Targeting Insulin-Like Growth Factor 1 Receptor Delays M-Phase Progression and Synergizes with Aurora B Inhibition to Suppress Cell Proliferation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
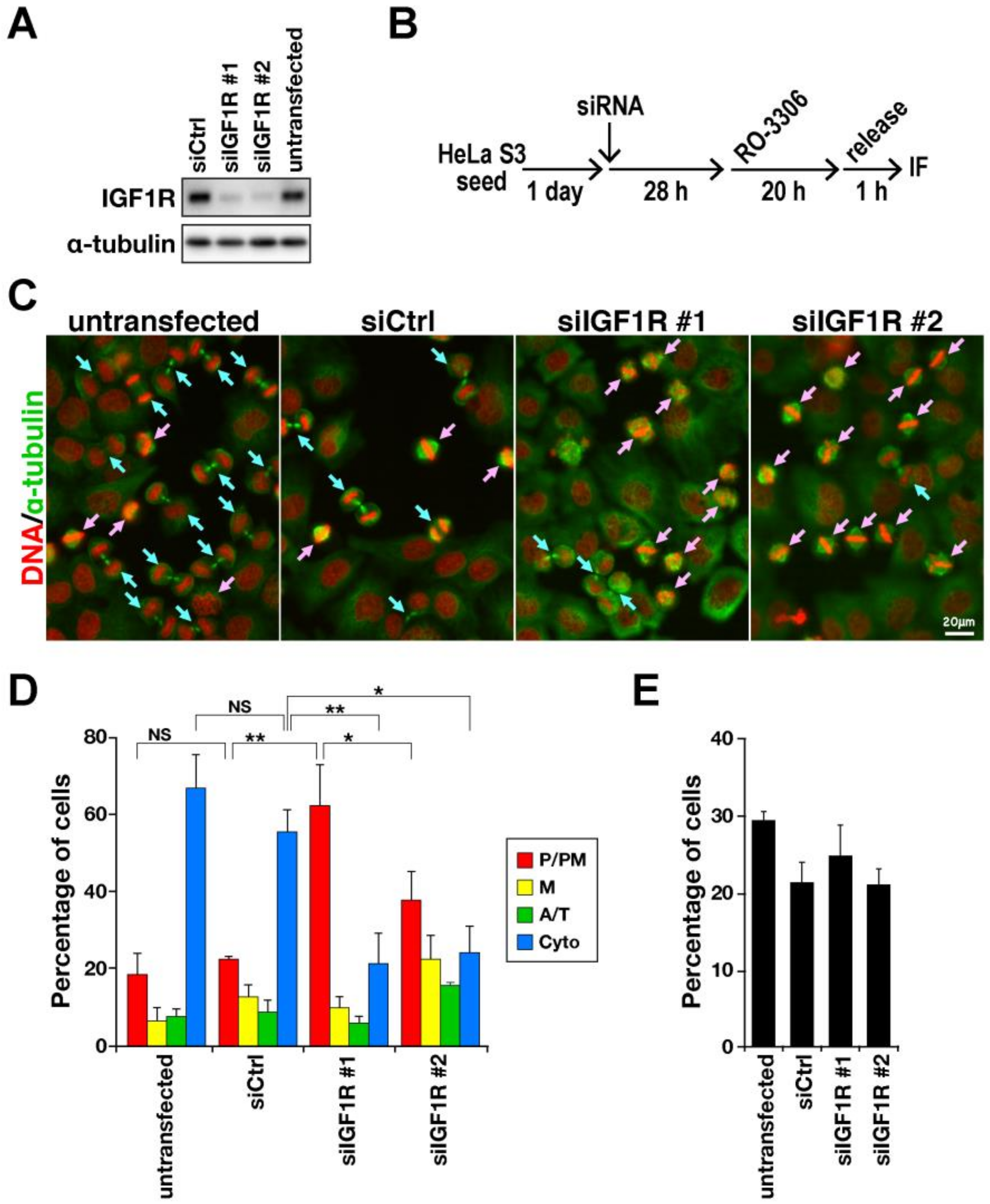
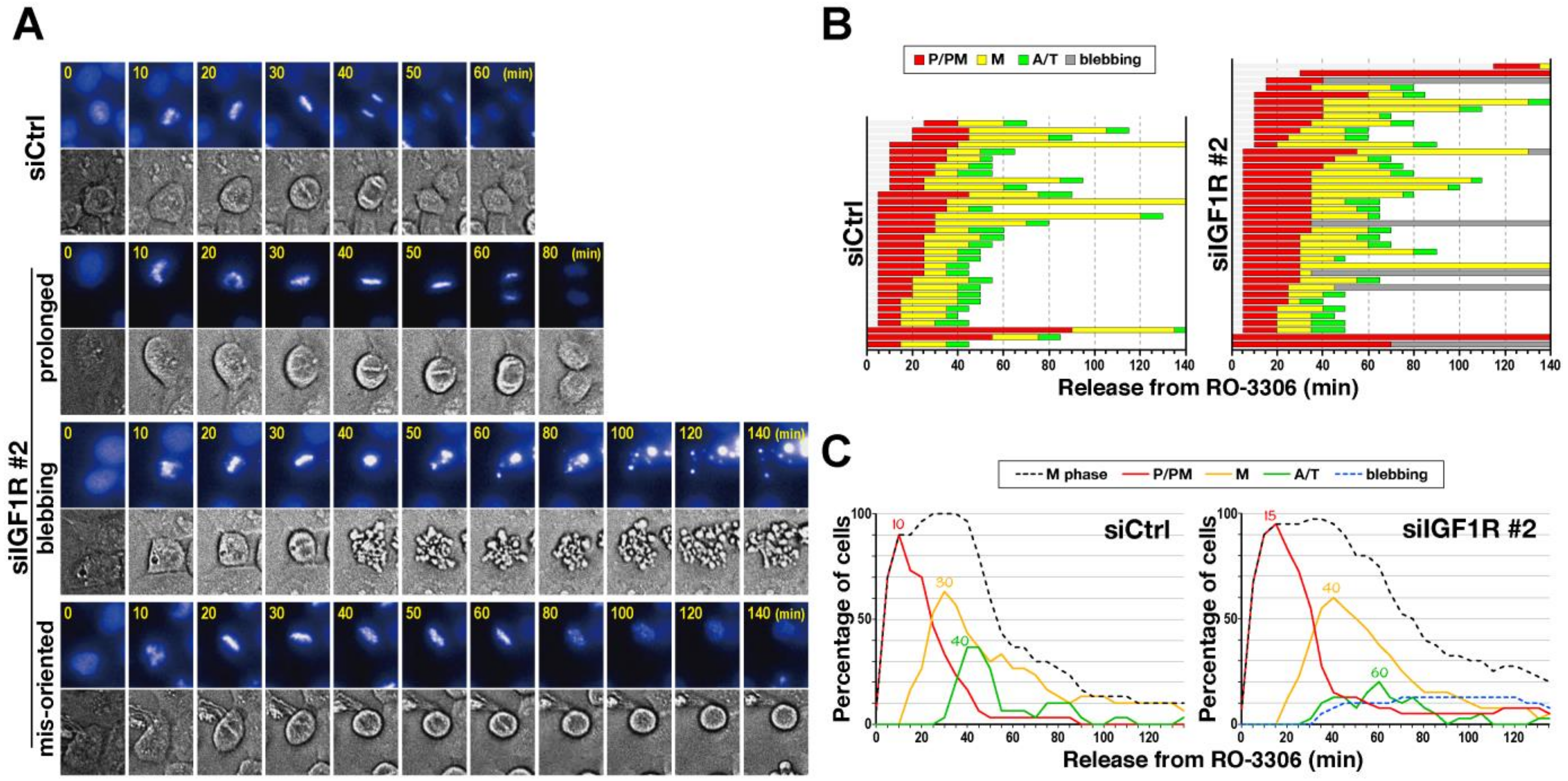
2.1. Delay in M-Phase Progression by IGF1R Knockdown
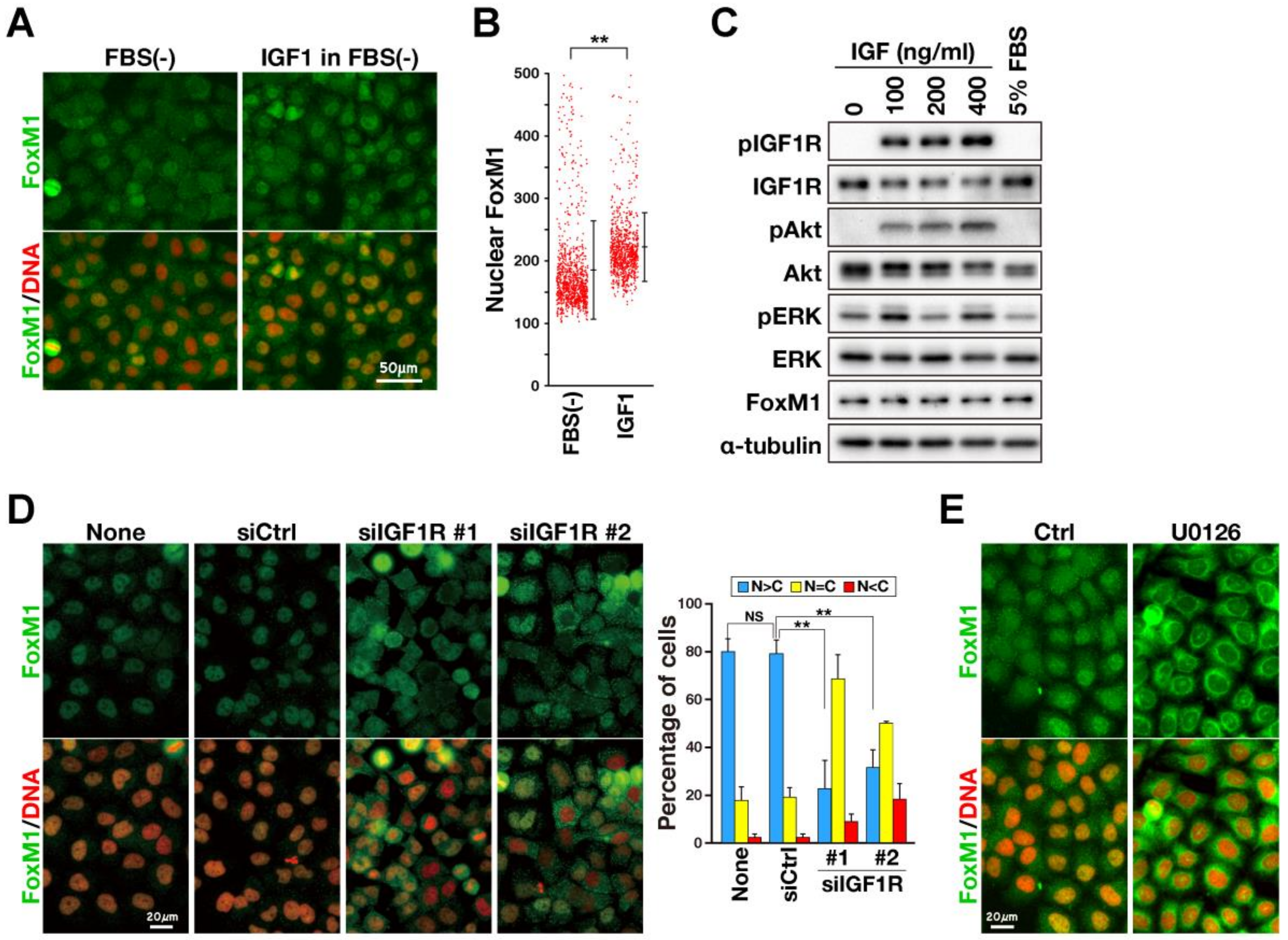
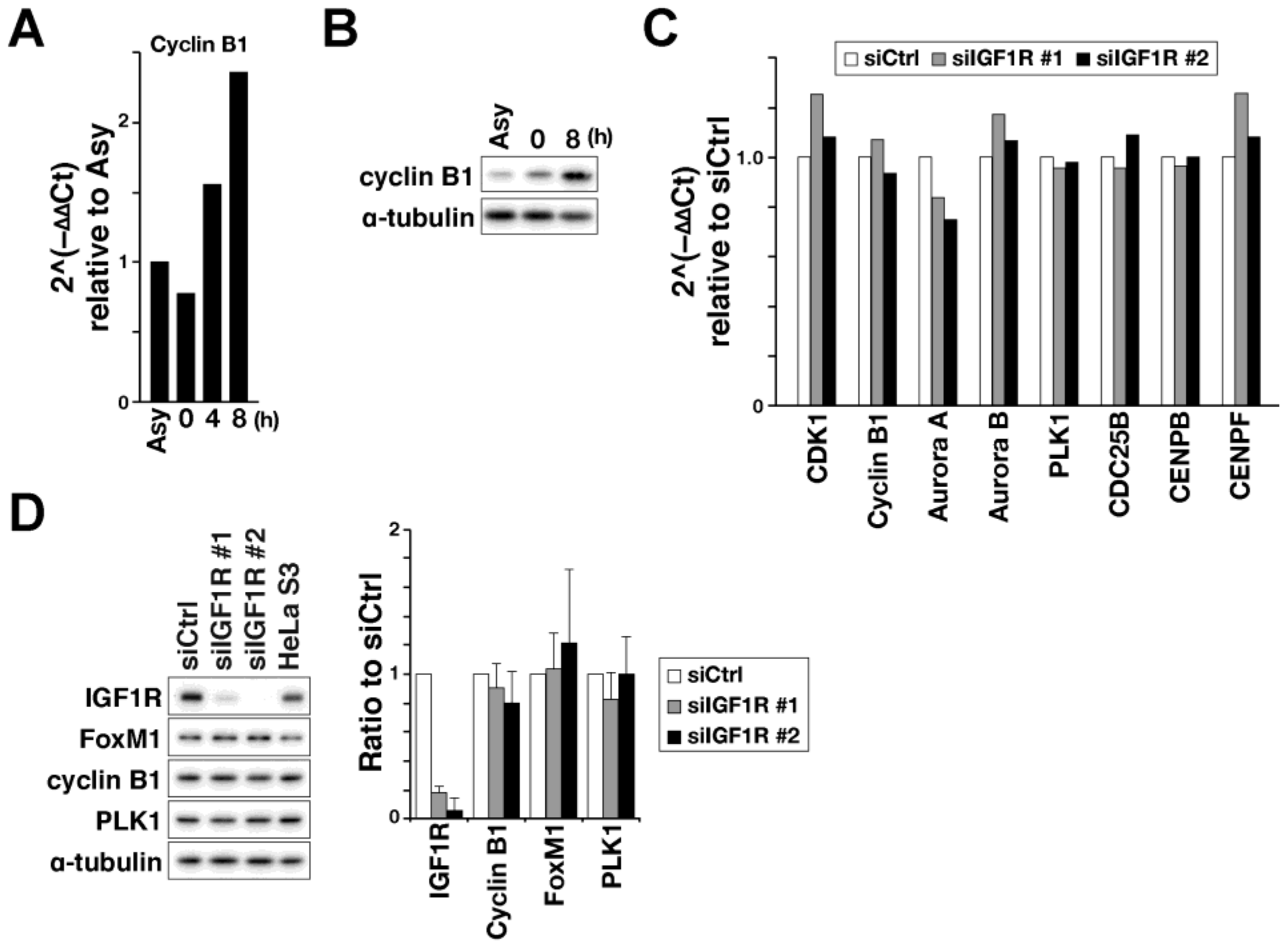
2.2. Effect on FoxM1-Mediated Transcription of M-Phase Regulators
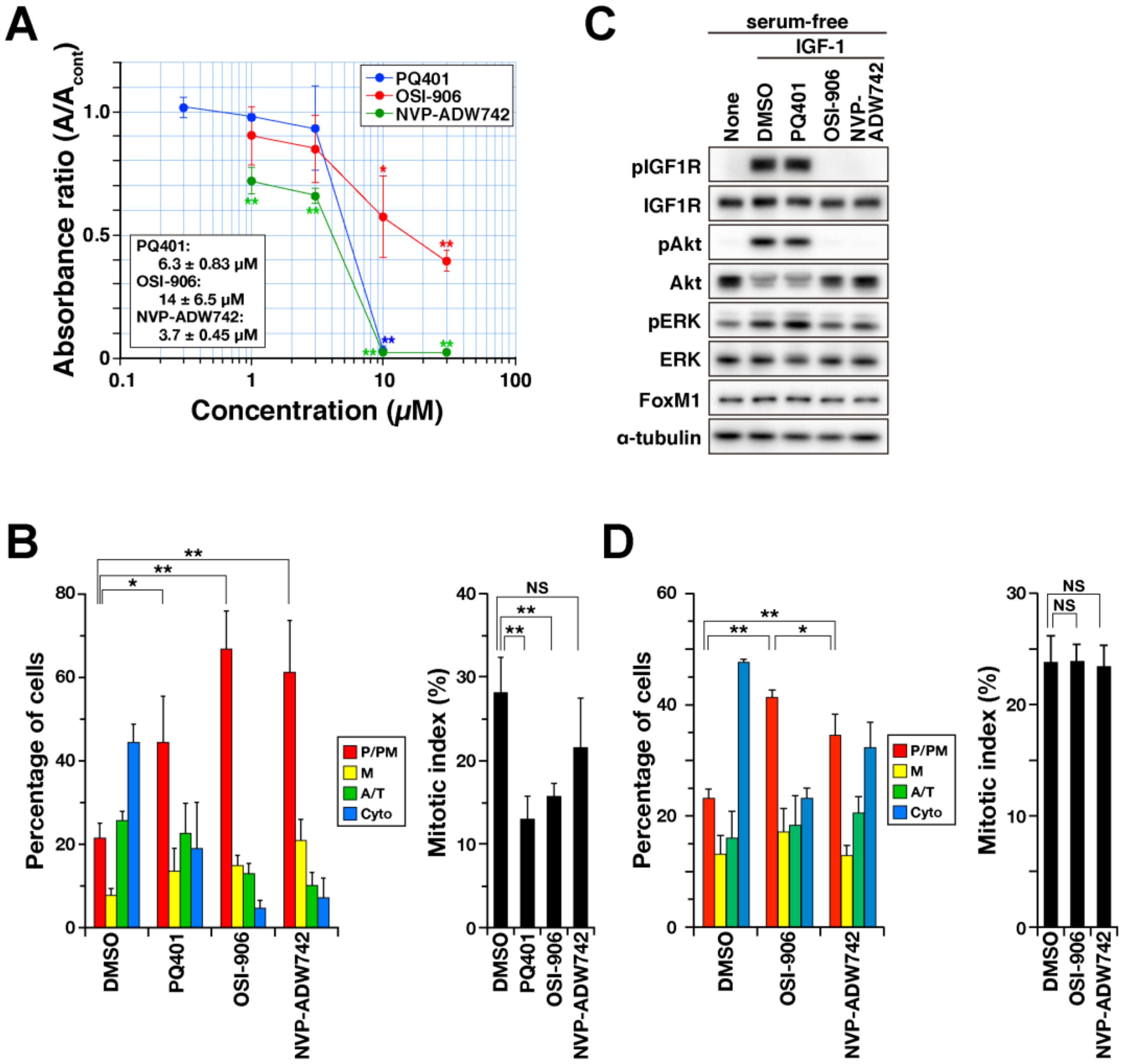
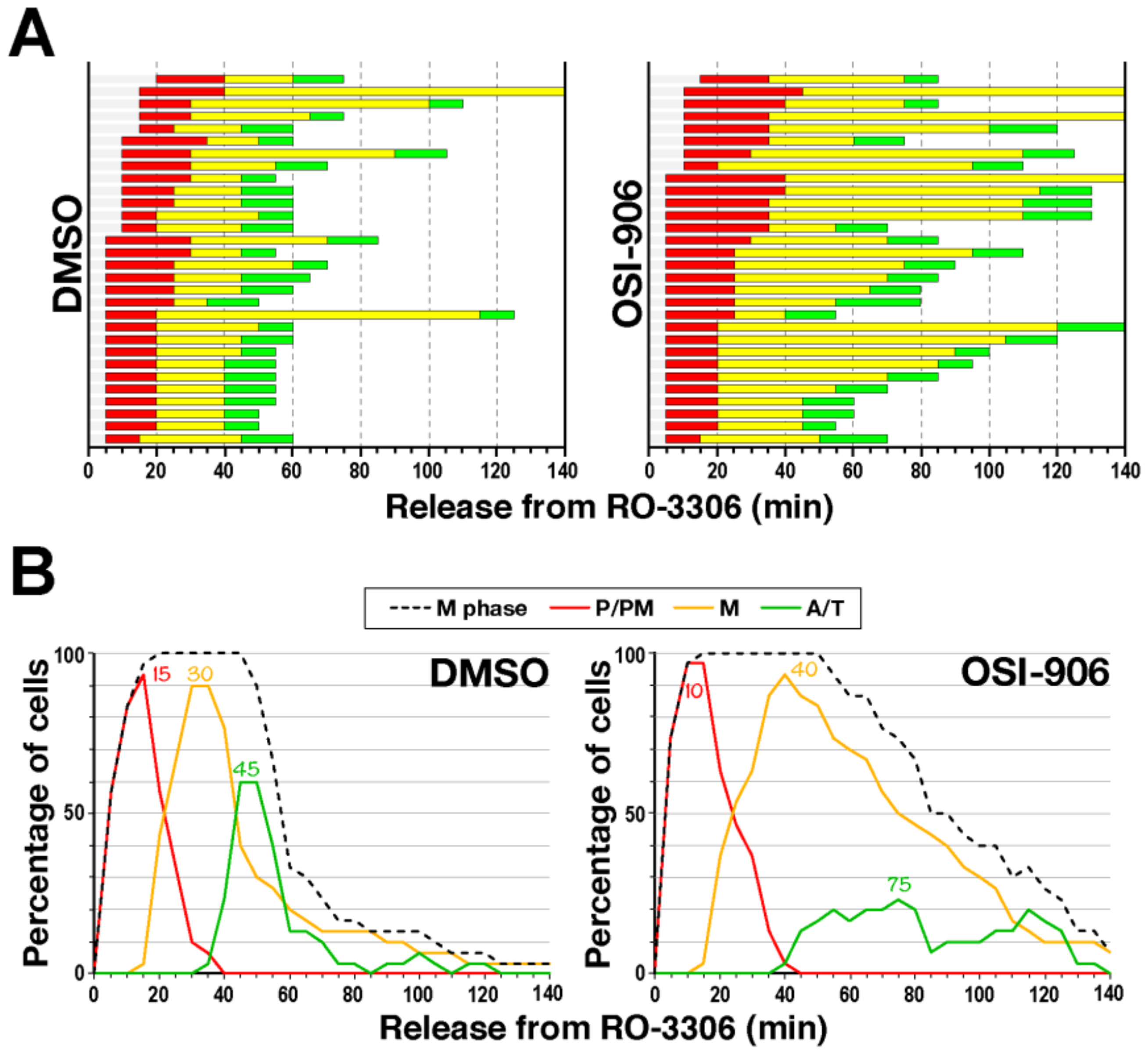
2.3. Delay in M-Phase Progression by IGF1R Inhibitors
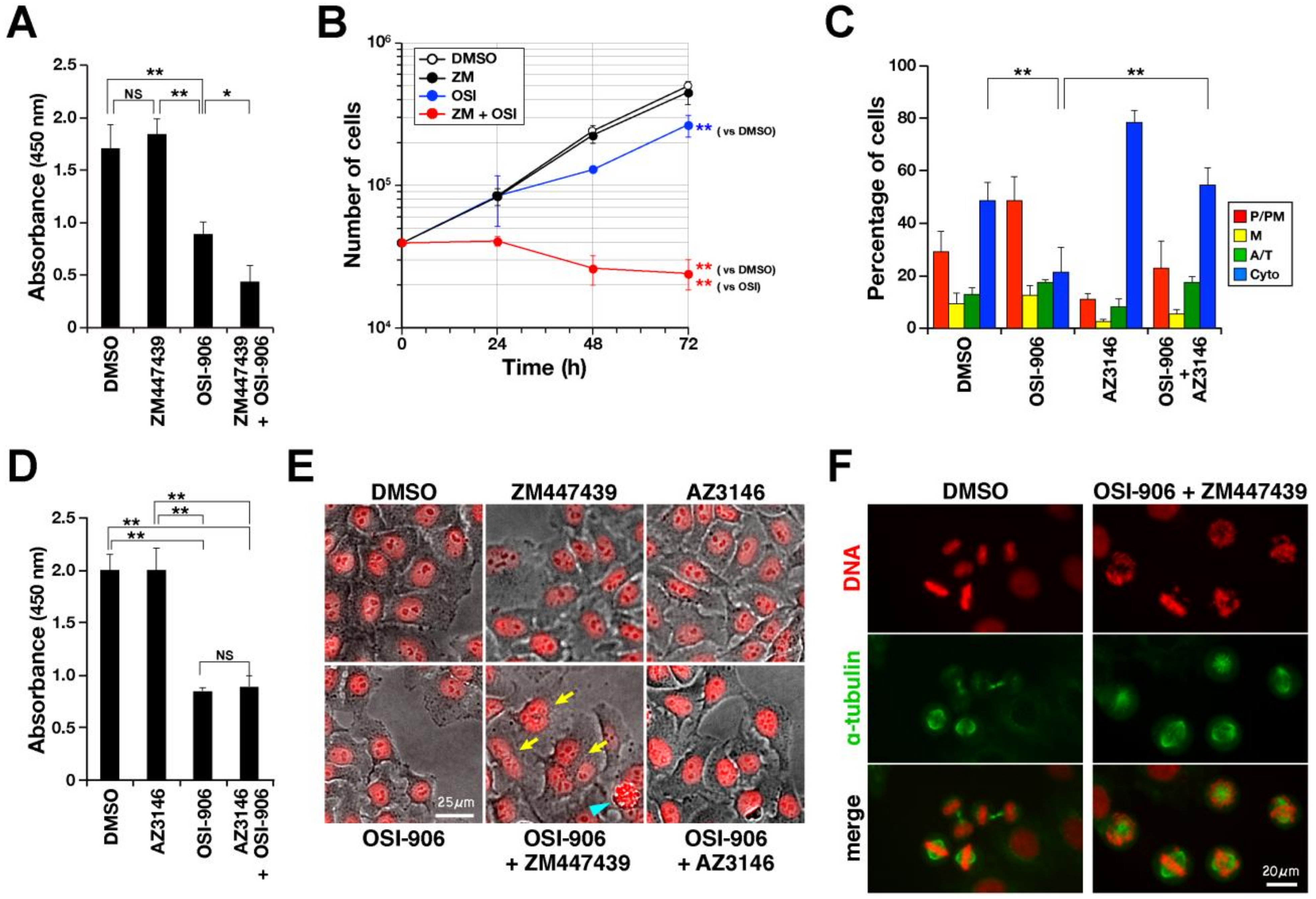
2.4. Aurora B Inhibitor Potentiates IGF1R Inhibitor-Induced Suppression of Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Chemicals
4.3. siRNA
4.4. Antibodies
4.5. Immunofluorescence Microscopy
4.6. Western Blotting
4.7. Cell-Cycle Synchronization
4.8. Time-Lapse Imaging
4.9. Proliferation Assay
4.10. Real-Time PCR
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A/T | anaphase/telophase |
| CDK1 | cyclin-dependent kinase 1 |
| CIN | chromosomal instability |
| Cyto | cytokinesis |
| DMSO | dimethyl sulfoxide |
| EGFR | epidermal growth factor receptor |
| FBS | fetal bovine serum |
| FoxM1 | Forkhead box M1 |
| IF | immunofluorescence |
| IGF1 | insulin-like growth factor 1 |
| IGF1R | IGF1 receptor |
| IRS1 | insulin receptor substrate 1 |
| HRP | horse radish peroxidase |
| M | metaphase |
| PBS | phosphate-buffered saline |
| PI3K | phosphatidylinositol 3-kinase |
| P/PM | prophase/prometaphase |
| SAC | spindle assembly checkpoint |
| WB | western blotting |
| WST | water-soluble tetrazolium |
References
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Spitz, M.R.; Mistry, J.; Gu, J.; Hong, W.K.; Wu, X. Plasma levels of insulin-like growth factor-I and lung cancer risk: A case-control analysis. J. Natl. Cancer Inst. 1999, 91, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.C.; Leeding, K.S.; Bach, L.A.; Baldwin, G.S.; Macrae, F.A.; Shulkes, A. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J. Natl. Cancer Inst. 2000, 92, 1947–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef]
- Hakam, A.; Yeatman, T.J.; Lu, L.; Mora, L.; Marcet, G.; Nicosia, S.V.; Karl, R.C.; Coppola, D. Expression of insulin-like growth factor-1 receptor in human colorectal cancer. Hum. Pathol. 1999, 30, 1128–1133. [Google Scholar] [CrossRef]
- Xie, Y.; Skytting, B.; Nilsson, G.; Brodin, B.; Larsson, O. Expression of insulin-like growth factor-1 receptor in synovial sarcoma: Association with an aggressive phenotype. Cancer Res. 1999, 59, 3588–3591. [Google Scholar]
- All-Ericsson, C.; Girnita, L.; Seregard, S.; Bartolazzi, A.; Jager, M.J.; Larsson, O. Insulin-like growth factor-1 receptor in uveal melanoma: A predictor for metastatic disease and a potential therapeutic target. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1–8. [Google Scholar]
- Jiang, Y.; Wang, L.; Gong, W.; Wei, D.; Le, X.; Yao, J.; Ajani, J.; Abbruzzese, J.L.; Huang, S.; Xie, K. A high expression level of insulin-like growth factor I receptor is associated with increased expression of transcription factor Sp1 and regional lymph node metastasis of human gastric cancer. Clin. Exp. Metastasis 2004, 21, 755–764. [Google Scholar] [CrossRef]
- Kornprat, P.; Rehak, P.; Rüschoff, J.; Langner, C. Expression of IGF-I, IGF-II, and IGF-IR in gallbladder carcinoma. A systematic analysis including primary and corresponding metastatic tumours. J. Clin. Pathol. 2006, 59, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Tennant, M.K.; Thrasher, J.B.; Twomey, P.A.; Drivdahl, R.H.; Birnbaum, R.S.; Plymate, S.R. Protein and messenger ribonucleic acid (mRNA) for the type 1 insulin-like growth factor (IGF) receptor is decreased and IGF-II mRNA is increased in human prostate carcinoma compared to benign prostate epithelium. J. Clin. Endocrinol. Metab. 1996, 81, 3774–3782. [Google Scholar] [PubMed] [Green Version]
- Figueroa, J.A.; De Raad, S.; Speights, V.O.; Rinehart, J.J. Gene expression of insulin-like growth factors and receptors in neoplastic prostate tissues: Correlation with clinico-pathological parameters. Cancer Invest. 2001, 19, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, C.; Hasegawa, T.; Tani, Y.; Takahashi, F.; Takeuchi, M.; Watanabe, T.; Ando, M.; Katsumata, N.; Fujiwara, Y. Expression of insulin-like growth factor 1 receptor in primary breast cancer: Immunohistochemical analysis. Hum. Pathol. 2004, 35, 1537–1542. [Google Scholar] [CrossRef]
- Shiono, S.; Ishii, G.; Nagai, K.; Murata, Y.; Tsuta, K.; Nitadori, J.; Kodama, T.; Ochiai, A. Immunohistochemical prognostic factors in resected colorectal lung metastases using tissue microarray analysis. Eur. J. Surg. Oncol. 2006, 32, 308–309. [Google Scholar] [CrossRef]
- Gadde, S.; Heald, R. Mechanisms and molecules of the mitotic spindle. Curr. Biol. 2004, 14, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Kunda, P.; Baum, B. The actin cytoskeleton in spindle assembly and positioning. Trends Cell Biol. 2009, 19, 174–179. [Google Scholar] [CrossRef]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Shirakawa, J.; Fernandez, M.; Takatani, T.; El Ouaamari, A.; Jungtrakoon, P.; Okawa, E.R.; Zhang, W.; Yi, P.; Doria, A.; Kulkarni, R.N. Insulin signaling regulates the FoxM1/PLK1/CENP-A pathway to promote adaptive pancreatic β cell proliferation. Cell Metab. 2017, 25, 868–882. [Google Scholar] [CrossRef] [Green Version]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Adesanya, O.O.; Zhou, J.; Samathanam, C.; Powell-Braxton, L.; Bondy, C.A. Insulin-like growth factor 1 is required for G2 progression in the estradiol-induced mitotic cycle. Proc. Natl. Acad. Sci. USA 1999, 96, 3287–3291. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.Y.M.; Tong, T.H.K.; Cheung, A.M.S.; Tsang, A.C.C.; Leung, W.Y.; Yao, K.-M. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J. Cell Sci. 2005, 118, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The spindle assembly checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkley, B.R.; Cartwright, J. Cold-Labile and Cold-Stable Microtubules in the Mitotic Spindle of Mammalian Cells. Ann. N. Y. Acad. Sci. 1975, 253, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Coppola, D.; Ferber, A.; Miura, M.; Sell, C.; D’Ambrosio, C.; Rubin, R.; Baserga, R. A functional insulin-like growth factor I receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 4588–4595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sell, C.; Dumenil, G.; Deveaud, C.; Miura, M.; Coppola, D.; DeAngelis, T.; Rubin, R.; Efstratiadis, A.; Baserga, R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell Biol. 1994, 14, 3604–3612. [Google Scholar] [CrossRef] [Green Version]
- Morrison, K.B.; Tognon, C.E.; Garnett, M.J.; Deal, C.; Sorensen, P.H.B. ETV6-NTRK3 transformation requires insulin-like growth factor 1 receptor signaling and is associated with constitutive IRS-1 tyrosine phosphorylation. Oncogene 2002, 21, 5684–5695. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, C.; Johnson, V.L.; Tighe, A.; Ellston, R.; Haworth, C.; Johnson, T.; Mortlock, A.; Keen, N.; Taylor, S.S. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 2003, 161, 267–280. [Google Scholar] [CrossRef]
- Hewitt, L.; Tighe, A.; Santaguida, S.; White, A.M.; Jones, C.D.; Musacchio, A.; Green, S.; Taylor, S.S. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 2010, 190, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [Green Version]
- Laoukili, J.; Alvarez-Fernandez, M.; Stahl, M.; Medema, R.H. FoxM1 is degraded at mitotic exit in a Cdh1-dependent manner. Cell Cycle 2008, 7, 2720–2726. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Lam, E.W.F. Role of the forkhead transcription factor FOXO-FOXM1 axis in cancer and drug resistance. Front. Med. China 2012, 6, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Yung, M.M.H.; Chan, D.W.; Liu, V.W.S.; Yao, K.M.; Ngan, H.Y.S. Activation of AMPK inhibits cervical cancer cell growth through AKT/FOXO3a/FOXM1 signaling cascade. BMC Cancer 2013, 13, 327. [Google Scholar] [CrossRef] [Green Version]
- Zha, J.; Lackner, M.R. Targeting the insulin-like growth factor receptor-1R pathway for cancer therapy. Clin. Cancer Res. 2010, 16, 2512–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.K.; Gaikwad, S.M.; Jinager, A.; Chaudhury, S.; Maheshwari, A.; Ray, P. IGF-1R inhibition potentiates cytotoxic effects of chemotherapeutic agents in early stages of chemoresistant ovarian cancer cells. Cancer Lett. 2014, 354, 254–262. [Google Scholar] [CrossRef]
- Ochnik, A.M.; Baxter, R.C. Combination therapy approaches to target insulin-like growth factor receptor signaling in breast cancer. Endocr. Relat. Cancer 2016, 23, R527–R550. [Google Scholar] [CrossRef]
- Saurin, A.T.; van der Waal, M.S.; Medema, R.H.; Lens, S.M.A.; Kops, G.J.P.L. Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat. Commun. 2011, 2, 316. [Google Scholar] [CrossRef]
- Maldonado, K.; Kapoor, T.M. Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat. Cell Biol. 2011, 13, 475–482. [Google Scholar] [CrossRef]
- Krenn, V.; Musacchio, A. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front. Oncol. 2015, 5, 225. [Google Scholar] [CrossRef] [Green Version]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009, 136, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, D.; Hagino, M.; Yamagishi, A.; Kaibori, Y.; Munira, S.; Saito, Y.; Nakayama, Y. Inhibitors of the VEGF Receptor Suppress HeLa S3 Cell Proliferation via Misalignment of Chromosomes and Rotation of the Mitotic Spindle, Causing a Delay in M-Phase Progression. Int. J. Mol. Sci. 2018, 19, 4014. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, M.; Kuga, T.; Saito, Y.; Nagano, M.; Adachi, J.; Tomonaga, T.; Yamaguchi, N.; Nakayama, Y. The tyrosine kinase v-Src causes mitotic slippage by phosphorylating an inhibitory tyrosine residue of Cdk1. J. Biol. Chem. 2018, 293, 15524–15537. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Saito, Y.; Soeda, S.; Iwamoto, E.; Ogawa, S.; Yamagishi, N.; Kuga, T.; Yamaguchi, N. Genistein induces cytokinesis failure through RhoA delocalization and anaphase chromosome bridging. J. Cell. Biochem. 2014, 115, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, E.; Ueta, N.; Matsui, Y.; Kamijo, K.; Kuga, T.; Saito, Y.; Yamaguchi, N.; Nakayama, Y. ERK plays a role in chromosome alignment and participates in M-phase progression. J. Cell. Biochem. 2016, 117, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Kakihana, A.; Oto, Y.; Saito, Y.; Nakayama, Y. Heat shock-induced mitotic arrest requires heat shock protein 105 for the activation of spindle assembly checkpoint. FASEB J. 2019, 33, 3936–3953. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamagishi, A.; Ikeda, Y.; Ikeuchi, M.; Yuki, R.; Saito, Y.; Nakayama, Y. Targeting Insulin-Like Growth Factor 1 Receptor Delays M-Phase Progression and Synergizes with Aurora B Inhibition to Suppress Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 1058. https://doi.org/10.3390/ijms21031058
Yamagishi A, Ikeda Y, Ikeuchi M, Yuki R, Saito Y, Nakayama Y. Targeting Insulin-Like Growth Factor 1 Receptor Delays M-Phase Progression and Synergizes with Aurora B Inhibition to Suppress Cell Proliferation. International Journal of Molecular Sciences. 2020; 21(3):1058. https://doi.org/10.3390/ijms21031058
Chicago/Turabian StyleYamagishi, Akane, Yuki Ikeda, Masayoshi Ikeuchi, Ryuzaburo Yuki, Youhei Saito, and Yuji Nakayama. 2020. "Targeting Insulin-Like Growth Factor 1 Receptor Delays M-Phase Progression and Synergizes with Aurora B Inhibition to Suppress Cell Proliferation" International Journal of Molecular Sciences 21, no. 3: 1058. https://doi.org/10.3390/ijms21031058