Gene-Specific Intron Retention Serves as Molecular Signature that Distinguishes Melanoma from Non-Melanoma Cancer Cells in Greek Patients

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
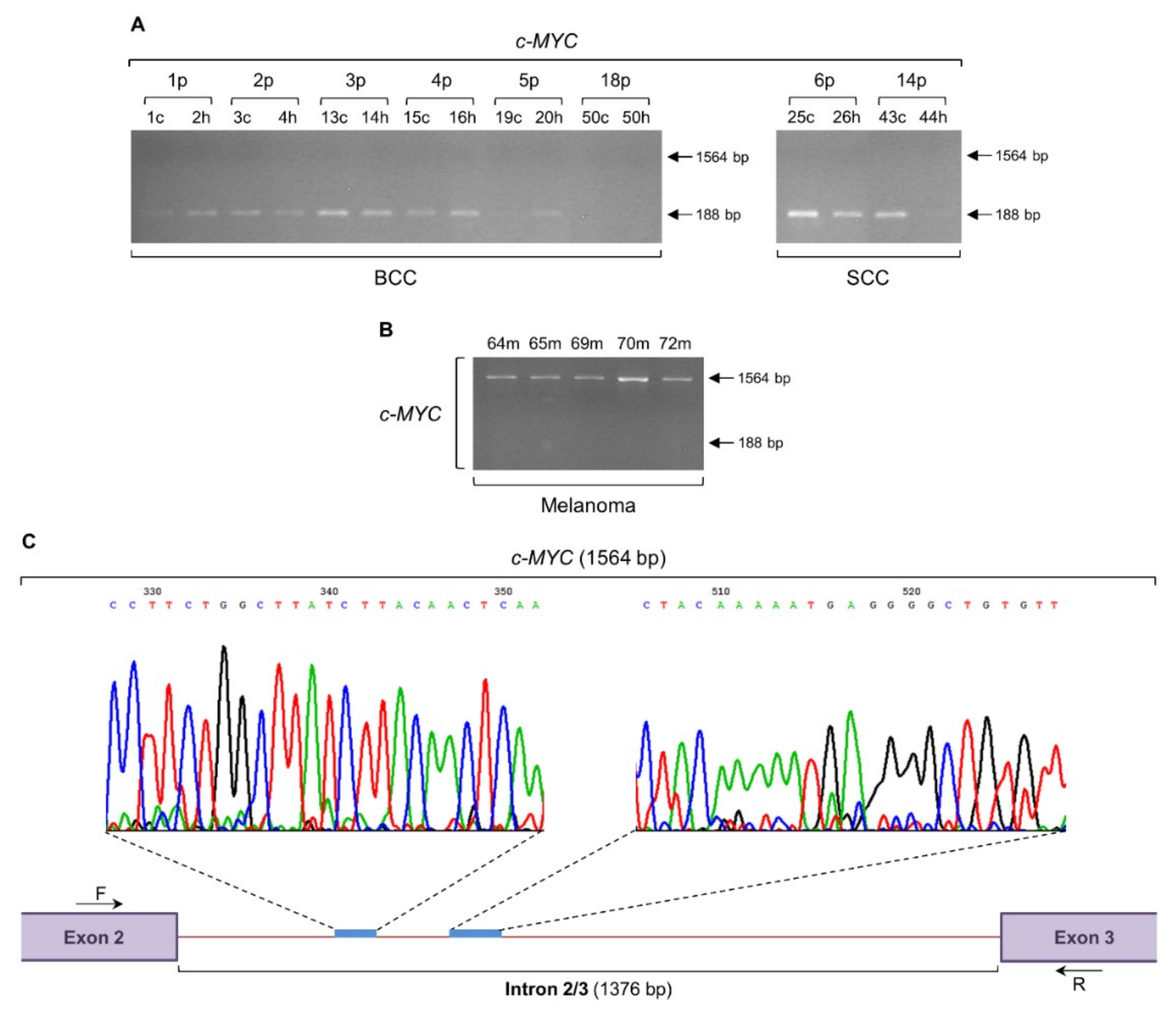
2.1. Melanoma-Specific Intron Retention of c-MYC Gene
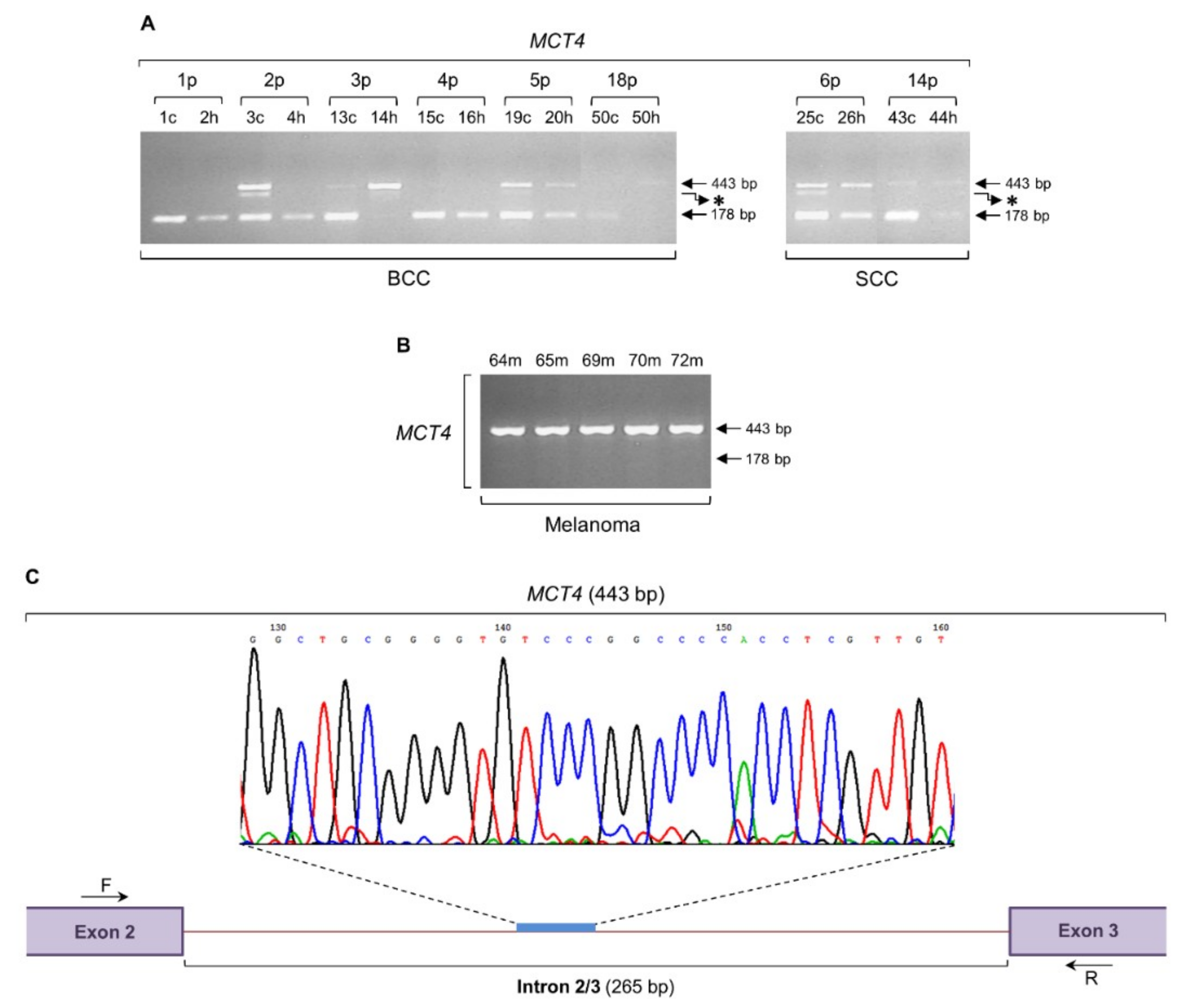
2.2. The MCT4 Gene is Subjected to an Intron Retention Process both in Non-Melanoma and Melanoma Biopsy Specimens
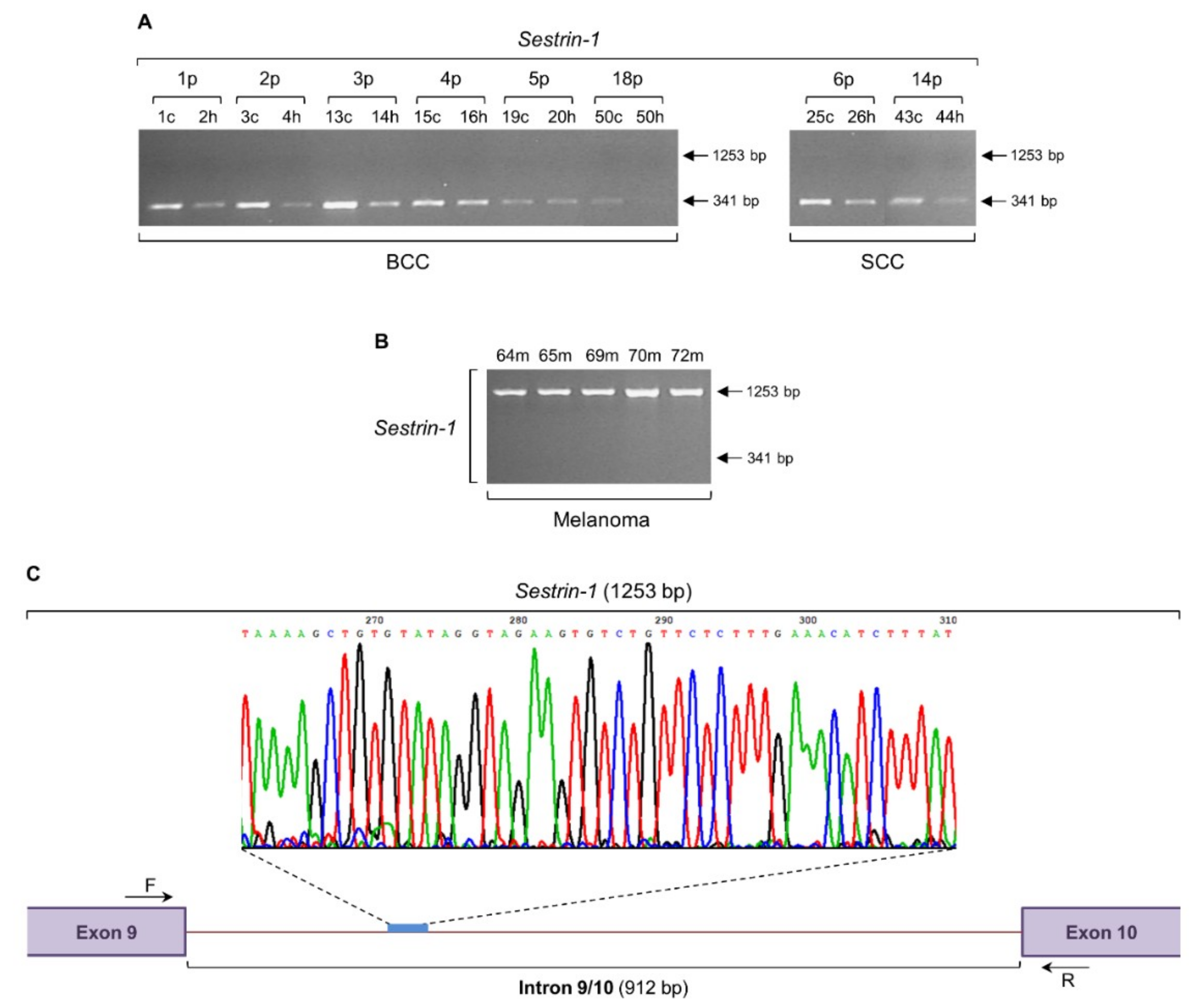
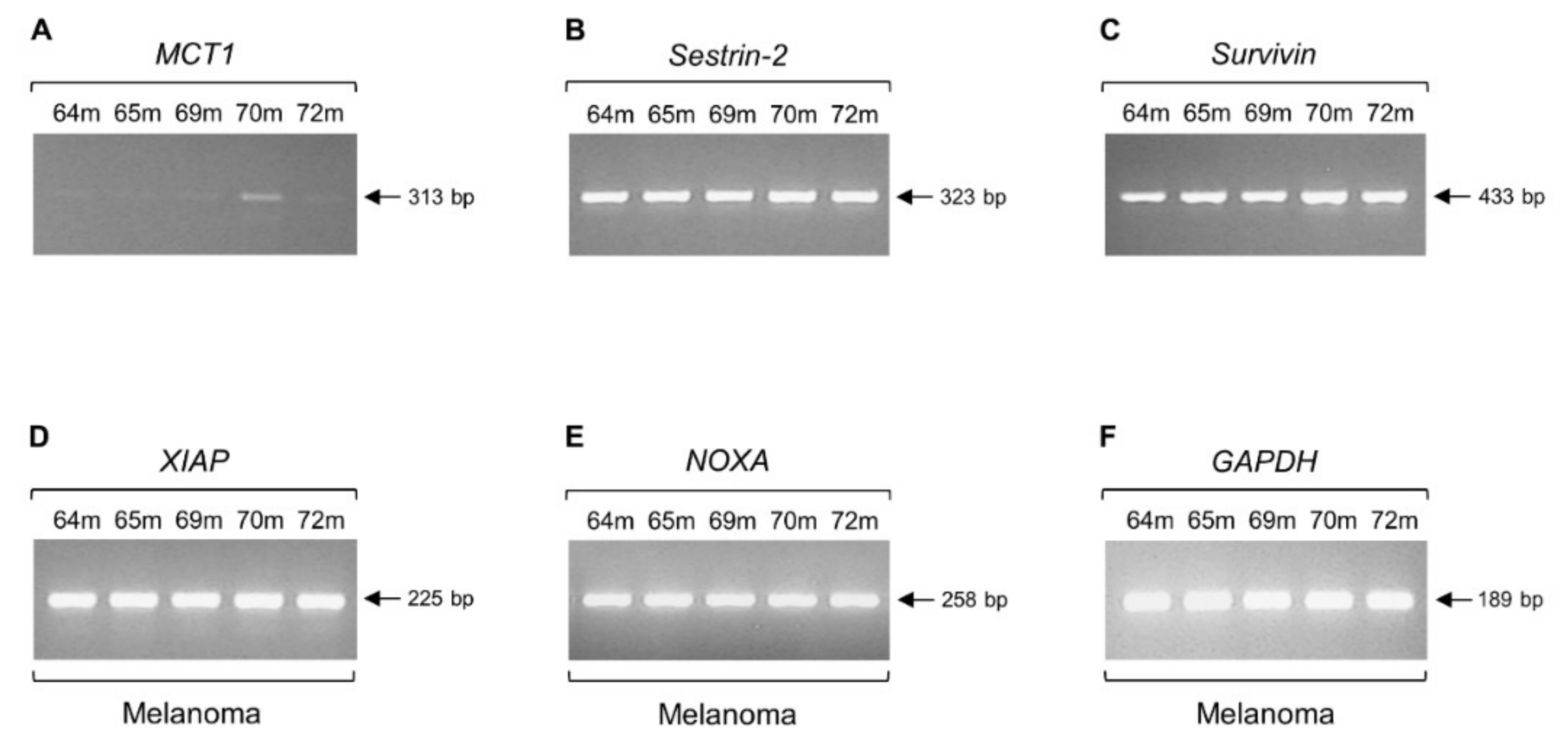
2.3. Intron Retention of Sestrin-1 Gene can Distinguish Non-Melanoma from Melanoma Tissue
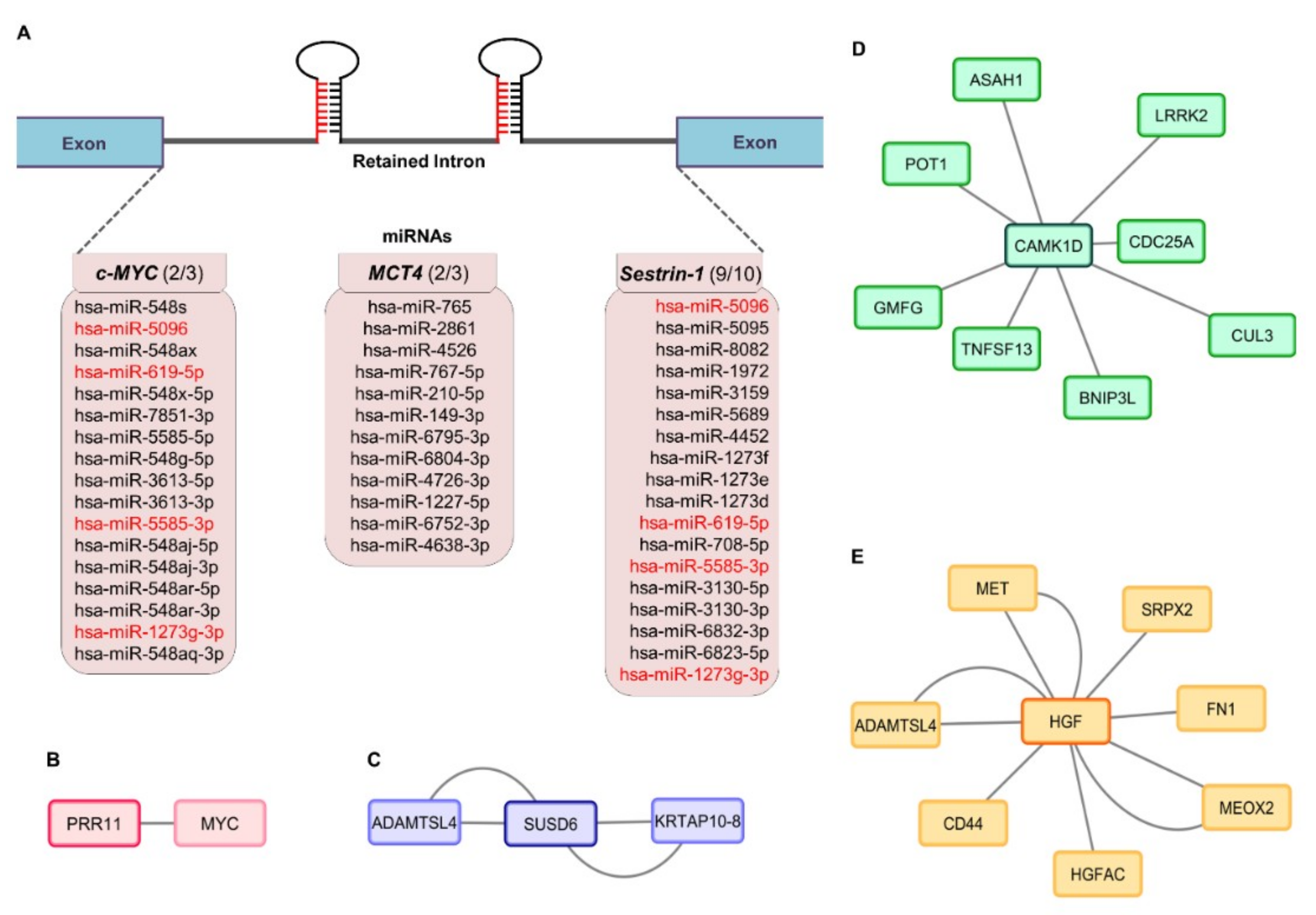
2.4. Mapping the miRNA Landscape of c-MYC, MCT4 and Sestrin-1 Retained Introns: from Target Genes to Protein Interactomes
2.5. Intronic miRNA Target Gene-Specific Silencing and Aberrant Splicing in Melanoma
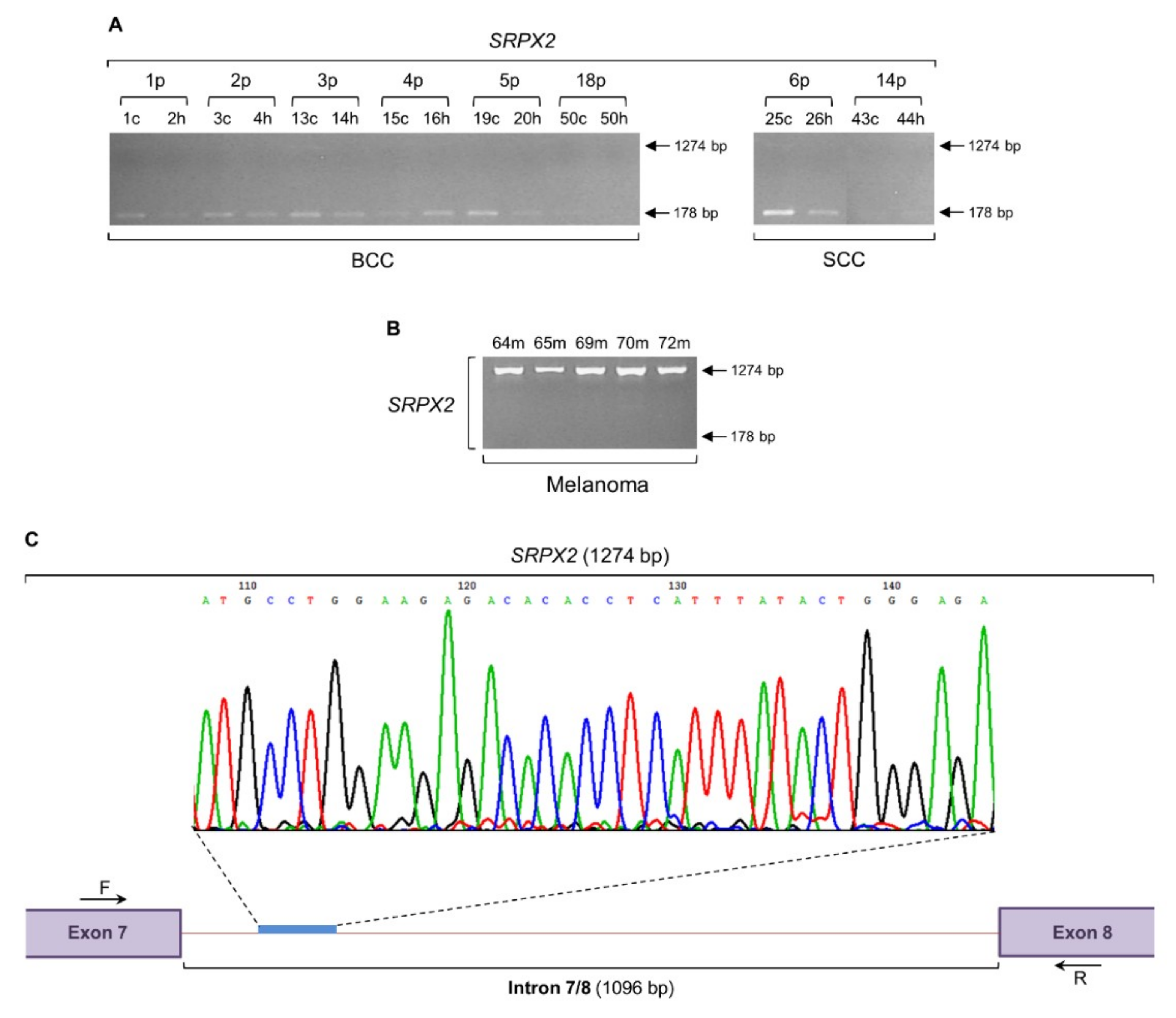
2.6. SRPX2 Gene Undergoes a Strong Intron Retention Process in Melanoma
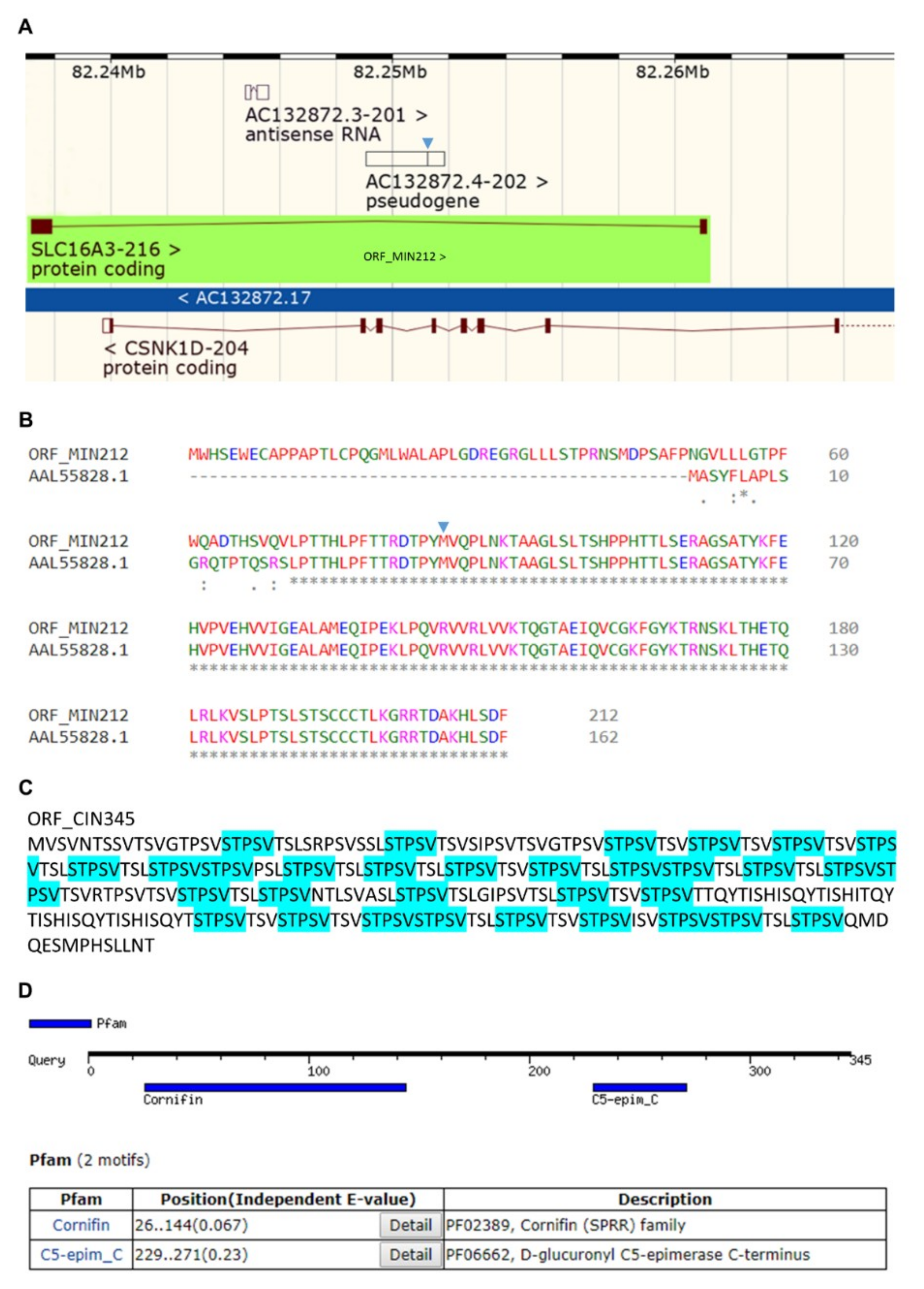
2.7. “ORFing” the c-MYC, MCT4, Sestrin-1, and SRPX2 Introns
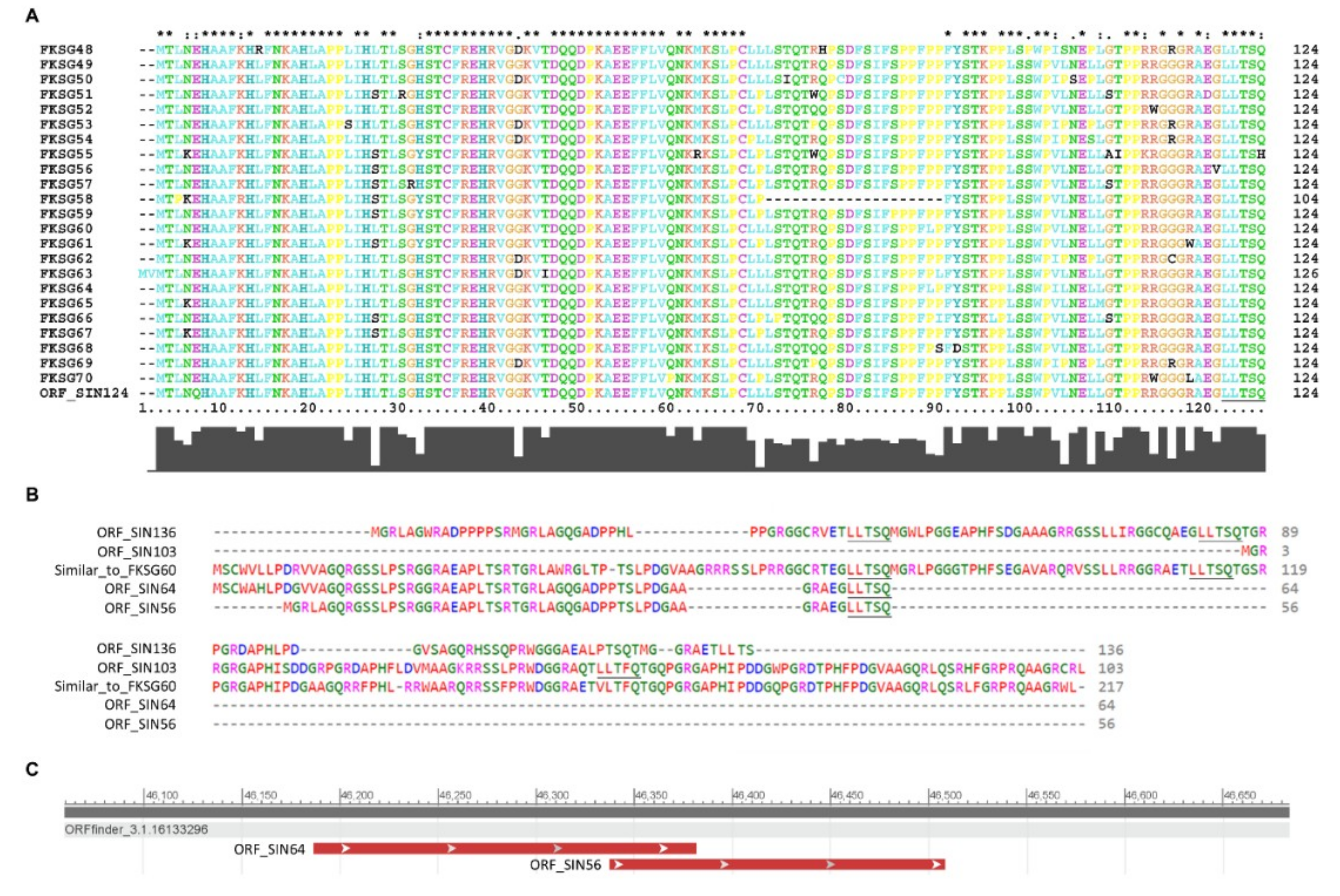
2.8. Molecular Modeling of “LLTSQ”- or “STPSV”-Rich Proteins
2.9. The “LLTSQ”-Rich Protein CA1ICL-861 Is Predicted to form Dimers and Tetramers, and to Also Recognize Acetylated Lysine
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Total RNA Extraction
4.3. RT-sqPCR
4.4. DNA Sequencing of PCR Products
4.5. Bioinformatics Analysis
4.5.1. miRNA Alignments to Intron Sequences—miRNA Target Predictions
4.5.2. Intronic Open Reading Frame (ORF) Identification
4.5.3. Protein Sequence Alignments
4.5.4. Molecular Assembly of Protein Interactomes
4.5.5. Protein Molecular Modeling—Structural Prediction of Protein–Protein Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BCC | basal cell carcinoma |
| NMD | nonsense-mediated mRNA decay |
| PCR | polymerase chain reaction |
| RT | reverse transcription |
| SCC | squamous cell carcinoma |
| sq | semi-quantitative |
| UV | ultraviolet |
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.A.; Fisher, D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Dlugosz, A.; Merlino, G.; Yuspa, S.H. Progress in cutaneous cancer research. J. Investig. Dermatol. Symp. Proc. 2002, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.T. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat. Rev. Cancer 2004, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Halliday, G.M.; Damian, D.L. Non-melanoma skin cancer: Carcinogenesis and chemoprevention. Pathology 2013, 45, 331–341. [Google Scholar] [CrossRef]
- Neville, J.A.; Welch, E.; Leffell, D.J. Management of nonmelanoma skin cancer in 2007. Nat. Clin. Pract. Oncol. 2007, 4, 462–469. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Stacey, S.N.; Helgason, H.; Gudjonsson, S.A.; Thorleifsson, G.; Zink, F.; Sigurdsson, A.; Kehr, B.; Gudmundsson, J.; Sulem, P.; Sigurgeirsson, B.; et al. New basal cell carcinoma susceptibility loci. Nat. Commun. 2015, 6, 6825. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Siegle, J.M.; Basin, A.; Sastre-Perona, A.; Yonekubo, Y.; Brown, J.; Sennett, R.; Rendl, M.; Tsirigos, A.; Carucci, J.A.; Schober, M. SOX2 is a cancer-specific regulator of tumour initiating potential in cutaneous squamous cell carcinoma. Nat. Commun. 2014, 5, 4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.S.; Bhaduri, A.; Mah, A.; Johnson, W.L.; Ungewickell, A.; Aros, C.J.; Nguyen, C.B.; Rios, E.J.; Siprashvili, Z.; Straight, A.; et al. Recurrent point mutations in the kinetochore gene KNSTRN in cutaneous squamous cell carcinoma. Nat. Genet. 2014, 46, 1060–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahal, H.S.; Lin, Y.; Ransohoff, K.J.; Hinds, D.A.; Wu, W.; Dai, H.J.; Qureshi, A.A.; Li, W.Q.; Kraft, P.; Tang, J.Y.; et al. Genome-wide association study identifies novel susceptibility loci for cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFbeta receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Ashford, B.G.; Clark, J.; Gupta, R.; Iyer, N.G.; Yu, B.; Ranson, M. Reviewing the genetic alterations in high-risk cutaneous squamous cell carcinoma: A search for prognostic markers and therapeutic targets. Head Neck 2017, 39, 1462–1469. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Woods, S.L.; Boyle, G.M.; Aoude, L.G.; MacGregor, S.; Zismann, V.; Gartside, M.; Cust, A.E.; Haq, R.; Harland, M.; et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 2011, 480, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvinge, H.; Bradley, R.K. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Au, A.Y.; Ritchie, W.; Rasko, J.E. Intron retention in mRNA: No longer nonsense: Known and putative roles of intron retention in normal and disease biology. Bioessays 2016, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, J.; Horvath, A.; Godbole, S.; Reddy, S.D.; Mudvari, P.; Ohshiro, K.; Cyanam, D.; Nair, S.; Fuqua, S.A.; Polyak, K.; et al. RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep. 2013, 3, 1689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, H.; Jin, H.; Tan, H.; Zhang, J.; Sheng, S. The global landscape of intron retentions in lung adenocarcinoma. BMC Med. Genom. 2014, 7, 15. [Google Scholar] [CrossRef]
- Ren, S.; Peng, Z.; Mao, J.H.; Yu, Y.; Yin, C.; Gao, X.; Cui, Z.; Zhang, J.; Yi, K.; Xu, W.; et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012, 22, 806–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.Y.; Simon, L.M.; Neill, N.J.; Marcotte, R.; Sayad, A.; Bland, C.S.; Echeverria, G.V.; Sun, T.; Kurley, S.J.; Tyagi, S.; et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 2015, 525, 384–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabo, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Kraehn, G.M.; Utikal, J.; Udart, M.; Greulich, K.M.; Bezold, G.; Kaskel, P.; Leiter, U.; Peter, R.U. Extra c-myc oncogene copies in high risk cutaneous malignant melanoma and melanoma metastases. Br. J. Cancer 2001, 84, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.S.; Joly, M.M.; Allen-Petersen, B.L.; Worth, P.J.; Lanciault, C.; Sauer, D.; Link, J.; Pelz, C.; Heiser, L.M.; Morton, J.P.; et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun. 2017, 8, 1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, T.R.; Sabo, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Xiu, R.; Ren, P.; Yue, M.; Su, H.; Guo, G.; Xiao, D.; Yu, J.; Jiang, H.; Liu, H.; et al. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene 2016, 35, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. Monocarboxylic acid transport. Compr. Physiol. 2013, 3, 1611–1643. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; de Moura, M.B.; Lin, Y.; Vincent, G.; Thorne, S.; Duncan, L.M.; Hui-Min, L.; Kirkwood, J.M.; Becker, D.; Van Houten, B.; et al. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol. Cancer 2012, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, C.; Miranda-Goncalves, V.; Longatto-Filho, A.; Vicente, A.L.; Berardinelli, G.N.; Scapulatempo-Neto, C.; Costa, R.F.; Viana, C.R.; Reis, R.M.; Baltazar, F.; et al. The metabolic microenvironment of melanomas: Prognostic value of MCT1 and MCT4. Cell Cycle 2016, 15, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Si, L.; Li, Y.; Wu, X.; Xu, X.; Dai, J.; Tang, H.; Ma, M.; Chi, Z.; Sheng, X.; et al. Analysis of mTOR Gene Aberrations in Melanoma Patients and Evaluation of Their Sensitivity to PI3K-AKT-mTOR Pathway Inhibitors. Clin. Cancer Res. 2016, 22, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Li, M.O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 2014, 159, 122–133. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef]
- Monterisi, S.; D’Ario, G.; Dama, E.; Rotmensz, N.; Confalonieri, S.; Tordonato, C.; Troglio, F.; Bertalot, G.; Maisonneuve, P.; Viale, G.; et al. Mining cancer gene expression databases for latent information on intronic microRNAs. Mol. Oncol. 2015, 9, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Iliopoulos, D.; Janas, M.M.; Schubert, S.; Pinner, S.; Chen, P.H.; Li, S.; Fletcher, A.L.; Yokoyama, S.; et al. Intronic miR-211 assumes the tumor suppressive function of its host gene in melanoma. Mol. Cell 2010, 40, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Gorski, S.A.; Vogel, J.; Doudna, J.A. RNA-based recognition and targeting: Sowing the seeds of specificity. Nat. Rev. Mol. Cell Biol. 2017, 18, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Mannavola, F.; Tucci, M.; Felici, C.; Stucci, S.; Silvestris, F. miRNAs in melanoma: A defined role in tumor progression and metastasis. Expert Rev. Clin. Immunol. 2016, 12, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Xie, M.; Lan, H.; Zhang, Y.; Long, Y.; Weng, H.; Li, D.; Cai, W.; Zhu, H.; Niu, Y.; et al. PRR11 is a novel gene implicated in cell cycle progression and lung cancer. Int. J. Biochem. Cell Biol. 2013, 45, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Y.; Li, Y.; Zhu, H.; Wang, Y.; Cai, W.; Zhu, J.; Ozaki, T.; Bu, Y. PRR11 regulates late-S to G2/M phase progression and induces premature chromatin condensation (PCC). Biochem. Biophys. Res. Commun. 2015, 458, 501–508. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, H.; Zhang, X.; Shen, Y.; Zheng, D.; Zhang, A.; Lai, Y.; Li, H. Proline-rich protein 11 regulates epithelial-to-mesenchymal transition to promote breast cancer cell invasion. Int. J. Clin. Exp. Pathol. 2014, 7, 8692–8699. [Google Scholar]
- Dimitrova, N.; Gocheva, V.; Bhutkar, A.; Resnick, R.; Jong, R.M.; Miller, K.M.; Bendor, J.; Jacks, T. Stromal Expression of miR-143/145 Promotes Neoangiogenesis in Lung Cancer Development. Cancer Discov. 2016, 6, 188–201. [Google Scholar] [CrossRef]
- Lawson, J.; Dickman, C.; MacLellan, S.; Towle, R.; Jabalee, J.; Lam, S.; Garnis, C. Selective secretion of microRNAs from lung cancer cells via extracellular vesicles promotes CAMK1D-mediated tube formation in endothelial cells. Oncotarget 2017, 8, 83913–83924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamaschi, A.; Kim, Y.H.; Kwei, K.A.; La Choi, Y.; Bocanegra, M.; Langerod, A.; Han, W.; Noh, D.Y.; Huntsman, D.G.; Jeffrey, S.S.; et al. CAMK1D amplification implicated in epithelial-mesenchymal transition in basal-like breast cancer. Mol. Oncol. 2008, 2, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A.; Salto-Tellez, M.; Laurent-Puig, P.; Bardelli, A.; Rolfo, C.; Tabernero, J.; Khawaja, H.A.; Lawler, M.; Johnston, P.G.; Van Schaeybroeck, S.; et al. Targeting c-MET in gastrointestinal tumours: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2017, 14, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8, 15016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascone, T.; Xu, L.; Lin, H.Y.; Liu, W.; Tran, H.T.; Liu, Y.; Howells, K.; Haddad, V.; Hanrahan, E.; Nilsson, M.B.; et al. The HGF/c-MET Pathway Is a Driver and Biomarker of VEGFR-inhibitor Resistance and Vascular Remodeling in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 5489–5501. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Staib, F.; Robles, A.I.; Varticovski, L.; Wang, X.W.; Zeeberg, B.R.; Sirotin, M.; Zhurkin, V.B.; Hofseth, L.J.; Hussain, S.P.; Weinstein, J.N.; et al. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005, 65, 10255–10264. [Google Scholar] [CrossRef]
- Polato, F.; Rusconi, P.; Zangrossi, S.; Morelli, F.; Boeri, M.; Musi, A.; Marchini, S.; Castiglioni, V.; Scanziani, E.; Torri, V.; et al. DRAGO (KIAA0247), a new DNA damage-responsive, p53-inducible gene that cooperates with p53 as oncosuppressor. [Corrected]. J. Natl. Cancer Inst. 2014, 106, dju053. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Huang, N.; Zhang, X.; Hu, J.; Cheng, S.; Pi, L.; Cheng, Y. KIAA0247 suppresses the proliferation, angiogenesis and promote apoptosis of human glioma through inactivation of the AKT and Stat3 signaling pathway. Oncotarget 2016, 7, 87100–87113. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kundu, R.K.; Ikeda, S.; Kobara, M.; Matsubara, H.; Quertermous, T. Glia maturation factor-gamma is preferentially expressed in microvascular endothelial and inflammatory cells and modulates actin cytoskeleton reorganization. Circ. Res. 2006, 99, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Zuo, P.; Ma, Y.; Huang, Y.; Ye, F.; Wang, P.; Wang, X.; Zhou, C.; Lu, W.; Kong, B.; Xie, X. High GMFG expression correlates with poor prognosis and promotes cell migration and invasion in epithelial ovarian cancer. Gynecol. Oncol. 2014, 132, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Z.; Chang, H.; Mu, X.; Deng, W.; Yuan, Z.; Yao, F.; Liu, Y.; Mai, R.; Wu, B. Expression of glia maturation factor gamma is associated with colorectal cancer metastasis and its downregulation suppresses colorectal cancer cell migration and invasion in vitro. Oncol. Rep. 2017, 37, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Leal, A.D.; Gorski, D.H. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res. 2005, 65, 1414–1424. [Google Scholar] [CrossRef]
- Chen, Y.; Banda, M.; Speyer, C.L.; Smith, J.S.; Rabson, A.B.; Gorski, D.H. Regulation of the expression and activity of the antiangiogenic homeobox gene GAX/MEOX2 by ZEB2 and microRNA-221. Mol. Cell. Biol. 2010, 30, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Avila-Moreno, F.; Armas-Lopez, L.; Alvarez-Moran, A.M.; Lopez-Bujanda, Z.; Ortiz-Quintero, B.; Hidalgo-Miranda, A.; Urrea-Ramirez, F.; Rivera-Rosales, R.M.; Vazquez-Manriquez, E.; Pena-Mirabal, E.; et al. Overexpression of MEOX2 and TWIST1 is associated with H3K27me3 levels and determines lung cancer chemoresistance and prognosis. PLoS ONE 2014, 9, e114104. [Google Scholar] [CrossRef]
- Armas-Lopez, L.; Pina-Sanchez, P.; Arrieta, O.; de Alba, E.G.; Ortiz-Quintero, B.; Santillan-Doherty, P.; Christiani, D.C.; Zuniga, J.; Avila-Moreno, F. Epigenomic study identifies a novel mesenchyme homeobox2-GLI1 transcription axis involved in cancer drug resistance, overall survival and therapy prognosis in lung cancer patients. Oncotarget 2017, 8, 67056–67081. [Google Scholar] [CrossRef]
- Roll, P.; Rudolf, G.; Pereira, S.; Royer, B.; Scheffer, I.E.; Massacrier, A.; Valenti, M.P.; Roeckel-Trevisiol, N.; Jamali, S.; Beclin, C.; et al. SRPX2 mutations in disorders of language cortex and cognition. Hum. Mol. Genet. 2006, 15, 1195–1207. [Google Scholar] [CrossRef] [Green Version]
- Sia, G.M.; Clem, R.L.; Huganir, R.L. The human language-associated gene SRPX2 regulates synapse formation and vocalization in mice. Science 2013, 342, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Reader, R.H.; Hoischen, A.; Veltman, J.A.; Simpson, N.H.; Francks, C.; Newbury, D.F.; Fisher, S.E. Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci. Rep. 2017, 7, 46105. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Arao, T.; Maegawa, M.; Matsumoto, K.; Kaneda, H.; Kudo, K.; Fujita, Y.; Yokote, H.; Yanagihara, K.; Yamada, Y.; et al. SRPX2 is overexpressed in gastric cancer and promotes cellular migration and adhesion. Int. J. Cancer 2009, 124, 1072–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Zhang, J.; Bi, M.; Han, X.; Han, Z.; Wang, H.; Ou, Y. SRPX2 promotes cell migration and invasion via FAK dependent pathway in pancreatic cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 4791–4798. [Google Scholar] [PubMed]
- Lin, X.; Chang, W.; Wang, Y.; Tian, M.; Yu, Z. SRPX2, an independent prognostic marker, promotes cell migration and invasion in hepatocellular carcinoma. Biomed. Pharmacother. 2017, 93, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Miljkovic-Licina, M.; Hammel, P.; Garrido-Urbani, S.; Bradfield, P.F.; Szepetowski, P.; Imhof, B.A. Sushi repeat protein X-linked 2, a novel mediator of angiogenesis. FASEB J. 2009, 23, 4105–4116. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Fan, J.; Wu, J. Sushi repeat-containing protein X-linked 2 promotes angiogenesis through the urokinase-type plasminogen activator receptor dependent integrin alphavbeta3/focal adhesion kinase pathways. Drug Discov. Ther. 2017, 11, 212–217. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, J.; Zhang, L.; Zhao, J.; Zhuang, Y.; Liang, P. SRPX2 Enhances the Epithelial-Mesenchymal Transition and Temozolomide Resistance in Glioblastoma Cells. Cell. Mol. Neurobiol. 2016, 36, 1067–1076. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef]
- Byun, J.K.; Choi, Y.K.; Kim, J.H.; Jeong, J.Y.; Jeon, H.J.; Kim, M.K.; Hwang, I.; Lee, S.Y.; Lee, Y.M.; Lee, I.K.; et al. A Positive Feedback Loop between Sestrin2 and mTORC2 Is Required for the Survival of Glutamine-Depleted Lung Cancer Cells. Cell Rep. 2017, 20, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Chawla-Sarkar, M.; Bae, S.I.; Reu, F.J.; Jacobs, B.S.; Lindner, D.J.; Borden, E.C. Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2004, 11, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.; Yoon, H.J.; Kang, M.C.; Kwon, J.; Lee, H.W. MiR-338-5p enhances the radiosensitivity of esophageal squamous cell carcinoma by inducing apoptosis through targeting survivin. Sci. Rep. 2017, 7, 10932. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Korneluk, R.G. XIAP, the guardian angel. Nat. Rev. Mol. Cell Biol. 2001, 2, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Charles, E.M.; Rehm, M. Key regulators of apoptosis execution as biomarker candidates in melanoma. Mol. Cell. Oncol. 2014, 1, e964037. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.M.; Busselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lu, X.; Tan, T.Z.; Chng, W.J. X-linked inhibitor of apoptosis inhibition sensitizes acute myeloid leukemia cell response to TRAIL and chemotherapy through potentiated induction of proapoptotic machinery. Mol. Oncol. 2018, 12, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.C.; Brinkmann, K.; Kashkar, H. Noxa and cancer therapy: Tuning up the mitochondrial death machinery in response to chemotherapy. Mol. Cell. Oncol. 2014, 1, e29906. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.M.; Mohana-Kumaran, N.; Lau, D.; Zhang, X.D.; Hersey, P.; Huang, D.C.; Weninger, W.; Haass, N.K.; Allen, J.D. Modulation of NOXA and MCL-1 as a strategy for sensitizing melanoma cells to the BH3-mimetic ABT-737. Clin. Cancer Res. 2012, 18, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, K.; Zigrino, P.; Witt, A.; Schell, M.; Ackermann, L.; Broxtermann, P.; Schull, S.; Andree, M.; Coutelle, O.; Yazdanpanah, B.; et al. Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Rep. 2013, 3, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Sirover, M.A. Subcellular dynamics of multifunctional protein regulation: Mechanisms of GAPDH intracellular translocation. J. Cell. Biochem. 2012, 113, 2193–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Liu, S.; Sun, M.Z. Novel insight into the role of GAPDH playing in tumor. Clin. Transl. Oncol. 2013, 15, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Carregaro, F.; Stefanini, A.C.; Henrique, T.; Tajara, E.H. Study of small proline-rich proteins (SPRRs) in health and disease: A review of the literature. Arch. Dermatol. Res. 2013, 305, 857–866. [Google Scholar] [CrossRef]
- Metri, R.; Mohan, A.; Nsengimana, J.; Pozniak, J.; Molina-Paris, C.; Newton-Bishop, J.; Bishop, D.; Chandra, N. Identification of a gene signature for discriminating metastatic from primary melanoma using a molecular interaction network approach. Sci. Rep. 2017, 7, 17314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, C.; Roy, S.; Namjoshi, S.; Malarkey, C.S.; Jones, D.N.; Kutateladze, T.G.; Churchill, M.E.; Tyler, J.K. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc. Natl. Acad. Sci. USA 2014, 111, E1072–E1081. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Ornaghi, P.; Yang, J.C.; Lowe, N.; Evans, P.R.; Ballario, P.; Neuhaus, D.; Filetici, P.; Travers, A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA Decay and Cancer. Curr. Opin. Genet. Dev. 2017, 48, 44–50. [Google Scholar] [CrossRef]
- Pimentel, H.; Parra, M.; Gee, S.L.; Mohandas, N.; Pachter, L.; Conboy, J.G. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 2016, 44, 838–851. [Google Scholar] [CrossRef]
- Mauger, O.; Lemoine, F.; Scheiffele, P. Targeted Intron Retention and Excision for Rapid Gene Regulation in Response to Neuronal Activity. Neuron 2016, 92, 1266–1278. [Google Scholar] [CrossRef]
- Liu, C.; Karam, R.; Zhou, Y.; Su, F.; Ji, Y.; Li, G.; Xu, G.; Lu, L.; Wang, C.; Song, M.; et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014, 20, 596–598. [Google Scholar] [CrossRef] [Green Version]
- Gunther, O.P.; Lin, D.; Balshaw, R.F.; Ng, R.T.; Hollander, Z.; Wilson-McManus, J.; McMaster, W.R.; McManus, B.M.; Keown, P.A.; Biomarkers, T. Effects of sample timing and treatment on gene expression in early acute renal allograft rejection. Transplantation 2011, 91, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Conery, A.R.; Centore, R.C.; Neiss, A.; Keller, P.J.; Joshi, S.; Spillane, K.L.; Sandy, P.; Hatton, C.; Pardo, E.; Zawadzke, L.; et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashchenko, A.; Berillo, O.; Pyrkova, A.; Niyazova, R.; Atambayeva, S. The properties of binding sites of miR-619-5p, miR-5095, miR-5096, and miR-5585-3p in the mRNAs of human genes. Biomed. Res. Int. 2014, 2014, 720715. [Google Scholar] [CrossRef]
- Ivashchenko, A.; Berillo, O.; Pyrkova, A.; Niyazova, R. Binding sites of miR-1273 family on the mRNA of target genes. Biomed. Res. Int. 2014, 2014, 620530. [Google Scholar] [CrossRef]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Berillo, O.; Regnier, M.; Ivashchenko, A. Binding of intronic miRNAs to the mRNAs of host genes encoding intronic miRNAs and proteins that participate in tumourigenesis. Comput. Biol. Med. 2013, 43, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Chanteloup, G.; Boucher, J.; Pernet, N.; Boudesco, C.; Jego, G.; Chatelier, A.; Bois, P.; Gobbo, J.; Cronier, L.; et al. Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget 2017, 8, 37681–37693. [Google Scholar] [CrossRef]
- Wong, J.J.; Ritchie, W.; Ebner, O.A.; Selbach, M.; Wong, J.W.; Huang, Y.; Gao, D.; Pinello, N.; Gonzalez, M.; Baidya, K.; et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.R.; Ritchie, W.; Wong, J.J.; Schmitz, U.; Middleton, R.; An, X.; Mohandas, N.; Rasko, J.E.; Blobel, G.A. A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood 2016. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Zhou, X.; Pendleton, K.E.; Hunter, O.V.; Kohler, J.J.; O’Donnell, K.A.; Conrad, N.K. A Conserved Splicing Silencer Dynamically Regulates O-GlcNAc Transferase Intron Retention and O-GlcNAc Homeostasis. Cell Rep. 2017, 20, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835.e814. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Gao, D.; Nguyen, T.V.; Kwok, C.T.; van Geldermalsen, M.; Middleton, R.; Pinello, N.; Thoeng, A.; Nagarajah, R.; Holst, J.; et al. Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat. Commun. 2017, 8, 15134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Shivakumar, M.; Han, S.; Sinclair, M.S.; Lee, Y.J.; Zheng, Y.; Olopade, O.I.; Kim, D.; Lee, Y. Population-dependent Intron Retention and DNA Methylation in Breast Cancer. Mol. Cancer Res. 2018, 16, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Kostaki, M.; Manona, A.D.; Stavraka, I.; Korkolopoulou, P.; Levidou, G.; Trigka, E.A.; Christofidou, E.; Champsas, G.; Stratigos, A.J.; Katsambas, A.; et al. High-frequency p16(INK) (4A) promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma. Exp. Dermatol. 2014, 23, 332–338. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Improving microRNA target prediction by modeling with unambiguously identified microRNA-target pairs from CLIP-ligation studies. Bioinformatics 2016, 32, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, S.J.; Bonvin, A.M. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs | Number of Genes | Target Genes | Intron Retained | Target Score |
|---|---|---|---|---|
| hsa-miR-5585-3p | 2 | TSTD3 | c-MYC + Sestrin-1 | 50 |
| HGF | 52 | |||
| hsa-miR-1273g-3p | 4 | PRR11 | c-MYC + Sestrin-1 | 50 |
| SUSD6 | 76 | |||
| CAMK1D | 87 | |||
| HGF | 89 | |||
| hsa-miR-619-5p | 4 | TSTD3 | c-MYC + Sestrin-1 | 58 |
| PRR11 | 65 | |||
| SUSD6 | 75 | |||
| CAMK1D | 81 | |||
| hsa-miR-5096 | 5 | TSTD3 | c-MYC + Sestrin-1 | 52 |
| HGF | 71 | |||
| PRR11 | 74 | |||
| SUSD6 | 85 | |||
| CAMK1D | 94 |
| Patient Number | Age Ranges | Lesion | Affected Tissue | Ulceration | Actinic (Solar) Elastosis (Neighboring Papillary Chorion Tissue) | Infiltration | Foci Number |
|---|---|---|---|---|---|---|---|
| 1 | 80–85 | BCC | Nose (Left Pterygium) | Yes | Yes | No | 2 |
| 2 | 70–75 | BCC | Cheek | Yes | Yes | No | 1 |
| 3 | 70–75 | BCC | Eye (Right Upper Eyelid/Inner Canthus) | No | Yes | No | 2 |
| 4 | 70–75 | BCC | Eye (Left Inner Canthus) | No | Yes | No | 1 |
| 5 | 80–85 | BCC | Cheek (Right) | No | No | Yes (Subcutaneous Fat Tissue) | 1 |
| 6 | 65–70 | SCC | Forehead | No | Yes | Yes (Subcutaneous Fat Tissue) | 1 |
| 14 | 85–90 | SCC | Cheek (Left) | Yes | Yes | No | 1 |
| 18 | 70–75 | BCC | Dorsum (Back)/Thoracic Wall | Yes | No | Yes | Several (Some with BCC-SCC Features) |
| Patient Number | Age Ranges | Lesion | Histogenic Type: Nodular Features | Growth Pattern | Lymphocyte Infiltration | Mitotic Index | Ulceration | Epidermal Infiltration | Neurotropism | Regression | Satellite Foci | Clark Staging: Melanoma Invasion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 64 | 80–85 | Malignant Nevus | No | Horizontal | Absent | 0–5 mitoses/mm3 | No | No | No | No | No | I–III: Early Stage |
| 65 | 60–65 | Melanoma | No | Horizontal | Absent | 0–5 mitoses/mm3 | No | No | No | No | No | I–III: Early Stage |
| 69 | 80–85 | Melanoma | No | Horizontal | Absent | 0–5 mitoses/mm3 | No | No | No | No | No | I–III: Early Stage |
| 70 | 45–50 | Melanoma | No | Horizontal | Brisk | 0–5 mitoses/mm3 | No | Yes | No | Yes | No | I–III: Early Stage |
| 72 | 80–85 | Melanoma | No | Horizontal | Brisk | 0–5 mitoses/mm3 | No | Yes | No | Yes | No | I–III: Early Stage |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannopoulou, A.F.; Konstantakou, E.G.; Velentzas, A.D.; Avgeris, S.N.; Avgeris, M.; Papandreou, N.C.; Zoi, I.; Filippa, V.; Katarachia, S.; Lampidonis, A.D.; et al. Gene-Specific Intron Retention Serves as Molecular Signature that Distinguishes Melanoma from Non-Melanoma Cancer Cells in Greek Patients. Int. J. Mol. Sci. 2019, 20, 937. https://doi.org/10.3390/ijms20040937
Giannopoulou AF, Konstantakou EG, Velentzas AD, Avgeris SN, Avgeris M, Papandreou NC, Zoi I, Filippa V, Katarachia S, Lampidonis AD, et al. Gene-Specific Intron Retention Serves as Molecular Signature that Distinguishes Melanoma from Non-Melanoma Cancer Cells in Greek Patients. International Journal of Molecular Sciences. 2019; 20(4):937. https://doi.org/10.3390/ijms20040937
Chicago/Turabian StyleGiannopoulou, Aikaterini F., Eumorphia G. Konstantakou, Athanassios D. Velentzas, Socratis N. Avgeris, Margaritis Avgeris, Nikos C. Papandreou, Ilianna Zoi, Vicky Filippa, Stamatia Katarachia, Antonis D. Lampidonis, and et al. 2019. "Gene-Specific Intron Retention Serves as Molecular Signature that Distinguishes Melanoma from Non-Melanoma Cancer Cells in Greek Patients" International Journal of Molecular Sciences 20, no. 4: 937. https://doi.org/10.3390/ijms20040937