DNAM-1 Activating Receptor and Its Ligands: How Do Viruses Affect the NK Cell-Mediated Immune Surveillance during the Various Phases of Infection?

 ,
,  , and
, and 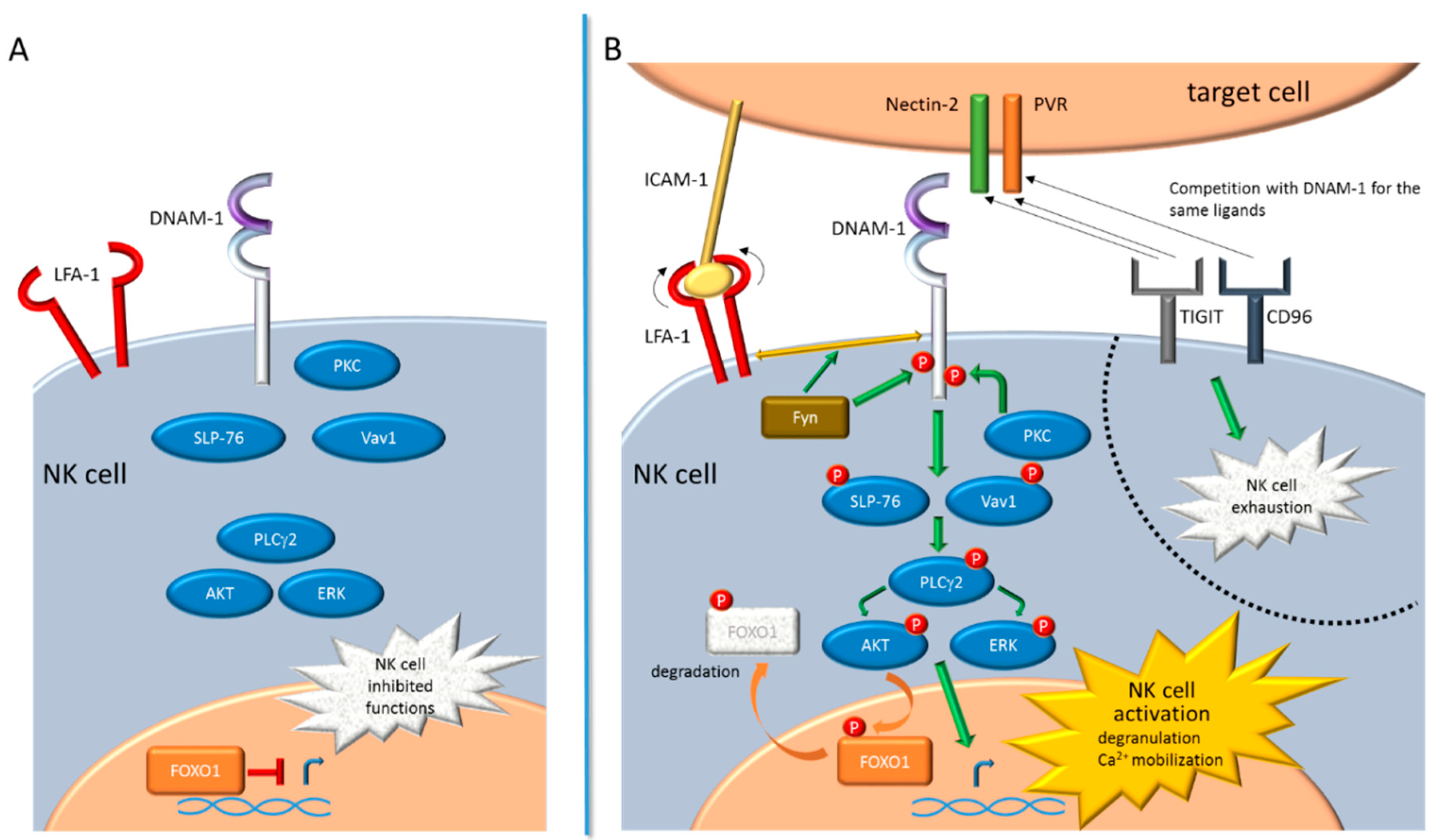{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DNAM-1 Signaling Pathway
3. Regulation and Function of DNAM-1 Ligands
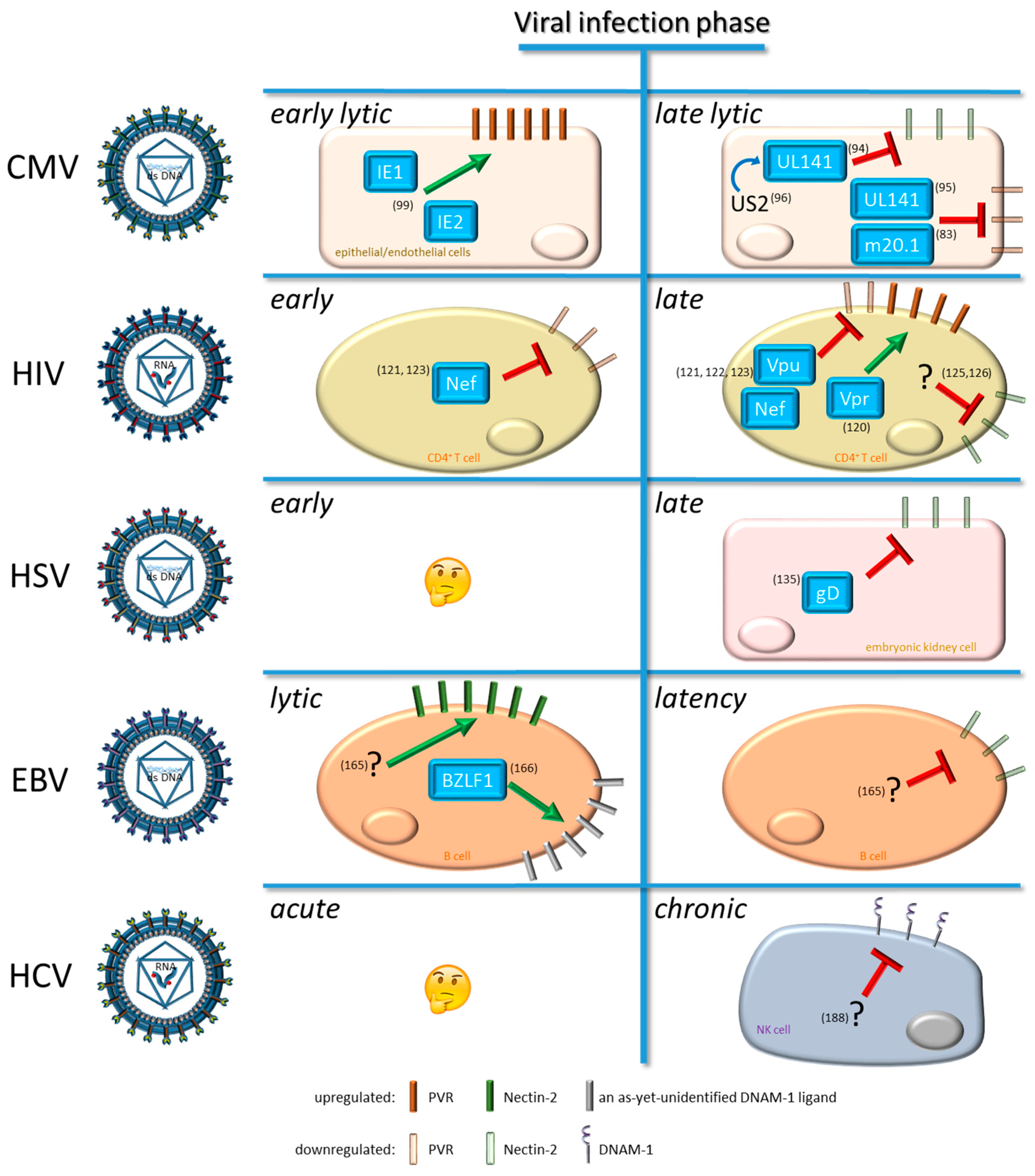
4. Viral Modulation of DNAM-1 and Its Ligands
4.1. Modulation by Cytomegalovirus (CMV)
4.2. Modulation by Human Immunodeficiency Virus Type 1 (HIV-1)
4.3. Modulation by Human Herpes Simplex Virus (HSV)
4.4. Modulation by Epstein-Barr Virus (EBV)
4.5. Modulation by Hepatitis C Virus (HCV)
5. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.L.; Morris, C.R.; Solheim, J.C. Virus evasion of mhc class i molecule presentation. J. Immunol. 2003, 171, 4473–4478. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural killer cells control tumor growth by sensing a growth factor. Cell 2018, 172, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. Antitumor immunity. A shed nkg2d ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble mic ligands impair expression of nkg2d and t-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Steinle, A.; Watzl, C.; Mandelboim, O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013, 34, 182–191. [Google Scholar] [CrossRef]
- Groth, A.; Kloss, S.; von Strandmann, E.P.; Koehl, U.; Koch, J. Mechanisms of tumor and viral immune escape from natural killer cell-mediated surveillance. J. Innate Immun. 2011, 3, 344–354. [Google Scholar] [CrossRef]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet. Glob Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Jonjic, S.; Babic, M.; Polic, B.; Krmpotic, A. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 2008, 20, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Li, X.; Kuang, E. Viral evasion of natural killer cell activation. Viruses 2016, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Roles of the nkg2d immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of pvr (cd155) and nectin-2 (cd112) as cell surface ligands for the human dnam-1 (cd226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Vilches, C.; Parham, P. Kir: Diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev. Immunol. 2002, 20, 217–251. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (nk) cell-mediated cytotoxicity: Differential use of trail and fas ligand by immature and mature primary human nk cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, H.S. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front. Immunol. 2018, 9, 2041. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Colonna, M. The role of nk cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin Cancer Biol. 2006, 16, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Presti, R.; Vermi, W.; Lavender, K.; Turnbull, E.; Ochsenbauer-Jambor, C.; Kappes, J.C.; Ferrari, G.; Kessels, L.; Williams, I.; et al. Loss of dnam-1 contributes to cd8+ t-cell exhaustion in chronic hiv-1 infection. Eur. J. Immunol. 2010, 40, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.J.; Teijaro, J.R.; Lewicki, H.A.; Colonna, M.; Oldstone, M.B. Cd8 t cell defect of tnf-alpha and il-2 in dnam-1 deficient mice delays clearance in vivo of a persistent virus infection. Virology 2012, 429, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Nabekura, T.; Kanaya, M.; Shibuya, A.; Fu, G.; Gascoigne, N.R.; Lanier, L.L. Costimulatory molecule dnam-1 is essential for optimal differentiation of memory natural killer cells during mouse cytomegalovirus infection. Immunity 2014, 40, 225–234. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, L.F.; Smyth, M.J.; Martinet, L. Dnam-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol. 2014, 92, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Bozzano, F.; Marras, F.; Ascierto, M.L.; Cantoni, C.; Cenderello, G.; Dentone, C.; Di Biagio, A.; Orofino, G.; Mantia, E.; Boni, S.; et al. ‘Emergency exit’ of bone-marrow-resident cd34(+)dnam-1(bright)cxcr4(+)-committed lymphoid precursors during chronic infection and inflammation. Nat. Commun. 2015, 6, 8109. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Georgoudaki, A.M.; Chambers, B.J.; Qiu, Q.; Kremmer, E.; Maier, M.K.; Czeloth, N.; Ravens, I.; Foerster, R.; Bernhardt, G. Heterogeneous expression of the adhesion receptor cd226 on murine nk and t cells and its function in nk-mediated killing of immature dendritic cells. J. Leukoc. Biol. 2009, 86, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Ferrari De Andrade, L.; Guillerey, C.; Lee, J.S.; Liu, J.; Souza-Fonseca-Guimaraes, F.; Hutchinson, D.S.; Kolesnik, T.B.; Nicholson, S.E.; Huntington, N.D.; et al. Dnam-1 expression marks an alternative program of nk cell maturation. Cell Rep. 2015, 11, 85–97. [Google Scholar] [CrossRef]
- Tahara-Hanaoka, S.; Shibuya, K.; Onoda, Y.; Zhang, H.; Yamazaki, S.; Miyamoto, A.; Honda, S.; Lanier, L.L.; Shibuya, A. Functional characterization of dnam-1 (cd226) interaction with its ligands pvr (cd155) and nectin-2 (prr-2/cd112). Int. Immunol. 2004, 16, 533–538. [Google Scholar] [CrossRef]
- Xiong, P.; Sang, H.W.; Zhu, M. Critical roles of co-activation receptor dnax accessory molecule-1 in natural killer cell immunity. Immunology 2015, 146, 369–378. [Google Scholar] [CrossRef]
- Casado, J.G.; Pawelec, G.; Morgado, S.; Sanchez-Correa, B.; Delgado, E.; Gayoso, I.; Duran, E.; Solana, R.; Tarazona, R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol. Immunother. 2009, 58, 1517–1526. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Corrias, M.V.; Lanino, E.; Pende, D.; Moretta, L.; Bottino, C.; Moretta, A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: Critical role of dnax accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004, 64, 9180–9184. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zheng, Q.; Xin, N.; Wang, W.; Zhao, C. Cd155, an onco-immunologic molecule in human tumors. Cancer Sci. 2017, 108, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Tahara-Hanaoka, S.; Shibuya, K.; Kai, H.; Miyamoto, A.; Morikawa, Y.; Ohkochi, N.; Honda, S.; Shibuya, A. Tumor rejection by the poliovirus receptor family ligands of the dnam-1 (cd226) receptor. Blood 2006, 107, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Sato, S.; Kato, J.; Ito, Y.; Watanabe, T.; Tsuji, I.; Hori, A.; Kurokawa, T.; Kokubo, T. Nectin-2 is a potential target for antibody therapy of breast and ovarian cancers. Mol. Cancer 2013, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Mateo, M.; Generous, A.; Sinn, P.L.; Cattaneo, R. Connections matter--how viruses use cell-cell adhesion components. J. Cell Sci. 2015, 128, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. Ncrs and dnam-1 mediate nk cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin Invest. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Shin, B.R.; Lee, H.K.; Lee, J.H.; Kim, K.H.; Choi, J.E.; Ji, A.Y.; Hong, J.T.; Kim, Y.; Han, S.B. Cd226(-/-) natural killer cells fail to establish stable contacts with cancer cells and show impaired control of tumor metastasis in vivo. Oncoimmunology 2017, 6, e1338994. [Google Scholar] [CrossRef]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. Dnam-1/cd155 interactions promote cytokine and nk cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, N.; Lu, Y.; Davidson, D.; Colonna, M.; Veillette, A. Dnam-1 controls nk cell activation via an itt-like motif. J. Exp. Med. 2015, 212, 2165–2182. [Google Scholar] [CrossRef]
- Gilfillan, S.; Chan, C.J.; Cella, M.; Haynes, N.M.; Rapaport, A.S.; Boles, K.S.; Andrews, D.M.; Smyth, M.J.; Colonna, M. Dnam-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J. Exp. Med. 2008, 205, 2965–2973. [Google Scholar] [CrossRef]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and nectin-like molecules: Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. Dnam-1, a novel adhesion molecule involved in the cytolytic function of t lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef]
- Shibuya, K.; Lanier, L.L.; Phillips, J.H.; Ochs, H.D.; Shimizu, K.; Nakayama, E.; Nakauchi, H.; Shibuya, A. Physical and functional association of lfa-1 with dnam-1 adhesion molecule. Immunity 1999, 11, 615–623. [Google Scholar] [CrossRef]
- Enqvist, M.; Ask, E.H.; Forslund, E.; Carlsten, M.; Abrahamsen, G.; Beziat, V.; Andersson, S.; Schaffer, M.; Spurkland, A.; Bryceson, Y.; et al. Coordinated expression of dnam-1 and lfa-1 in educated nk cells. J. Immunol. 2015, 194, 4518–4527. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; de Almeida, P.; Manieri, N.; de Almeida Nagata, D.; Wu, T.D.; Harden Bowles, K.; Arumugam, V.; Schartner, J.; Cubas, R.; Mittman, S.; et al. Cd226 regulates natural killer cell antitumor responses via phosphorylation-mediated inactivation of transcription factor foxo1. Proc. Natl. Acad. Sci. USA 2018, 115, E11731–E11740. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Kerdiles, Y.; Chu, J.; Yuan, S.; Wang, Y.; Chen, X.; Mao, H.; Zhang, L.; Zhang, J.; Hughes, T.; et al. Transcription factor foxo1 is a negative regulator of natural killer cell maturation and function. Immunity 2015, 42, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of age: Cd96 emerges as modulator of immune responses. Front Immunol. 2018, 9, 1072. [Google Scholar] [CrossRef]
- Manieri, N.A.; Chiang, E.Y.; Grogan, J.L. Tigit: A key inhibitor of the cancer immunity cycle. Trends Immunol. 2017, 38, 20–28. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor tigit prevents nk cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors cd96 and cd226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef]
- Holmes, V.M.; Maluquer de Motes, C.; Richards, P.T.; Roldan, J.; Bhargava, A.K.; Orange, J.S.; Krummenacher, C. Interaction between nectin-1 and the human natural killer cell receptor cd96. PLoS ONE 2019, 14, e0212443. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Cella, M.; Giurisato, E.; Shaw, A.S.; Colonna, M. Cutting edge: Cd96 (tactile) promotes nk cell-target cell adhesion by interacting with the poliovirus receptor (cd155). J. Immunol. 2004, 172, 3994–3998. [Google Scholar] [CrossRef] [PubMed]
- Deuss, F.A.; Gully, B.S.; Rossjohn, J.; Berry, R. Recognition of nectin-2 by the natural killer cell receptor t cell immunoglobulin and itim domain (tigit). J. Biol. Chem. 2017, 292, 11413–11422. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of tigit with pvr and pvrl2 inhibits human nk cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Yin, X.; Liu, T.; Wang, Z.; Ma, M.; Lei, J.; Zhang, Z.; Fu, S.; Fu, Y.; Hu, Q.; Ding, H.; et al. Expression of the inhibitory receptor tigit is up-regulated specifically on nk cells with cd226 activating receptor from hiv-infected individuals. Front Immunol. 2018, 9, 2341. [Google Scholar] [CrossRef]
- Takai, Y.; Nakanishi, H. Nectin and afadin: Novel organizers of intercellular junctions. J. Cell Sci. 2003, 116, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Vendome, J.; Brasch, J.; Jin, X.; Hong, S.; Katsamba, P.S.; Ahlsen, G.; Troyanovsky, R.B.; Troyanovsky, S.M.; Honig, B.; et al. Nectin ectodomain structures reveal a canonical adhesive interface. Nat. Struct. Mol. Biol. 2012, 19, 906–915. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K.; Takai, Y. Nectin spot: A novel type of nectin-mediated cell adhesion apparatus. Biochem. J. 2016, 473, 2691–2715. [Google Scholar] [CrossRef]
- Mandai, K.; Rikitake, Y.; Mori, M.; Takai, Y. Nectins and nectin-like molecules in development and disease. Curr. Top Dev. Biol. 2015, 112, 197–231. [Google Scholar]
- Takai, Y.; Irie, K.; Shimizu, K.; Sakisaka, T.; Ikeda, W. Nectins and nectin-like molecules: Roles in cell adhesion, migration, and polarization. Cancer Sci. 2003, 94, 655–667. [Google Scholar] [CrossRef]
- Lopez, M.; Aoubala, M.; Jordier, F.; Isnardon, D.; Gomez, S.; Dubreuil, P. The human poliovirus receptor related 2 protein is a new hematopoietic/endothelial homophilic adhesion molecule. Blood 1998, 92, 4602–4611. [Google Scholar] [PubMed]
- Bouchard, M.J.; Dong, Y.; McDermott, B.M., Jr.; Lam, D.H.; Brown, K.R.; Shelanski, M.; Bellve, A.R.; Racaniello, V.R. Defects in nuclear and cytoskeletal morphology and mitochondrial localization in spermatozoa of mice lacking nectin-2, a component of cell-cell adherens junctions. Mol. Cell Biol. 2000, 20, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Nakanishi, H.; Kimura, K.; Matsubara, K.; Ozaki-Kuroda, K.; Katata, T.; Honda, T.; Kiyohara, Y.; Heo, K.; Higashi, M.; et al. Nectin: An adhesion molecule involved in formation of synapses. J. Cell Biol. 2002, 156, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.S.; Geraghty, R.J.; Martinez, W.M.; Montgomery, R.I.; Whitbeck, J.C.; Xu, R.; Eisenberg, R.J.; Cohen, G.H.; Spear, P.G. A cell surface protein with herpesvirus entry activity (hveb) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 1998, 246, 179–189. [Google Scholar] [CrossRef]
- Martinez, W.M.; Spear, P.G. Structural features of nectin-2 (hveb) required for herpes simplex virus entry. J. Virol. 2001, 75, 11185–11195. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Lee, B.; Choi, Y.J.; Jeon, S.A.; Kim, J.H.; Lee, H.K.; Kwon, S.M.; Cho, J.Y. Nectin-2 (cd112) is expressed on outgrowth endothelial cells and regulates cell proliferation and angiogenic function. PLoS ONE 2016, 11, e0163301. [Google Scholar] [CrossRef]
- Satomi-Kobayashi, S.; Ueyama, T.; Mueller, S.; Toh, R.; Masano, T.; Sakoda, T.; Rikitake, Y.; Miyoshi, J.; Matsubara, H.; Oh, H.; et al. Deficiency of nectin-2 leads to cardiac fibrosis and dysfunction under chronic pressure overload. Hypertension 2009, 54, 825–831. [Google Scholar] [CrossRef]
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaide, J.; Dubreuil, P.; Lopez, M. Nectin4/prr4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/prr1 through v domain interaction. J. Biol. Chem. 2001, 276, 43205–43215. [Google Scholar] [CrossRef]
- Reymond, N.; Imbert, A.M.; Devilard, E.; Fabre, S.; Chabannon, C.; Xerri, L.; Farnarier, C.; Cantoni, C.; Bottino, C.; Moretta, A.; et al. Dnam-1 and pvr regulate monocyte migration through endothelial junctions. J. Exp. Med. 2004, 199, 1331–1341. [Google Scholar] [CrossRef]
- Sloan, K.E.; Stewart, J.K.; Treloar, A.F.; Matthews, R.T.; Jay, D.G. Cd155/pvr enhances glioma cell dispersal by regulating adhesion signaling and focal adhesion dynamics. Cancer Res. 2005, 65, 10930–10937. [Google Scholar] [CrossRef]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. Atm-atr-dependent up-regulation of dnam-1 and nkg2d ligands on multiple myeloma cells by therapeutic agents results in enhanced nk-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [PubMed]
- Ardolino, M.; Zingoni, A.; Cerboni, C.; Cecere, F.; Soriani, A.; Iannitto, M.L.; Santoni, A. Dnam-1 ligand expression on ag-stimulated t lymphocytes is mediated by ros-dependent activation of DNA-damage response: Relevance for nk-t cell interaction. Blood 2011, 117, 4778–4786. [Google Scholar] [CrossRef]
- Fionda, C.; Soriani, A.; Zingoni, A.; Santoni, A.; Cippitelli, M. Nkg2d and dnam-1 ligands: Molecular targets for nk cell-mediated immunotherapeutic intervention in multiple myeloma. Biomed. Res. Int. 2015, 2015, 178698. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Irie, K.; Okamoto, R.; Ikeda, W.; Takai, Y. Transcriptional activation of the mouse necl-5/tage4/pvr/cd155 gene by fibroblast growth factor or oncogenic ras through the raf-mek-erk-ap-1 pathway. Oncogene 2005, 24, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Solecki, D.J.; Gromeier, M.; Mueller, S.; Bernhardt, G.; Wimmer, E. Expression of the human poliovirus receptor/cd155 gene is activated by sonic hedgehog. J. Biol. Chem. 2002, 277, 25697–25702. [Google Scholar] [CrossRef] [PubMed]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma cells control antimelanoma ctl responses via interaction between tigit and cd155 in the effector phase. J. Invest. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Al-Herz, W.; Essa, S. Spectrum of viral infections among primary immunodeficient children: Report from a national registry. Front Immunol. 2019, 10, 1231. [Google Scholar] [CrossRef] [PubMed]
- Gianella, S.; Letendre, S. Cytomegalovirus and hiv: A dangerous pas de deux. J. Infect. Dis. 2016, 214, S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Boeckh, M.; Murphy, W.J.; Peggs, K.S. Recent advances in cytomegalovirus: An update on pharmacologic and cellular therapies. Biol. Blood Marrow Transplant 2015, 21, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of nk cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory nk cell receptors. Science 2002, 296, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Angulo, A.; Vilches, C.; Gomez-Lozano, N.; Malats, N.; Lopez-Botet, M. Imprint of human cytomegalovirus infection on the nk cell receptor repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Lenac Rovis, T.; Kucan Brlic, P.; Kaynan, N.; Juranic Lisnic, V.; Brizic, I.; Jordan, S.; Tomic, A.; Kvestak, D.; Babic, M.; Tsukerman, P.; et al. Inflammatory monocytes and nk cells play a crucial role in dnam-1-dependent control of cytomegalovirus infection. J. Exp. Med. 2016, 213, 1835–1850. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Gruhler, A.; Galocha, B.; Jones, T.R.; Wiertz, E.J.; Ploegh, H.L.; Peterson, P.A.; Yang, Y.; Fruh, K. The er-luminal domain of the hcmv glycoprotein us6 inhibits peptide translocation by tap. Immunity 1997, 6, 613–621. [Google Scholar] [CrossRef]
- Furman, M.H.; Dey, N.; Tortorella, D.; Ploegh, H.L. The human cytomegalovirus us10 gene product delays trafficking of major histocompatibility complex class i molecules. J. Virol. 2002, 76, 11753–11756. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.R.; Wiertz, E.J.; Sun, L.; Fish, K.N.; Nelson, J.A.; Ploegh, H.L. Human cytomegalovirus us3 impairs transport and maturation of major histocompatibility complex class i heavy chains. Proc. Natl. Acad. Sci. USA 1996, 93, 11327–11333. [Google Scholar] [CrossRef] [PubMed]
- Trgovcich, J.; Cebulla, C.; Zimmerman, P.; Sedmak, D.D. Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class i cell surface expression. J. Virol. 2006, 80, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. Ulbps, novel mhc class i-related molecules, bind to cmv glycoprotein ul16 and stimulate nk cytotoxicity through the nkg2d receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Kubin, M.; Cassiano, L.; Chalupny, J.; Chin, W.; Cosman, D.; Fanslow, W.; Mullberg, J.; Rousseau, A.M.; Ulrich, D.; Armitage, R. Ulbp1, 2, 3: Novel mhc class i-related molecules that bind to human cytomegalovirus glycoprotein ul16, activate nk cells. Eur. J. Immunol. 2001, 31, 1428–1437. [Google Scholar] [CrossRef]
- Welte, S.A.; Sinzger, C.; Lutz, S.Z.; Singh-Jasuja, H.; Sampaio, K.L.; Eknigk, U.; Rammensee, H.G.; Steinle, A. Selective intracellular retention of virally induced nkg2d ligands by the human cytomegalovirus ul16 glycoprotein. Eur. J. Immunol. 2003, 33, 194–203. [Google Scholar] [CrossRef]
- Chalupny, N.J.; Rein-Weston, A.; Dosch, S.; Cosman, D. Down-regulation of the nkg2d ligand mica by the human cytomegalovirus glycoprotein ul142. Biochem. Biophys. Res. Commun. 2006, 346, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host immune system gene targeting by a viral mirna. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Ashiru, O.; Reeves, M.B.; Okecha, G.; Trowsdale, J.; Tomasec, P.; Wilkinson, G.W.; Sinclair, J.; Sissons, J.G. Human cytomegalovirus encodes an mhc class i-like molecule (ul142) that functions to inhibit nk cell lysis. J. Immunol. 2005, 175, 7457–7465. [Google Scholar] [CrossRef] [PubMed]
- Tomasec, P.; Wang, E.C.; Davison, A.J.; Vojtesek, B.; Armstrong, M.; Griffin, C.; McSharry, B.P.; Morris, R.J.; Llewellyn-Lacey, S.; Rickards, C.; et al. Downregulation of natural killer cell-activating ligand cd155 by human cytomegalovirus ul141. Nat. Immunol. 2005, 6, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Prod’homme, V.; Sugrue, D.M.; Stanton, R.J.; Nomoto, A.; Davies, J.; Rickards, C.R.; Cochrane, D.; Moore, M.; Wilkinson, G.W.; Tomasec, P. Human cytomegalovirus ul141 promotes efficient downregulation of the natural killer cell activating ligand cd112. J. Gen. Virol. 2010, 91, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; van den Boomen, D.J.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J.; et al. Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by hcmv us2 in cooperation with ul141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.P.; Frame, F.M.; Rogoff, H.A.; Pickering, M.T.; Yurochko, A.D.; Kowalik, T.F. Human cytomegalovirus ie1-72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J. Virol. 2005, 79, 11467–11475. [Google Scholar] [CrossRef] [PubMed]
- E, X.; Pickering, M.T.; Debatis, M.; Castillo, J.; Lagadinos, A.; Wang, S.; Lu, S.; Kowalik, T.F. An e2f1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011, 7, e1001342. [Google Scholar] [CrossRef]
- Pignoloni, B.; Fionda, C.; Dell’Oste, V.; Luganini, A.; Cippitelli, M.; Zingoni, A.; Landolfo, S.; Gribaudo, G.; Santoni, A.; Cerboni, C. Distinct roles for human cytomegalovirus immediate early proteins ie1 and ie2 in the transcriptional regulation of mica and pvr/cd155 expression. J. Immunol. 2016, 197, 4066–4078. [Google Scholar] [CrossRef]
- Fauci, A.S.; Desrosiers, R.C. Pathogenesis of hiv and siv. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune activation, inflammation, and non-aids co-morbidities in hiv-infected patients under long-term art. Viruses 2019, 11, 200. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of cd4+ t cells provides a mechanism for lifelong persistence of hiv-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.R.; et al. International aids society global scientific strategy: Towards an hiv cure 2016. Nat. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Siliciano, R.F. Targeting the latent reservoir for hiv-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]
- Persaud, D.; Luzuriaga, K. Absence of hiv-1 after treatment cessation in an infant. N. Engl. J. Med. 2014, 370, 678. [Google Scholar] [PubMed]
- Violari, A.; Cotton, M.F.; Kuhn, L.; Schramm, D.B.; Paximadis, M.; Loubser, S.; Shalekoff, S.; Da Costa Dias, B.; Otwombe, K.; Liberty, A.; et al. A child with perinatal hiv infection and long-term sustained virological control following antiretroviral treatment cessation. Nat. Commun. 2019, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Gay, H.; Ziemniak, C.; Sanborn, K.B.; Somasundaran, M.; Rainwater-Lovett, K.; Mellors, J.W.; Rosenbloom, D.; Persaud, D. Viremic relapse after hiv-1 remission in a perinatally infected child. N. Engl. J. Med. 2015, 372, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Rocca, S.; Zangari, P.; Cotugno, N.; De Rossi, A.; Ferns, B.; Petricone, D.; Rinaldi, S.; Giaquinto, C.; Bernardi, S.; Rojo, P.; et al. Human immunodeficiency virus (hiv)-antibody repertoire estimates reservoir size and time of antiretroviral therapy initiation in virally suppressed perinatally hiv-infected children. J. Pediatric. Infect. Dis. Soc. 2018. [Google Scholar] [CrossRef]
- Pantaleo, G.; Fauci, A.S. Immunopathogenesis of hiv infection. Annu Rev. Microbiol. 1996, 50, 825–854. [Google Scholar] [CrossRef]
- Fauci, A.S.; Mavilio, D.; Kottilil, S. Nk cells in hiv infection: Paradigm for protection or targets for ambush. Nat. Rev. Immunol. 2005, 5, 835–843. [Google Scholar] [CrossRef]
- Scully, E.; Alter, G. Nk cells in hiv disease. Curr. HIV/AIDS Rep. 2016, 13, 85–94. [Google Scholar] [CrossRef]
- Hu, P.F.; Hultin, L.E.; Hultin, P.; Hausner, M.A.; Hirji, K.; Jewett, A.; Bonavida, B.; Detels, R.; Giorgi, J.V. Natural killer cell immunodeficiency in hiv disease is manifest by profoundly decreased numbers of cd16+cd56+ cells and expansion of a population of cd16dimcd56- cells with low lytic activity. J. Acquir. Immune Defic Syndr. Hum Retrovirol. 1995, 10, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Mavilio, D.; Lombardo, G.; Benjamin, J.; Kim, D.; Follman, D.; Marcenaro, E.; O’Shea, M.A.; Kinter, A.; Kovacs, C.; Moretta, A.; et al. Characterization of cd56-/cd16+ natural killer (nk) cells: A highly dysfunctional nk subset expanded in hiv-infected viremic individuals. Proc. Natl. Acad. Sci. USA 2005, 102, 2886–2891. [Google Scholar] [CrossRef] [PubMed]
- Mavilio, D.; Benjamin, J.; Daucher, M.; Lombardo, G.; Kottilil, S.; Planta, M.A.; Marcenaro, E.; Bottino, C.; Moretta, L.; Moretta, A.; et al. Natural killer cells in hiv-1 infection: Dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc. Natl. Acad. Sci. USA 2003, 100, 15011–15016. [Google Scholar] [CrossRef] [PubMed]
- De Maria, A.; Fogli, M.; Mazza, S.; Basso, M.; Picciotto, A.; Costa, P.; Congia, S.; Mingari, M.C.; Moretta, L. Increased natural cytotoxicity receptor expression and relevant il-10 production in nk cells from chronically infected viremic hcv patients. Eur. J. Immunol. 2007, 37, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Nolting, A.; Dugast, A.S.; Rihn, S.; Luteijn, R.; Carrington, M.F.; Kane, K.; Jost, S.; Toth, I.; Nagami, E.; Faetkenheuer, G.; et al. Mhc class i chain-related protein a shedding in chronic hiv-1 infection is associated with profound nk cell dysfunction. Virology 2010, 406, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Mavilio, D.; Lombardo, G.; Kinter, A.; Fogli, M.; La Sala, A.; Ortolano, S.; Farschi, A.; Follmann, D.; Gregg, R.; Kovacs, C.; et al. Characterization of the defective interaction between a subset of natural killer cells and dendritic cells in hiv-1 infection. J. Exp. Med. 2006, 203, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhang, Z.; Jiang, Y.; Han, X.; Wang, Y.; Zhang, M.; Liu, J.; Geng, W.; Dai, D.; Shi, W.; et al. Expression of human cd226 on t cells and natural killer cells and of soluble cd226 in plasma of hiv-1-infected chinese patients. Viral Immunol. 2006, 19, 576–581. [Google Scholar] [CrossRef]
- Mahapatra, S.; Shearer, W.T.; Minard, C.G.; Mace, E.; Paul, M.; Orange, J.S. Nk cells in treated hiv-infected children display altered phenotype and function. J. Allergy Clin Immunol. 2019. [Google Scholar] [CrossRef]
- Vassena, L.; Giuliani, E.; Matusali, G.; Cohen, E.A.; Doria, M. The human immunodeficiency virus type 1 vpr protein upregulates pvr via activation of the atr-mediated DNA damage response pathway. J. Gen Virol. 2013, 94, 2664–2669. [Google Scholar] [CrossRef]
- Matusali, G.; Potesta, M.; Santoni, A.; Cerboni, C.; Doria, M. The human immunodeficiency virus type 1 nef and vpu proteins downregulate the natural killer cell-activating ligand pvr. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef]
- Bolduan, S.; Reif, T.; Schindler, M.; Schubert, U. Hiv-1 vpu mediated downregulation of cd155 requires alanine residues 10, 14 and 18 of the transmembrane domain. Virology 2014, 464–465, 375–384. [Google Scholar] [CrossRef]
- Galaski, J.; Ahmad, F.; Tibroni, N.; Pujol, F.M.; Muller, B.; Schmidt, R.E.; Fackler, O.T. Cell surface downregulation of nk cell ligands by patient-derived hiv-1 vpu and nef alleles. J. Acquir Immune Defic Syndr. 2016, 72, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.B.; Sowrirajan, B.; Cogswell, A.; Ward, J.P.; Planelles, V.; Barker, E. Cd155 on hiv-infected cells is not modulated by hiv-1 vpu and nef but synergizes with nkg2d ligands to trigger nk cell lysis of autologous primary hiv-infected cells. AIDS Res. Hum. Retroviruses 2017, 33, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Bisio, F.; Bozzano, F.; Marras, F.; Di Biagio, A.; Moretta, L.; De Maria, A. Successfully treated hiv-infected patients have differential expression of nk cell receptors (nkp46 and nkp30) according to aids status at presentation. Immunol. Lett. 2013, 152, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Tremblay-McLean, A.; Bruneau, J.; Lebouche, B.; Lisovsky, I.; Song, R.; Bernard, N.F. Expression profiles of ligands for activating natural killer cell receptors on hiv infected and uninfected cd4(+) t cells. Viruses 2017, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Bradley, H.; Markowitz, L.E.; Gibson, T.; McQuillan, G.M. Seroprevalence of herpes simplex virus types 1 and 2--united states, 1999-2010. J. Infect Dis. 2014, 209, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Reske, A.; Pollara, G.; Krummenacher, C.; Chain, B.M.; Katz, D.R. Understanding hsv-1 entry glycoproteins. Rev. Med. Virol. 2007, 17, 205–215. [Google Scholar] [CrossRef]
- Subramanian, R.P.; Geraghty, R.J. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins d, h, l, and b. Proc. Natl. Acad. Sci. USA 2007, 104, 2903–2908. [Google Scholar] [CrossRef]
- Simpson, S.A.; Manchak, M.D.; Hager, E.J.; Krummenacher, C.; Whitbeck, J.C.; Levin, M.J.; Freed, C.R.; Wilcox, C.L.; Cohen, G.H.; Eisenberg, R.J.; et al. Nectin-1/hvec mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J. Neurovirol. 2005, 11, 208–218. [Google Scholar] [CrossRef]
- Krummenacher, C.; Nicola, A.V.; Whitbeck, J.C.; Lou, H.; Hou, W.; Lambris, J.D.; Geraghty, R.J.; Spear, P.G.; Cohen, G.H.; Eisenberg, R.J. Herpes simplex virus glycoprotein d can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 1998, 72, 7064–7074. [Google Scholar] [PubMed]
- Delboy, M.G.; Patterson, J.L.; Hollander, A.M.; Nicola, A.V. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via ph-independent fusion with the plasma membrane of chinese hamster ovary cells. Virol. J. 2006, 3, 105. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Grauwet, K.; Cantoni, C.; Parodi, M.; De Maria, A.; Devriendt, B.; Pende, D.; Moretta, L.; Vitale, M.; Favoreel, H.W. Modulation of cd112 by the alphaherpesvirus gd protein suppresses dnam-1-dependent nk cell-mediated lysis of infected cells. Proc. Natl. Acad. Sci. USA 2014, 111, 16118–16123. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, H.; Cai, S.; Perng, G.C.; Nesburn, A.B.; Wechsler, S.L. The role of natural killer cells in protection of mice against death and corneal scarring following ocular hsv-1 infection. Antiviral Res. 2000, 45, 33–45. [Google Scholar] [CrossRef]
- Halford, W.P.; Maender, J.L.; Gebhardt, B.M. Re-evaluating the role of natural killer cells in innate resistance to herpes simplex virus type 1. Virol. J. 2005, 2, 56. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Thackray, A.M.; Bujdoso, R. Reduced herpes simplex virus type 1 latency in flt-3 ligand-treated mice is associated with enhanced numbers of natural killer and dendritic cells. Immunology 2001, 102, 352–358. [Google Scholar] [CrossRef]
- Ashkar, A.A.; Rosenthal, K.L. Interleukin-15 and natural killer and nkt cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J. Virol. 2003, 77, 10168–10171. [Google Scholar] [CrossRef]
- Nandakumar, S.; Woolard, S.N.; Yuan, D.; Rouse, B.T.; Kumaraguru, U. Natural killer cells as novel helpers in anti-herpes simplex virus immune response. J. Virol. 2008, 82, 10820–10831. [Google Scholar] [CrossRef]
- Kassim, S.H.; Rajasagi, N.K.; Ritz, B.W.; Pruett, S.B.; Gardner, E.M.; Chervenak, R.; Jennings, S.R. Dendritic cells are required for optimal activation of natural killer functions following primary infection with herpes simplex virus type 1. J. Virol. 2009, 83, 3175–3186. [Google Scholar] [CrossRef]
- Long, B.R.; Erickson, A.E.; Chapman, J.M.; Barbour, J.D.; Vu, B.A.; Ho, E.L.; Lanier, L.L.; Sauer, M.M.; Carvalho, K.I.; Nixon, D.F.; et al. Increased number and function of natural killer cells in human immunodeficiency virus 1-positive subjects co-infected with herpes simplex virus 2. Immunology 2010, 129, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Ando, T.; Lee, J.R.; Kim, G.; Kawakami, Y.; Nakasaki, T.; Nakasaki, M.; Matsumoto, K.; Choi, Y.S.; Kawakami, T. Defective natural killer cell activity in a mouse model of eczema herpeticum. J. Allergy Clin Immunol. 2017, 139, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.; Savic, S.; Cole, J.; Wood, P. Natural killer cell receptor expression in patients with severe and recurrent herpes simplex virus-1 (hsv-1) infections. Cell Immunol. 2007, 246, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Bjorkstrom, N.K.; Svensson, A.; Malmberg, K.J.; Eriksson, K.; Ljunggren, H.G. Characterization of natural killer cell phenotype and function during recurrent human hsv-2 infection. PLoS ONE 2011, 6, e27664. [Google Scholar] [CrossRef] [PubMed]
- Schepis, D.; D’Amato, M.; Studahl, M.; Bergstrom, T.; Karre, K.; Berg, L. Herpes simplex virus infection downmodulates nkg2d ligand expression. Scand J. Immunol. 2009, 69, 429–436. [Google Scholar] [CrossRef]
- Krummenacher, C.; Baribaud, I.; Eisenberg, R.J.; Cohen, G.H. Cellular localization of nectin-1 and glycoprotein d during herpes simplex virus infection. J. Virol. 2003, 77, 8985–8999. [Google Scholar] [CrossRef]
- Lu, G.; Zhang, N.; Qi, J.; Li, Y.; Chen, Z.; Zheng, C.; Gao, G.F.; Yan, J. Crystal structure of herpes simplex virus 2 gd bound to nectin-1 reveals a conserved mode of receptor recognition. J. Virol. 2014, 88, 13678–13688. [Google Scholar] [CrossRef]
- Bhargava, A.K.; Rothlauf, P.W.; Krummenacher, C. Herpes simplex virus glycoprotein d relocates nectin-1 from intercellular contacts. Virology 2016, 499, 267–277. [Google Scholar] [CrossRef]
- Zhang, N.; Yan, J.; Lu, G.; Guo, Z.; Fan, Z.; Wang, J.; Shi, Y.; Qi, J.; Gao, G.F. Binding of herpes simplex virus glycoprotein d to nectin-1 exploits host cell adhesion. Nat. Commun. 2011, 2, 577. [Google Scholar] [CrossRef]
- Stiles, K.M.; Milne, R.S.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein d. Virology 2008, 373, 98–111. [Google Scholar] [CrossRef]
- Stiles, K.M.; Krummenacher, C. Glycoprotein d actively induces rapid internalization of two nectin-1 isoforms during herpes simplex virus entry. Virology 2010, 399, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Hikita, S.I.; Takeda, K.; Ozaki, K.; Inoue, H.; Takakuwa, H.; Sonoda, K.H.; Ono, E. Evaluation of the antiviral potential of the soluble forms of glycoprotein d receptors on ocular herpes caused by hsv-1 and hsv-2 infections in a transgenic mouse model. J. Med. Virol. 2019, 91, 820–828. [Google Scholar] [CrossRef]
- Fu, X.; Tao, L.; Wang, P.Y.; Cripe, T.P.; Zhang, X. Comparison of infectivity and spread between hsv-1 and hsv-2 based oncolytic viruses on tumor cells with different receptor expression profiles. Oncotarget 2018, 9, 21348–21358. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Jaime-Ramirez, A.C.; Bolyard, C.; Dai, H.; Nallanagulagari, T.; Wojton, J.; Hurwitz, B.S.; Relation, T.; Lee, T.J.; Lotze, M.T.; et al. Bortezomib treatment sensitizes oncolytic hsv-1-treated tumors to nk cell immunotherapy. Clin Cancer Res. 2016, 22, 5265–5276. [Google Scholar] [CrossRef] [PubMed]
- Burnard, S.; Lechner-Scott, J.; Scott, R.J. Ebv and ms: Major cause, minor contribution or red-herring? Mult. Scler. Relat. Disord 2017, 16, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the burkitt’s lymphoma epstein-barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef]
- Young, L.S.; Dawson, C.W. Epstein-barr virus and nasopharyngeal carcinoma. Chin J. Cancer 2014, 33, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Winter, S. Inherited immunodeficiencies with high predisposition to epstein-barr virus-driven lymphoproliferative diseases. Front Immunol. 2018, 9, 1103. [Google Scholar] [CrossRef]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of epstein-barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef]
- Chijioke, O.; Landtwing, V.; Munz, C. Nk cell influence on the outcome of primary epstein-barr virus infection. Front Immunol. 2016, 7, 323. [Google Scholar] [CrossRef]
- Williams, H.; McAulay, K.; Macsween, K.F.; Gallacher, N.J.; Higgins, C.D.; Harrison, N.; Swerdlow, A.J.; Crawford, D.H. The immune response to primary ebv infection: A role for natural killer cells. Br. J. Haematol. 2005, 129, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Dunmire, S.K.; Grimm, J.M.; Schmeling, D.O.; Balfour, H.H., Jr.; Hogquist, K.A. The incubation period of primary epstein-barr virus infection: Viral dynamics and immunologic events. PLoS Pathog. 2015, 11, e1005286. [Google Scholar] [CrossRef] [PubMed]
- Azzi, T.; Lunemann, A.; Murer, A.; Ueda, S.; Beziat, V.; Malmberg, K.J.; Staubli, G.; Gysin, C.; Berger, C.; Munz, C.; et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 2014, 124, 2533–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in epstein-barr virus-infected b cells is associated with sensitization to nk cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.R.; Quinn, L.L.; Rowe, M.; Zuo, J. Induction of the lytic cycle sensitizes epstein-barr virus-infected b cells to nk cell killing that is counteracted by virus-mediated nk cell evasion mechanisms in the late lytic cycle. J. Virol. 2016, 90, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Meazza, R.; Marcenaro, S.; Arico, M.; Bottino, C. 2b4 dysfunction in xlp1 nk cells: More than inability to control ebv infection. Clin Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Grossman, L.; Chang, C.; Dai, J.; Nikitin, P.A.; Jima, D.D.; Dave, S.S.; Luftig, M.A. Epstein-barr virus induces adhesion receptor cd226 (dnam-1) expression during primary b-cell transformation into lymphoblastoid cell lines. mSphere 2017, 2, e00305–e00317. [Google Scholar] [CrossRef] [PubMed]
- Chigbu, D.I.; Loonawat, R.; Sehgal, M.; Patel, D.; Jain, P. Hepatitis c virus infection: Host(-)virus interaction and mechanisms of viral persistence. Cells 2019, 8, 376. [Google Scholar] [CrossRef]
- Kanwal, F.; Hoang, T.; Kramer, J.R.; Asch, S.M.; Goetz, M.B.; Zeringue, A.; Richardson, P.; El-Serag, H.B. Increasing prevalence of hcc and cirrhosis in patients with chronic hepatitis c virus infection. Gastroenterology 2011, 140, 1182–1188. [Google Scholar] [CrossRef]
- Nattermann, J.; Feldmann, G.; Ahlenstiel, G.; Langhans, B.; Sauerbruch, T.; Spengler, U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis c. Gut 2006, 55, 869–877. [Google Scholar] [CrossRef]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M.U. Natural killer cell functional dichotomy in chronic hepatitis b and chronic hepatitis c virus infections. Gastroenterology 2009, 137, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, G.; Titerence, R.H.; Koh, C.; Edlich, B.; Feld, J.J.; Rotman, Y.; Ghany, M.G.; Hoofnagle, J.H.; Liang, T.J.; Heller, T.; et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis c in an interferon-alfa-dependent manner. Gastroenterology 2010, 138, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Madrigal-Estebas, L.; McGrath, E.; Conroy, M.J.; Ryan, E.J.; Hegarty, J.E.; O’Farrelly, C.; Doherty, D.G. Altered natural killer cell subset distributions in resolved and persistent hepatitis c virus infection following single source exposure. Gut 2008, 57, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Huang, C.X.; Ye, L.; Wang, X.; Song, L.; Wang, Y.J.; Liang, H.; Huang, X.Y.; Ho, W.Z. Natural killer cells suppress full cycle hcv infection of human hepatocytes. J. Viral Hepat. 2008, 15, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, S.I.; Thio, C.L.; Martin, M.P.; Brooks, C.R.; Gao, X.; Astemborski, J.; Cheng, J.; Goedert, J.J.; Vlahov, D.; Hilgartner, M.; et al. Hla and nk cell inhibitory receptor genes in resolving hepatitis c virus infection. Science 2004, 305, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, G.; Martin, M.P.; Gao, X.; Carrington, M.; Rehermann, B. Distinct kir/hla compound genotypes affect the kinetics of human antiviral natural killer cell responses. J. Clin Invest. 2008, 118, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Cariani, E.; Pilli, M.; Zerbini, A.; Rota, C.; Olivani, A.; Zanelli, P.; Zanetti, A.; Trenti, T.; Ferrari, C.; Missale, G. Hla and killer immunoglobulin-like receptor genes as outcome predictors of hepatitis c virus-related hepatocellular carcinoma. Clin Cancer Res. 2013, 19, 5465–5473. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Cox, A.L.; Randall, J.A.; Cheng, L.; Rosen, H.R. Increased natural killer cell cytotoxicity and nkp30 expression protects against hepatitis c virus infection in high-risk individuals and inhibits replication in vitro. Hepatology 2010, 52, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, K.A.; Bjorkstrom, N.K.; Veber, H.; Ciesek, S.; Riese, P.; Wiegand, J.; Hadem, J.; Suneetha, P.V.; Jaroszewicz, J.; Wang, C.; et al. Interferon-alpha-induced trail on natural killer cells is associated with control of hepatitis c virus infection. Gastroenterology 2010, 138, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Cheent, K.; Khakoo, S.I. Natural killer cells and hepatitis c: Action and reaction. Gut 2011, 60, 268–278. [Google Scholar] [CrossRef]
- Yoon, J.C.; Lim, J.B.; Park, J.H.; Lee, J.M. Cell-to-cell contact with hepatitis c virus-infected cells reduces functional capacity of natural killer cells. J. Virol. 2011, 85, 12557–12569. [Google Scholar] [CrossRef] [PubMed]
- Crotta, S.; Stilla, A.; Wack, A.; D’Andrea, A.; Nuti, S.; D’Oro, U.; Mosca, M.; Filliponi, F.; Brunetto, R.M.; Bonino, F.; et al. Inhibition of natural killer cells through engagement of cd81 by the major hepatitis c virus envelope protein. J. Exp. Med. 2002, 195, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.T.; Klimpel, G.R. Binding of the hepatitis c virus envelope protein e2 to cd81 inhibits natural killer cell functions. J. Exp. Med. 2002, 195, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; He, X.; Ma, H.; Hou, N.; Wei, C.; Song, T.; Zhang, Y.; Sun, L.; Ma, Q.; Zhong, H. Hepatitis c virus infection downregulates the ligands of the activating receptor nkg2d. Cell Mol. Immunol. 2008, 5, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Sene, D.; Levasseur, F.; Abel, M.; Lambert, M.; Camous, X.; Hernandez, C.; Pene, V.; Rosenberg, A.R.; Jouvin-Marche, E.; Marche, P.N.; et al. Hepatitis c virus (hcv) evades nkg2d-dependent nk cell responses through ns5a-mediated imbalance of inflammatory cytokines. PLoS Pathog. 2010, 6, e1001184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, X.M.; Li, S.R.; Twelkmeyer, T.; Wang, W.H.; Zhang, S.Y.; Wang, S.F.; Chen, J.Z.; Jin, X.; Wu, Y.Z.; et al. Nkg2a is a nk cell exhaustion checkpoint for hcv persistence. Nat. Commun. 2019, 10, 1507. [Google Scholar] [CrossRef] [PubMed]
- Bozzano, F.; Picciotto, A.; Costa, P.; Marras, F.; Fazio, V.; Hirsch, I.; Olive, D.; Moretta, L.; De Maria, A. Activating nk cell receptor expression/function (nkp30, nkp46, dnam-1) during chronic viraemic hcv infection is associated with the outcome of combined treatment. Eur. J. Immunol. 2011, 41, 2905–2914. [Google Scholar] [CrossRef]
- Stegmann, K.A.; Bjorkstrom, N.K.; Ciesek, S.; Lunemann, S.; Jaroszewicz, J.; Wiegand, J.; Malinski, P.; Dustin, L.B.; Rice, C.M.; Manns, M.P.; et al. Interferon alpha-stimulated natural killer cells from patients with acute hepatitis c virus (hcv) infection recognize hcv-infected and uninfected hepatoma cells via dnax accessory molecule-1. J. Infect. Dis. 2012, 205, 1351–1362. [Google Scholar] [CrossRef]
- de Oliveria Andrade, L.J.; D’Oliveira, A.; Melo, R.C.; De Souza, E.C.; Costa Silva, C.A.; Parana, R. Association between hepatitis c and hepatocellular carcinoma. J. Glob. Infect Dis. 2009, 1, 33–37. [Google Scholar] [CrossRef]
- Hammer, Q.; Ruckert, T.; Romagnani, C. Natural killer cell specificity for viral infections. Nat. Immunol. 2018, 19, 800–808. [Google Scholar] [CrossRef]
- Brandstadter, J.D.; Yang, Y. Natural killer cell responses to viral infection. J. Innate Immun. 2011, 3, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Moses, A.; Fruh, K. Evasion of adaptive and innate immune response mechanisms by gamma-herpesviruses. Curr. Opin. Virol. 2013, 3, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Iguchi-Manaka, A.; Okumura, G.; Kojima, H.; Cho, Y.; Hirochika, R.; Bando, H.; Sato, T.; Yoshikawa, H.; Hara, H.; Shibuya, A.; et al. Increased soluble cd155 in the serum of cancer patients. PLoS ONE 2016, 11, e0152982. [Google Scholar] [CrossRef] [PubMed]
- Krenzlin, H.; Behera, P.; Lorenz, V.; Passaro, C.; Zdioruk, M.; Nowicki, M.O.; Grauwet, K.; Zhang, H.; Skubal, M.; Ito, H.; et al. Cytomegalovirus promotes murine glioblastoma growth via pericyte recruitment and angiogenesis. J. Clin Invest. 2019, 130, 1671–1683. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting nk cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Strauss-Albee, D.M.; Blish, C.A. Human nk cell diversity in viral infection: Ramifications of ramification. Front Immunol. 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Clutton, G.T.; Jones, R.B. Diverse impacts of hiv latency-reversing agents on cd8+ t-cell function: Implications for hiv cure. Front Immunol. 2018, 9, 1452. [Google Scholar] [CrossRef]
- Ke, R.; Conway, J.M.; Margolis, D.M.; Perelson, A.S. Determinants of the efficacy of hiv latency-reversing agents and implications for drug and treatment design. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Garrido, C.; Spivak, A.M.; Soriano-Sarabia, N.; Checkley, M.A.; Barker, E.; Karn, J.; Planelles, V.; Margolis, D.M. Hiv latency-reversing agents have diverse effects on natural killer cell function. Front Immunol. 2016, 7, 356. [Google Scholar] [CrossRef]
- Desimio, M.G.; Giuliani, E.; Ferraro, A.S.; Adorno, G.; Doria, M. In vitro exposure to prostratin but not bryostatin-1 improves natural killer cell functions including killing of cd4(+) t cells harboring reactivated human immunodeficiency virus. Front Immunol. 2018, 9, 1514. [Google Scholar] [CrossRef]
- Giuliani, E.; Desimio, M.G.; Doria, M. Hexamethylene bisacetamide impairs nk cell-mediated clearance of acute t lymphoblastic leukemia cells and hiv-1-infected t cells that exit viral latency. Sci Rep 2019, 9, 4373. [Google Scholar] [CrossRef] [PubMed]
- Cifaldi, L.; Locatelli, F.; Marasco, E.; Moretta, L.; Pistoia, V. Boosting natural killer cell-based immunotherapy with anticancer drugs: A perspective. Trends Mol. Med. 2017, 23, 1156–1175. [Google Scholar] [CrossRef] [PubMed]
- Garro, H.A.; Pungitore, C.R. DNA related enzymes as molecular targets for antiviral and antitumoral chemotherapy. A natural overview of the current perspectives. Curr. Drug Targets 2019, 20, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Schmidt, S.; Tramsen, L.; Rais, B.; Ullrich, E.; Lehrnbecher, T. Natural killer cells as a therapeutic tool for infectious diseases - current status and future perspectives. Oncotarget 2018, 9, 20891–20907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, A.; Kamata, M.; Rezek, V.; Rick, J.; Levin, B.; Kasparian, S.; Chen, I.S.; Yang, O.O.; Zack, J.A.; Kitchen, S.G. Hiv-specific immunity derived from chimeric antigen receptor-engineered stem cells. Mol. Ther. 2015, 23, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Mazarzaei, A.; Vafaei, M.; Ghasemian, A.; Mirforughi, S.A.; Rajabi Vardanjani, H.; Alwan, N.A.S. Memory and car-nk cell-based novel approaches for hiv vaccination and eradication. J. Cell Physiol. 2019, 234, 14812–14817. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cifaldi, L.; Doria, M.; Cotugno, N.; Zicari, S.; Cancrini, C.; Palma, P.; Rossi, P. DNAM-1 Activating Receptor and Its Ligands: How Do Viruses Affect the NK Cell-Mediated Immune Surveillance during the Various Phases of Infection? Int. J. Mol. Sci. 2019, 20, 3715. https://doi.org/10.3390/ijms20153715
Cifaldi L, Doria M, Cotugno N, Zicari S, Cancrini C, Palma P, Rossi P. DNAM-1 Activating Receptor and Its Ligands: How Do Viruses Affect the NK Cell-Mediated Immune Surveillance during the Various Phases of Infection? International Journal of Molecular Sciences. 2019; 20(15):3715. https://doi.org/10.3390/ijms20153715
Chicago/Turabian StyleCifaldi, Loredana, Margherita Doria, Nicola Cotugno, Sonia Zicari, Caterina Cancrini, Paolo Palma, and Paolo Rossi. 2019. "DNAM-1 Activating Receptor and Its Ligands: How Do Viruses Affect the NK Cell-Mediated Immune Surveillance during the Various Phases of Infection?" International Journal of Molecular Sciences 20, no. 15: 3715. https://doi.org/10.3390/ijms20153715