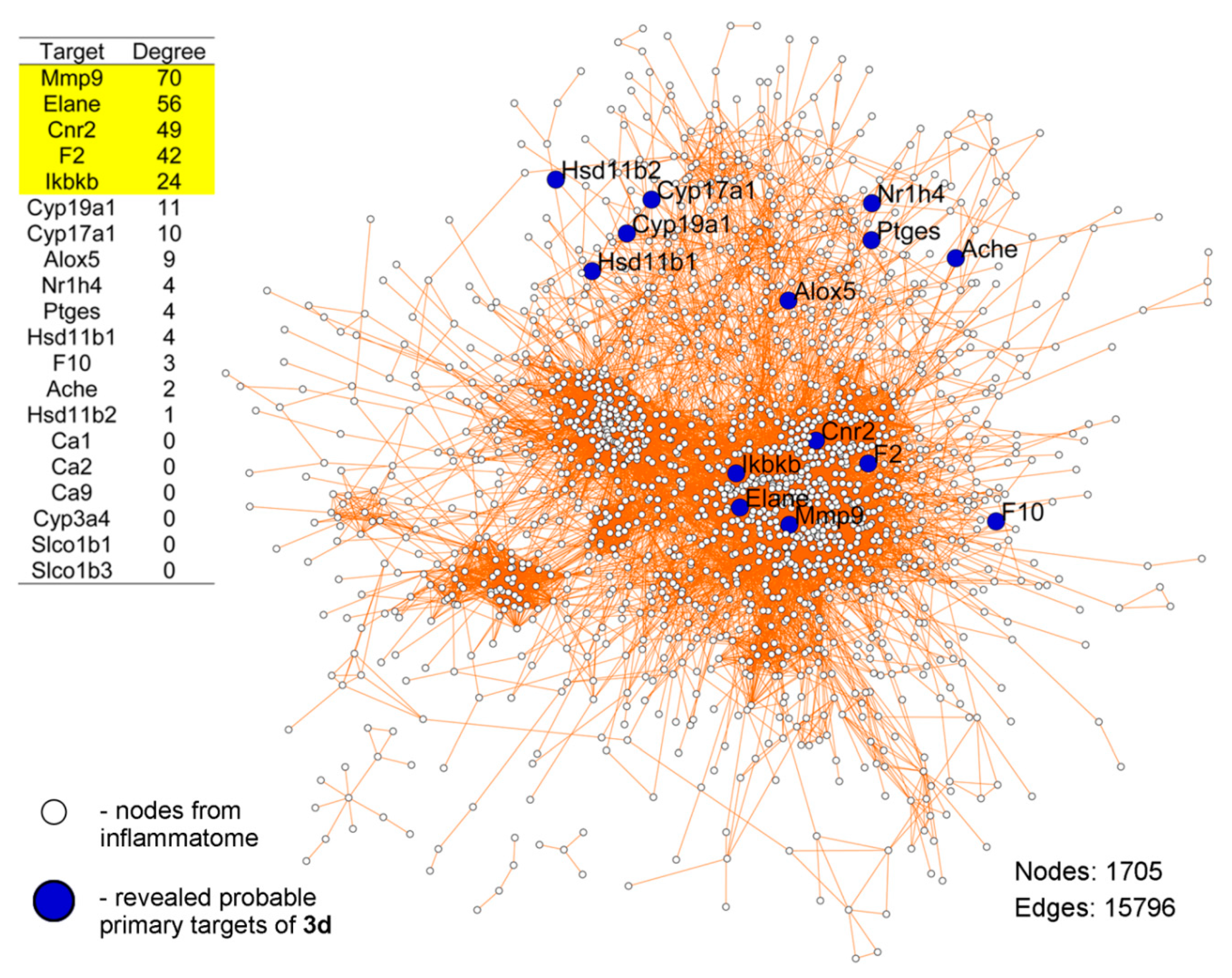
4.47. Prediction of Probable Primary Targets of 3d
4.48. PPI Network Reconstruction
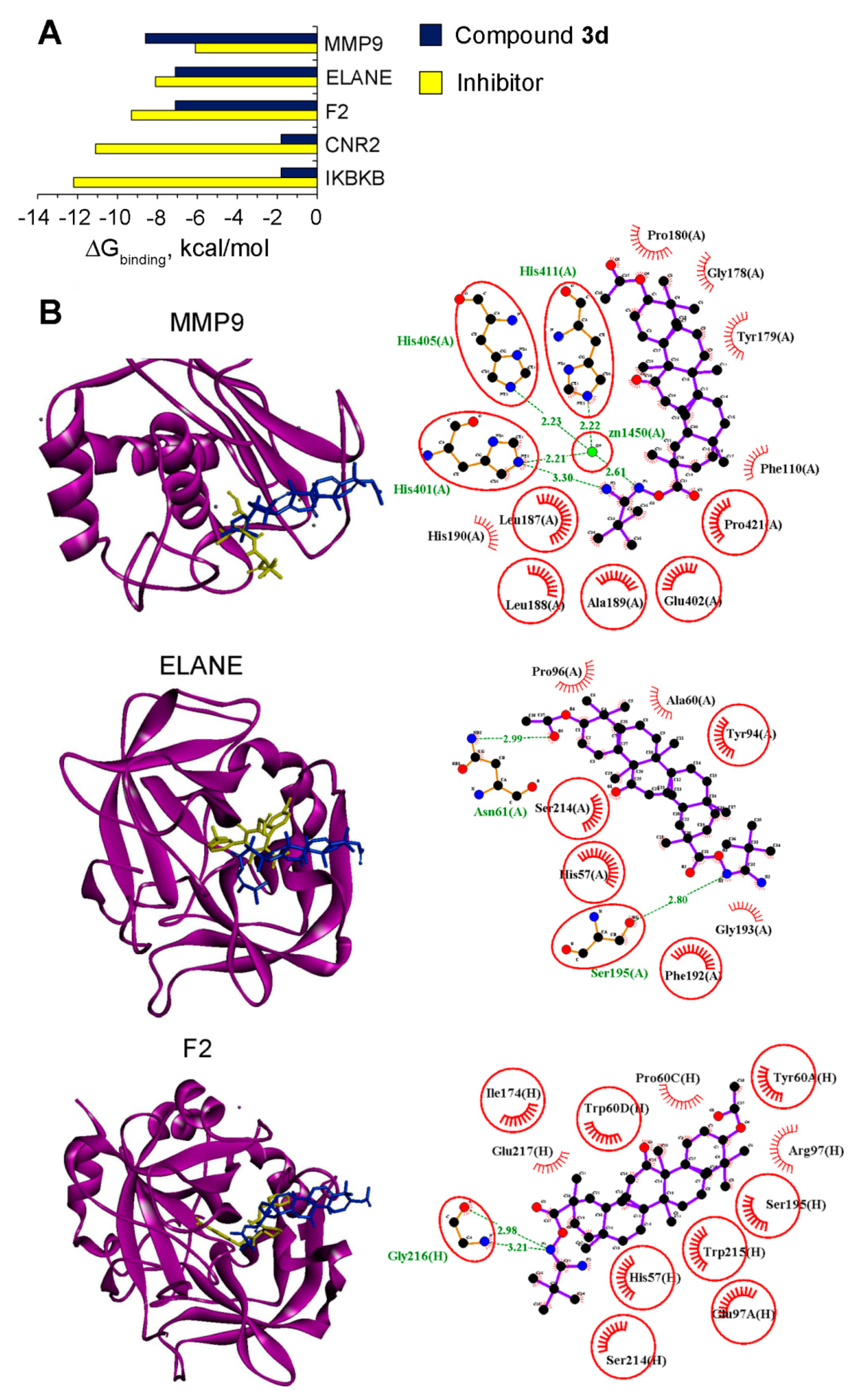
4.49. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDI | N,N′-carbonyldiimidazole |
| ELANE | Neutrophil elastase |
| F2 | Thrombin |
| GA | 18βH-glycyrrhetinic acid |
| IFNγ | Interferon γ |
| MII | Metastatic inhibition index |
| MMP9 | Matrix metalloproteinase 9 |
| NN | Nearest neighbor |
| PI | Propidium iodide |
| SI | Selectivity index |
| TBAF | tetrabutylammonium fluoride |
References
- Salassa, G.; Terenzi, A. Metal complexes of oxadiazole ligands: An overview. Int. J. Mol. Sci. 2019, 20, 3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanady, B.; Song, W.; Peng, R.; Wu, T.; Ge, Z. Efficiency enhancement of organic solar cells enabled by interface engineering of sol-gel zinc oxide with an oxadiazole-based material. Org. Electron. 2020, 76, 105483. [Google Scholar] [CrossRef]
- Wu, Q.; Braveenth, R.; Bae, I.J.; Zhang, H.Q.; Jung, H.; Kim, M.; Chai, K.Y. Oxadiazole- and indolocarbazole-based bipolar materials for green and yellow phosphorescent organic light emitting diodes. Dyes Pigment. 2020, 174, 108052. [Google Scholar] [CrossRef]
- Pace, A.; Pierro, P. The new era of 1,2,4-oxadiazoles. Org. Biomol. Chem. 2009, 7, 4337–4348. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in medicinal chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, N.M.M.; De Oliveira, S.P.; Srivastava, R.M.; Da Silva, J.R. Synthesis of 3-aryl-5-decapentyl-1,2,4-oxadiazoles possessing antiinflammatory and antitumor properties. Farmaco 2005, 60, 955–960. [Google Scholar] [CrossRef]
- Srivastava, R.M.; de Almeida Lima, A.; Viana, O.S.; da Costa Silva, M.J.; Catanho, M.T.J.A.; de Morais, J.O.F. Antiinflammatory Property of 3-Aryl-5-(n-propyl)-1,2,4-oxadiazoles and Antimicrobial Property of 3-Aryl-5-(n-propyl)-4,5-dihydro-1,2,4-oxadiazoles: Their Syntheses and Spectroscopic Studies. Bioorg. Med. Chem. 2003, 11, 1821–1827. [Google Scholar] [CrossRef]
- Benltifa, M.; Vidal, S.; Fenet, B.; Msaddek, M.; Goekjian, P.G.; Praly, J.-P.; Brunyánszki, A.; Docsa, T.; Gergely, P. In Search of Glycogen Phosphorylase Inhibitors: 5-Substituted 3-C-Glucopyranosyl-1,2,4-oxadiazoles from β-D-Glucopyranosyl Cyanides upon Cyclization of O-Acylamidoxime Intermediates. European J. Org. Chem. 2006, 2006, 4242–4256. [Google Scholar] [CrossRef]
- Janardhanan, J.; Chang, M.; Mobashery, S. The oxadiazole antibacterials. Curr. Opin. Microbiol. 2016, 33, 13–17. [Google Scholar] [CrossRef] [Green Version]
- De, S.S.; Khambete, M.P.; Degani, M.S. Oxadiazole scaffolds in anti-tuberculosis drug discovery. Bioorg. Med. Chem. Lett. 2019, 29, 1999–2007. [Google Scholar] [CrossRef]
- Rasool, I.; Ahmad, M.; Khan, Z.A.; Mansha, A.; Maqbool, T.; Zahoor, A.F.; Aslam, S. Recent advancements in oxadiazole-based anticancer agents. Trop. J. Pharm. Res. 2017, 16, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Liu, G.; Li, H.; Zhang, Y.; Song, D.; Li, C.; Wang, R.; Liu, B.; Liang, W.; Jing, Y.; et al. Novel oxadiazole analogues derived from ethacrynic acid: Design, synthesis, and structure—Activity relationships in inhibiting the activity of glutathione S-transferase P1-1 and cancer cell proliferation. J. Med. Chem. 2010, 53, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Z.; Kasibhatla, S.; Kuemmerle, J.; Kemnitzer, W.; Ollis-Mason, K.; Qiu, L.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery and structure-activity relationship of 3-aryl-5-aryl-1,2,4- oxadiazoles as a new series of apoptosis inducers and potential anticancer agents. J. Med. Chem. 2005, 48, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Amin Mohsin, N.U.; Irfan, M. Review recent approaches for the synthesis of hybrid resveratrol molecules and their biological activities: A review. J. Chil. Chem. Soc. 2018, 63, 4135–4149. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi-Khanaposhtani, M.; Ahangar, N.; Sobhani, S.; Masihi, P.H.; Shakiba, A.; Saeedi, M.; Akbarzadeh, T. Design, synthesis, in vivo, and in silico evaluation of new coumarin-1,2,4-oxadiazole hybrids as anticonvulsant agents. Bioorg. Chem. 2019, 89, 102989. [Google Scholar] [CrossRef]
- Krishna, C.; Bhargavi, M.V.; Rao, C.P.; Krupadanam, G.L.D. Synthesis and antimicrobial assessment of novel coumarins featuring 1,2,4-oxadiazole. Med. Chem. Res. 2015, 24, 3743–3751. [Google Scholar] [CrossRef]
- Gonda, T.; Bérdi, P.; Zupkó, I.; Fülöp, F.; Szakonyi, Z. Stereoselective synthesis, synthetic and pharmacological application of monoterpene-based 1,2,4- and 1,3,4-oxadiazoles. Int. J. Mol. Sci. 2018, 19, 81. [Google Scholar] [CrossRef] [Green Version]
- Kovács, D.; Mótyán, G.; Wölfling, J.; Kovács, I.; Zupkó, I.; Frank, É. A facile access to novel steroidal 17-2′-(1′,3′,4′) -oxadiazoles, and an evaluation of their cytotoxic activities in vitro. Bioorg. Med. Chem. Lett. 2014, 24, 1265–1268. [Google Scholar] [CrossRef]
- Kovács, D.; Wölfling, J.; Szabó, N.; Szécsi, M.; Kovács, I.; Zupkó, I.; Frank, É. An efficient approach to novel 17-5′-(1′,2′,4′)- oxadiazolyl androstenes via the cyclodehydration of cytotoxic O-steroidacylamidoximes, and an evaluation of their inhibitory action on 17α-hydroxylase/C17,20-lyase. Eur. J. Med. Chem. 2013, 70, 649–660. [Google Scholar] [CrossRef]
- Antimonova, A.N.; Petrenko, N.I.; Shakirov, M.M.; Pokrovskii, M.A.; Pokrovskii, A.G.; Shul’ts, E.E. Synthesis and cytotoxic activity of lupane triterpenoids containing 1,3,4-oxadiazoles. Chem. Nat. Compd. 2014, 50, 1016–1023. [Google Scholar] [CrossRef]
- Popov, S.A.; Semenova, M.D.; Baev, D.S.; Sorokina, I.V.; Zhukova, N.A.; Frolova, T.S.; Tolstikova, T.G.; Shults, E.E.; Turks, M. Lupane-type conjugates with aminoacids, 1,3,4- oxadiazole and 1,2,5-oxadiazole-2-oxide derivatives: Synthesis, anti-inflammatory activity and in silico evaluation of target affinity. Steroids 2019, 150, 108443. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.A.; Semenova, M.D.; Baev, D.S.; Frolova, T.S.; Shults, E.E.; Wang, C.; Turks, M. Synthesis of cytotoxic urs-12-ene- and 28-norurs-12-ene- type conjugates with amino- and mercapto-1,3,4-oxadiazoles and mercapto-1,2,4-triazoles. Steroids 2020, 153, 108524. [Google Scholar] [CrossRef]
- Gu, W.; Jin, X.Y.; Li, D.D.; Wang, S.F.; Tao, X.B.; Chen, H. Design, synthesis and in vitro anticancer activity of novel quinoline and oxadiazole derivatives of ursolic acid. Bioorg. Med. Chem. Lett. 2017, 27, 4128–4132. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.Y.; Chen, H.; Li, D.D.; Li, A.L.; Wang, W.Y.; Gu, W. Design, synthesis, and anticancer evaluation of novel quinoline derivatives of ursolic acid with hydrazide, oxadiazole, and thiadiazole moieties as potent MEK inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 955–972. [Google Scholar] [CrossRef] [Green Version]
- Challa, K.; Bhargavi, M.V.; Krupadanam, G.L.D. Design, semisynthesis and cytotoxic activity of novel ester derivatives of betulinic acid-1,2,4 oxadiazoles. J. Asian Nat. Prod. Res. 2016, 18, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Green, I.R.; Shamraiz, U.; Saleem, M.; Badshah, A.; Abbas, G.; Rehman, N.U.; Irshad, M. Therapeutic potential of glycyrrhetinic acids: A patent review (2010-2017). Expert Opin. Ther. Pat. 2018, 28, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Cancer and Inflammation: An Old Intuition with Rapidly Evolving New Concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef]
- Kayukova, L.A. Synthesis of 1,2,4-oxadiazoles (a review). Pharm. Chem. J. 2005, 39, 539–547. [Google Scholar] [CrossRef]
- Bora, R.; Dar, B.; Pradhan, V.; Farooqui, M. [1, 2, 4]-Oxadiazoles: Synthesis and Biological Applications. Mini-Rev. Med. Chem. 2014, 14, 355–369. [Google Scholar] [CrossRef]
- Bacchi, A.; Carcelli, M.; Compari, C.; Fisicaro, E.; Pala, N.; Rispoli, G.; Rogolino, D.; Sanchez, T.W.; Sechi, M.; Sinisi, V.; et al. Investigating the role of metal chelation in HIV-1 integrase strand transfer inhibitors. J. Med. Chem. 2011, 54, 8407–8420. [Google Scholar] [CrossRef]
- Kumpan, K.; Nathubhai, A.; Zhang, C.; Wood, P.J.; Lloyd, M.D.; Thompson, A.S.; Haikarainen, T.; Lehtiö, L.; Threadgill, M.D. Structure-based design, synthesis and evaluation in vitro of arylnaphthyridinones, arylpyridopyrimidinones and their tetrahydro derivatives as inhibitors of the tankyrases. Bioorg. Med. Chem. 2015, 23, 3013–3032. [Google Scholar] [CrossRef] [Green Version]
- Gangloff, A.R.; Litvak, J.; Shelton, E.J.; Sperandio, D.; Wang, V.R.; Rice, K.D. Tetrabutylammonium Fluoride As a Mild and Efficient Catalyst. Tetrahedron 2001, 42, 1441–1443. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.V.; Zenkova, M.A.; Logashenko, E.B. Modulation of Tumour-Related Signaling Pathways by Natural Pentacyclic Triterpenoids and their Semisynthetic Derivatives. Curr. Med. Chem. 2017, 24, 1277–1320. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Martínez, S.M.; Piedrafita, F.J.; Cannata-Andía, J.B.; López-Novoa, J.M.; López-Hernández, F.J. Necrotic concentrations of cisplatin activate the apoptotic machinery but inhibit effector caspases and interfere with the execution of apoptosis. Toxicol. Sci. 2011, 122, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Nemade, H.; Chaudhari, U.; Acharya, A.; Hescheler, J.; Hengstler, J.G.; Papadopoulos, S.; Sachinidis, A. Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch. Toxicol. 2018, 92, 1507–1524. [Google Scholar] [CrossRef] [Green Version]
- Ghafari, F.; Rajabi, M.R.; Mazoochi, T.; Taghizadeh, M.; Nikzad, H.; Atlasi, M.A.; Taherian, A. Comparing apoptosis and necrosis effects of Arctium lappa root extract and doxorubicin on MCF7 and MDA-MB-231 cell lines. Asian Pac. J. Cancer Prev. 2017, 18, 795–802. [Google Scholar]
- Hsieh, S.C.; Wu, C.C.; Hsu, S.L.; Yen, J.H. Molecular mechanisms of gallic acid-induced growth inhibition, apoptosis, and necrosis in hypertrophic scar fibroblasts. Life Sci. 2017, 179, 130–138. [Google Scholar] [CrossRef]
- Srivastava, S.; Somasagara, R.R.; Hegde, M.; Nishana, M.; Tadi, S.K.; Srivastava, M.; Choudhary, B.; Raghavan, S.C. Quercetin, a natural flavonoid interacts with DNA, arrests cell cycle and causes tumor regression by activating mitochondrial pathway of apoptosis. Sci. Rep. 2016, 6, 24049. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.H.; Wu, H.Y.; Chang, W.H. Dosage effects of curcumin on cell death types in a human osteoblast cell line. Food Chem. Toxicol. 2006, 44, 1362–1371. [Google Scholar] [CrossRef]
- Kitazumi, I.; Tsukahara, M. Regulation of DNA fragmentation: The role of caspases and phosphorylation. FEBS J. 2011, 278, 427–441. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chen, X.; Zhang, J.; Wang, J. 18β-glycyrrhetinic acid inhibits migration and invasion of human gastric cancer cells via the ROS/PKC-α/ERK pathway. J. Nat. Med. 2018, 72, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shen, Y.; Qiu, R.; Chen, Z.; Chen, Z.; Chen, W. 18 Β-Glycyrrhetinic Acid Exhibits Potent Antitumor Effects Against Colorectal Cancer Via Inhibition of Cell Proliferation and Migration. Int. J. Oncol. 2017, 51, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Wang, N.; Liu, Y.; Wang, S.; Wang, H.; Li, A.; Ren, S. Synthesis and discovery of 18β-glycyrrhetinic acid derivatives inhibiting cancer stem cell properties in ovarian cancer cells. RSC Adv. 2019, 9, 27294–27304. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Dai, F.J.; Cui, H.W.; Peng, S.H.; He, Y.; Wang, X.; Yi, Z.F.; Qiu, W.W. Synthesis of novel heterocyclic ring-fused 18β-glycyrrhetinic acid derivatives with antitumor and antimetastatic activity. Chem. Biol. Drug Des. 2014, 84, 223–233. [Google Scholar] [CrossRef]
- Schwartzberg, L.S.; Navari, R.M. Safety of Polysorbate 80 in the Oncology Setting. Adv. Ther. 2018, 35, 754–767. [Google Scholar] [CrossRef] [Green Version]
- Mizushina, Y.; Ishida, T.; Yagi, S.; Irino, Y.; Nishiumi, S.; Miki, I.; Kondo, Y.; Mizuno, S.; Yoshida, H.; Azuma, T.; et al. Inhibitory effects of glycyrrhetinic acid on DNA polymerase and inflammatory activities. Evid. Based Complement. Altern. Med. 2012, 2012, 650514. [Google Scholar]
- Wang, C.Y.; Kao, T.C.; Lo, W.H.; Yen, G.C. Glycyrrhizic acid and 18β-glycyrrhetinic acid modulate lipopolysaccharide-induced inflammatory response by suppression of NF-κB through PI3K p110δ and p110γ inhibitions. J. Agric. Food Chem. 2011, 59, 7726–7733. [Google Scholar] [CrossRef]
- Zhou, J.-X.; Wink, M. Evidence for Anti-Inflammatory Activity of Isoliquiritigenin, 18β Glycyrrhetinic Acid, Ursolic Acid, and the Traditional Chinese Medicine Plants Glycyrrhiza glabra and Eriobotrya japonica, at the Molecular Level. Medicines 2019, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Nishibu, A.; Yoshida, N.; Yasoshima, M.; Anzawa, K.; Watanabe, Y.; Nagai, Y.; Takatsu, K.; Ogawa, K.; Mochizuki, T. Glycyrrhetinic acid inhibits contact hypersensitivity induced by trichophytin via dectin-1. Exp. Dermatol. 2016, 25, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Qiu, L.; Wang, F. 18α-Glycyrrhetinic acid (GA) ameliorates fructose-induced nephropathy in mice by suppressing oxidative stress, dyslipidemia and inflammation. Biomed. Pharmacother. 2020, 125, 109702. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.D.; Kang, S.H.; Bang, K.S.; Chang, Y.N.; Lee, J.H.; Jin, J.S. Glycyrrhetic acid ameliorates dextran sulfate sodium-induced ulcerative colitis in vivo. Molecules 2016, 21, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Hong, J.H.; Lee, J.E.; Lee, Y.C. 18β-Glycyrrhetinic acid, the major bioactive component of Glycyrrhizae Radix, attenuates airway inflammation by modulating Th2 cytokines, GATA-3, STAT6, and Foxp3 transcription factors in an asthmatic mouse model. Environ. Toxicol. Pharmacol. 2017, 52, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kuang, G.; Gong, X.; Jiang, R.; Xie, T.; Tie, H.; Wu, S.; Wang, T.; Wan, J.; Wang, B. Glycyrrhetinic acid pretreatment attenuates liver ischemia/reperfusion injury via inhibiting TLR4 signaling cascade in mice. Int. Immunopharmacol. 2019, 76, 105870. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, D.E.; Lee, S.H. Effects of 18β-glycyrrhetinic acid on fungal protease-induced airway inflammatory responses. Mediat. Inflamm. 2018, 2018, 6461032. [Google Scholar] [CrossRef] [Green Version]
- Maitraie, D.; Hung, C.F.; Tu, H.Y.; Liou, Y.T.; Wei, B.L.; Yang, S.C.; Wang, J.P.; Lin, C.N. Synthesis, anti-inflammatory, and antioxidant activities of 18β-glycyrrhetinic acid derivatives as chemical mediators and xanthine oxidase inhibitors. Bioorg. Med. Chem. 2009, 17, 2785–2792. [Google Scholar] [CrossRef]
- Markov, A.V.; Sen’kova, A.V.; Zenkova, M.A.; Logashenko, E.B. Novel Glycyrrhetinic Acid Derivative Soloxolone Methyl Inhibits the Inflammatory Response and Tumor Growth in vivo. Mol. Biol. 2018, 52, 262–268. [Google Scholar] [CrossRef]
- Markov, A.V.; Sen’kova, A.V.; Warszycki, D.; Salomatina, O.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Logashenko, E.B. Soloxolone methyl inhibits influenza virus replication and reduces virus-induced lung inflammation. Sci. Rep. 2017, 7, 13968. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, Y.; Chen, L.; Chen, S.; Zhang, J.; Tang, W. 18α-Glycyrrhetinic acid monoglucuronide as an anti-inflammatory agent through suppression of the NF-κB and MAPK signaling pathway. Medchemcomm 2017, 8, 1498–1504. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, L.; Han, Y.; Wu, Z.; Chen, P.; Zhang, H.; Zhou, J. Protective effects of hepatocyte-specific glycyrrhetic derivatives against carbon tetrachloride-induced liver damage in mice. Bioorg. Chem. 2017, 72, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the pass online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Morris, C.J. Carrageenan-induced paw edema in the rat and mouse. In Inflammation Protocols; Humana Press: Totowa, NJ, USA, 2003; Volume 225, pp. 115–121. [Google Scholar]
- Jeong, G.-S.; Bae, J.-S. Anti-Inflammatory Effects of Triterpenoids; Naturally Occurring and Synthetic Agents. Mini. Rev. Org. Chem. 2014, 11, 316–329. [Google Scholar] [CrossRef]
- Xiao, S.; Tian, Z.; Wang, Y.; Si, L.; Zhang, L.; Zhou, D. Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Med. Res. Rev. 2018, 38, 951–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, P.Y. Novel targets of pentacyclic triterpenoids in Staphylococcus aureus: A systematic review. Phytomedicine 2019, 152933. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Hamid, K.; Kam, A.; Wong, K.H.; Abdelhak, Z.; Razmovski-Naumovski, V.; Chan, K.; Li, K.M.; Groundwater, P.W.; Li, G.Q. The Pentacyclic Triterpenoids in Herbal Medicines and Their Pharmacological Activities in Diabetes and Diabetic Complications. Curr. Med. Chem. 2013, 20, 908–931. [Google Scholar]
- Kvasnica, M.; Urban, M.; Dickinson, N.J.; Sarek, J. Pentacyclic triterpenoids with nitrogen- and sulfur-containing heterocycles: Synthesis and medicinal significance. Nat. Prod. Rep. 2015, 32, 1303–1330. [Google Scholar] [CrossRef]
- Ng, Y.P.; Chen, Y.; Hu, Y.; Ip, F.C.F.; Ip, N.Y. Olean-12-Eno[2,3-c] [1,2,5]Oxadiazol-28-Oic Acid (OEOA) Induces G1 Cell Cycle Arrest and Differentiation in Human Leukemia Cell Lines. PLoS ONE 2013, 8, e63580. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.Y.; Chi, K.Q.; Wang, K.S.; Wu, J.; Liu, L.P.; Piao, H.R. Design, synthesis, evaluation, and molecular docking of ursolic acid derivatives containing a nitrogen heterocycle as anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2018, 28, 1797–1803. [Google Scholar] [CrossRef]
- Deepthi, A.; Krishnan, D.; Sanju, A. Semisynthesis of ursolic acid-2-(2-thienylidene)-oxadiazole hybrid molecule and an evaluation of its COX inhibition property. J. Heterocycl. Chem. 2020, 57, 2048–2055. [Google Scholar] [CrossRef]
- Sidova, V.; Zoufaly, P.; Pokorny, J.; Dzubak, P.; Hajduch, M.; Popa, I.; Urban, M. Cytotoxic conjugates of betulinic acid and substituted triazoles prepared by Huisgen Cycloaddition from 30-azidoderivatives. PLoS ONE 2017, 12, e0171621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokorny, J.; Krajcovicova, S.; Hajduch, M.; Holoubek, M.; Gurska, S.; Dzubak, P.; Volna, T.; Popa, I.; Urban, M. Triterpenic azines, a new class of compounds with selective cytotoxicity to leukemia cells CCRF-CEM. Future Med. Chem. 2018, 10, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, H.; Liu, J.; Liu, Y.; Wang, Y.; Xu, S.; Zhu, Z.; Xu, J. Synthesis, biological evaluation and mechanism studies of C-23 modified 23-hydroxybetulinic acid derivatives as anticancer agents. Eur. J. Med. Chem. 2019, 182, 111659. [Google Scholar] [CrossRef] [PubMed]
- Awale, M.; Reymond, J.L. Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning. J. Chem. Inf. Model. 2019, 59, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.M.; Zhang, B.; Yang, X.; Zhu, J.; Stepaniants, S.; Zhang, C.; Meng, Q.; Peters, M.; He, Y.; Ni, C.; et al. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 2012, 8, 1–16. [Google Scholar] [CrossRef]
- Tochowicz, A.; Maskos, K.; Huber, R.; Oltenfreiter, R.; Dive, V.; Yiotakis, A.; Zanda, M.; Bode, W.; Goettig, P. Crystal Structures of MMP-9 Complexes with Five Inhibitors: Contribution of the Flexible Arg424 Side-chain to Selectivity. J. Mol. Biol. 2007, 371, 989–1006. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.X.; Li, S.Y.; Zhang, Q.; Chen, H.; Xia, Z.N.; Yang, F.Q. Characterization of phenolic acids binding to thrombin using frontal affinity chromatography and molecular docking. Anal. Methods 2017, 9, 5174–5180. [Google Scholar] [CrossRef]
- Pereira, R.C.C.; Lourenço, A.L.; Terra, L.; Abreu, P.A.; Teixeira, V.L.; Castro, H.C. Marine diterpenes: Molecular modeling of thrombin inhibitors with potential biotechnological application as an antithrombotic. Mar. Drugs 2017, 15, 79. [Google Scholar] [CrossRef] [Green Version]
- Vijayalakshmi, J.; Padmanabhan, K.P.; Tulinsky, A.; Mann, K.G. The isomorphous structures of prethrombin2, hirugen–, and PPACK–thrombin: Changes accompanying activation and exosite binding to thrombin. Protein Sci. 1994, 3, 2254–2271. [Google Scholar] [CrossRef] [Green Version]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Stoeckle, C.; Conus, S.; Simon, H.U. Living and dying for inflammation: Neutrophils, eosinophils, basophils. Trends Immunol. 2013, 34, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dorling, A. The roles of thrombin and protease-activated receptors in inflammation. Semin. Immunopathol. 2012, 34, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padrines, M.; Wolf, M.; Walz, A.; Baggiolini, M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 1994, 352, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Manon-Jensen, T.; Multhaupt, H.A.B.; Couchman, J.R. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J. 2013, 280, 2320–2331. [Google Scholar] [CrossRef]
- Mydel, P.; Shipley, J.M.; Adair-Kirk, T.L.; Kelley, D.G.; Broekelmann, T.J.; Mecham, R.P.; Senior, R.M. Neutrophil elastase cleaves laminin-332 (laminin-5) generating peptides that are chemotactic for neutrophils. J. Biol. Chem. 2008, 283, 9513–9522. [Google Scholar] [CrossRef] [Green Version]
- Kurtagic, E.; Rich, C.B.; Buczek-Thomas, J.A.; Nugent, M.A. Neutrophil elastase-generated fragment of vascular endothelial growth factor-A stimulates macrophage and endothelial progenitor cell migration. PLoS ONE 2015, 10, e0145115. [Google Scholar] [CrossRef] [Green Version]
- Tsen, A.; Kirschenbaum, L.A.; Larow, C.; Khan, R.; Kurtz, S.; Bansal, S.; Astiz, M.E. The effect of anticoagulants and the role of thrombin on neutrophil-endothelial cell interactions in septic shock. Shock 2009, 31, 120–124. [Google Scholar] [CrossRef]
- Hagiwara, S.; Iwasaka, H.; Hidaka, S.; Hasegawa, A.; Noguchi, T. Neutrophil elastase inhibitor (sivelestat) reduces the levels of inflammatory mediators by inhibiting NF-κB. Inflamm. Res. 2009, 58, 198–203. [Google Scholar] [CrossRef]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50, µM | |||||
|---|---|---|---|---|---|---|
| Duodenum | Cervix | Cervix | Melanoma | Lung | Fibroblast | |
| HuTu-80 | HeLa | KB-3-1 | B16 | A549 | hFF3 | |
| 1 | 2.9 ± 0.1 | 12.7 ± 1.1 | 7.4 ± 0.6 | 11.5 ± 1.5 | 15.9 ± 2.3 | 13.6 ± 2.2 |
| GA-Me | 1.6 ± 0.5 | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.9 ± 0.1 | 1.2 ± 0.1 | 9.8 ± 1.0 |
| 3a | 2.3 ± 0.1 | 2.6 ± 0.1 | 5.9 ± 0.2 | 6.8 ± 0.2 | 2.6 ± 0.1 | 1.7 ± 0.1 |
| 3b | 2.4 ± 0.2 | 3.3 ± 0.2 | 6.7 ± 0.7 | 7.2 ± 0.6 | 7.3 ± 3.0 | 2.9 ± 0.3 |
| 3c | 2.1 ± 0.2 | 3.0 ± 0.2 | 4.9 ± 0.4 | 5.6 ± 0.2 | 2.6 ± 0.1 | 2.6 ± 0.1 |
| 3d | >50 | >50 | >50 | >50 | >50 | >50 |
| 3e | 7.5 ± 0.1 | 5.8 ± 0.8 | 16.0 ± 1.4 | 16.4 ± 0.7 | 4.9 ± 0.4 | 7.3 ± 0.3 |
| 3f | 6.9 ± 0.3 | 6.4 ± 1.0 | 10.9 ± 2.2 | 22.5 ± 3.1 | 7.7 ± 1.8 | 7.6 ± 1.0 |
| 3g | 4.9 ± 0.3 | 6.1 ± 1.0 | 10.0 ± 0.9 | 8.9 ± 1.1 | 5.6 ± 0.9 | 8.5 ± 0.3 |
| 3h | 5.6 ± 0.1 | 7.1 ± 1.4 | 7.0 ± 1.4 | 7.8 ± 1.9 | 7.1 ± 1.6 | 11.1 ± 0.5 |
| 4f | 11.4 ± 1.0 | 17.9 ± 7.7 | 10.3 ± 1.2 | 6.0 ± 2.3 | 40.3 ± 4.1 | 11.5 ± 1.0 |
| 4g | 5.8 ± 0.5 | >50 | 9.5 ± 0.5 | >50 | 49.7 ± 4.5 | >50 |
| 4h | 49.7 ± 2.4 | >50 | >50 | >50 | >50 | >50 |
| 5c | 9.5 ± 0.5 | 2.6 ± 0.1 | 5.9 ± 0.1 | 6.6 ± 0.1 | 4.5 ± 1.1 | 5.6 ± 1.0 |
| 5d | 8.8 ± 1.1 | 3.7 ± 0.9 | 22.3 ± 3.7 | 11.3 ± 1.9 | 23.5 ± 6.3 | 21.6 ± 3.4 |
| 5e | >50 | 20.3 ± 2.2 | >50 | >50 | >50 | >50 |
| 5f | 3.8 ± 0.3 | 3.6 ± 0.5 | 4.7 ± 0.1 | 9.4 ± 0.4 | >50 | >50 |
| 5g | 4.7 ± 0.2 | 4.6 ± 0.3 | 16.9 ± 1.4 | >50 | >50 | >50 |
| 5h | 4.7 ± 0.4 | 3.5 ± 0.1 | 4.9 ± 0.3 | >50 | >50 | >50 |
| ID | Name |
|---|---|
| Ache | Acetylcholinesterase |
| Alox5 | Arachidonate 5-Lipoxygenase |
| Ca1 | Carbonic Anhydrase 1 |
| Ca2 | Carbonic Anhydrase 2 |
| Ca9 | Carbonic Anhydrase 9 |
| Cnr2 | Cannabinoid Receptor 2 |
| Cyp17a1 | Cytochrome P450 Family 17 Subfamily A Member 1 |
| Cyp19a1 | Cytochrome P450 Family 19 Subfamily A Member 1 |
| Cyp3a4 | Cytochrome P450 Family 3 Subfamily A Member 4 |
| Elane | Neutrophil Elastase |
| F10 | Coagulation Factor X |
| F2 | Thrombin |
| Hsd11b1 | Hydroxysteroid 11-Beta Dehydrogenase 1 |
| Hsd11b2 | Hydroxysteroid 11-Beta Dehydrogenase 2 |
| Ikbkb | Inhibitor Of Nuclear Factor Kappa B Kinase Subunit Beta |
| Mmp9 | Matrix Metalloproteinase 9 |
| Nr1h4 | Farnesoid X Receptor |
| Ptges | Prostaglandin E Synthase |
| Slco1b1 | Solute Carrier Organic Anion Transporter Family Member 1B1 |
| Slco1b3 | Solute Carrier Organic Anion Transporter Family Member 1B3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markov, A.V.; Sen’kova, A.V.; Popadyuk, I.I.; Salomatina, O.V.; Logashenko, E.B.; Komarova, N.I.; Ilyina, A.A.; Salakhutdinov, N.F.; Zenkova, M.A. Novel 3′-Substituted-1′,2′,4′-Oxadiazole Derivatives of 18βH-Glycyrrhetinic Acid and Their O-Acylated Amidoximes: Synthesis and Evaluation of Antitumor and Anti-Inflammatory Potential In Vitro and In Vivo. Int. J. Mol. Sci. 2020, 21, 3511. https://doi.org/10.3390/ijms21103511
Markov AV, Sen’kova AV, Popadyuk II, Salomatina OV, Logashenko EB, Komarova NI, Ilyina AA, Salakhutdinov NF, Zenkova MA. Novel 3′-Substituted-1′,2′,4′-Oxadiazole Derivatives of 18βH-Glycyrrhetinic Acid and Their O-Acylated Amidoximes: Synthesis and Evaluation of Antitumor and Anti-Inflammatory Potential In Vitro and In Vivo. International Journal of Molecular Sciences. 2020; 21(10):3511. https://doi.org/10.3390/ijms21103511
Chicago/Turabian StyleMarkov, Andrey V., Aleksandra V. Sen’kova, Irina I. Popadyuk, Oksana V. Salomatina, Evgeniya B. Logashenko, Nina I. Komarova, Anna A. Ilyina, Nariman F. Salakhutdinov, and Marina A. Zenkova. 2020. "Novel 3′-Substituted-1′,2′,4′-Oxadiazole Derivatives of 18βH-Glycyrrhetinic Acid and Their O-Acylated Amidoximes: Synthesis and Evaluation of Antitumor and Anti-Inflammatory Potential In Vitro and In Vivo" International Journal of Molecular Sciences 21, no. 10: 3511. https://doi.org/10.3390/ijms21103511