1. Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death worldwide [
1]. The development and progression of HCC is driven by both intrinsic epigenetic alterations and extrinsic tumor/T-cell crosstalk [
2,
3]. Accumulating evidence has demonstrated that tumor immune microenvironment facilitates tumor cells to immune escape by the infiltrating suppressive immune cells [
1,
4]. In recent years, inhibitory checkpoints—including cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), programmed cell death protein-1 (PD-1), and programmed cell death ligand 1 (PD-L1)—have been recognized to suppress anti-tumor immune responses in solid tumors [
5]. Blockade of these immune checkpoints has been translated into effective strategies for cancer immunotherapy. Despite that therapeutic blockade has revealed durable antitumor responses and long-term remissions, immune escape, or resistance to immunotherapy contributes to the relatively low response rates in HCC patients [
2,
6]. Thus, the best candidates that benefit from immune checkpoint therapy are much warranted but still unclear.
It is well known that T cells play a critical role in regulating immune responses and are activated by two classical signals: antigen recognition (signal 1) characterized by T cell receptors (TCRs) engaging major histocompatibility complex (MHC)-embedded antigen peptides (pMHCs) within the contact region; and co-stimulation (signal 2) which requires a combination of several co-regulators consist of both co-stimulatory and co-inhibitory molecules [
7]. Therapeutic modulation of these signal transduction pathways has been translated into effective strategies for cancer immunotherapy [
8]. At present, the co-stimulatory and co-inhibitory pathways mainly contain two major families: the B7 family of immunoglobulins3 and the tumor necrosis factor receptor (TNFR) superfamily [
9,
10].
The B7 family has 10 reported members including CD80 (B7-1), CD86 (B7-2), B7-DC, PD-L1 (B7-H1), B7-H2, B7-H3, B7-H4, B7-H5, B7-H6, and B7-H7 [
9]. Moreover, CD80 (B7-1), B7-2 (CD86)/CTLA-4, and B7-H1 (PD-L1)/PD-1 have been identified as promising targets and their inhibitors have achieved great success in cancer immunotherapy [
11]. The TNFR superfamily proteins are expressed by antigen-presenting cells (APC) or tumor cells, including TNFSF4 (OX40L), TNFRSF5 (CD40), TNFSF7 (CD70), TNFSF9 (CD137L), TNFRSF14 (HVEM), and TNFSF18 (GITRL) [
10]. TNFR superfamily proteins function as secondary T cell activation signals and may serve as novel candidates in cancer immunotherapy. However, the systemic alterations of these families have not been defined in HCC.
The B7 and TNFR family members and their receptors exhibit great potential as therapeutic targets in HCC immunotherapy. In this study, we elucidate comprehensive molecular profiling of the 10 B7 and 6 TNFR family genes across 6 HCC studies by TCGA data from cBioPortal for Cancer Genomics. Herein, we analyzed the mutation, amplification, copy number data, mRNA dysregulation, DNA methylation and the relevant clinical profiles to understand the molecular landscape and immunotherapeutic implications of the B7 and TNFR families in HCC. By bioinformatics, we focused on B7-H6 and further investigated the effect of B7-H6 on the metastasis and prognosis of HCC, aiming to provide novel efficacious therapeutic targets that stratify patients responsive to HCC immunotherapy.
3. Discussion
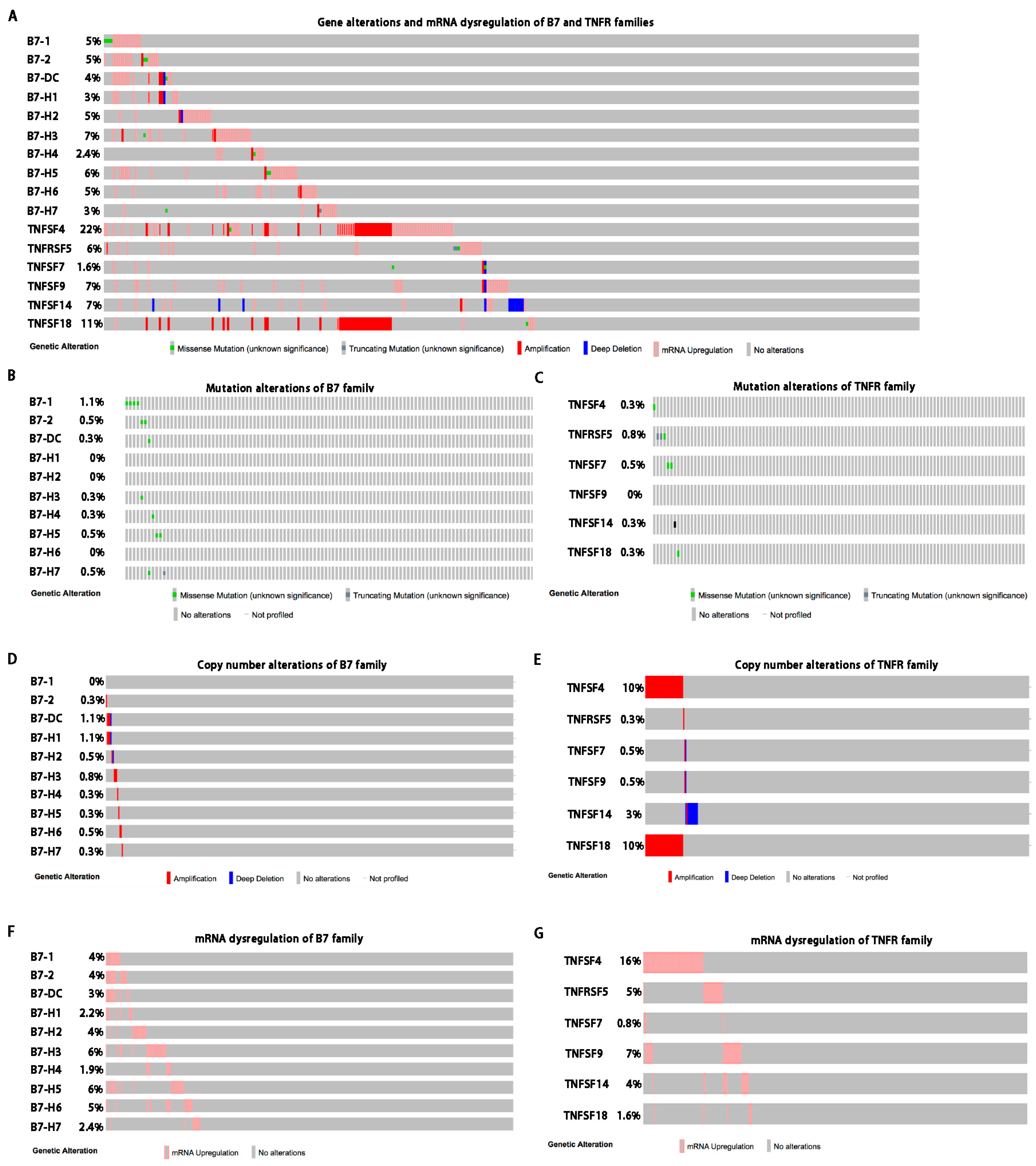
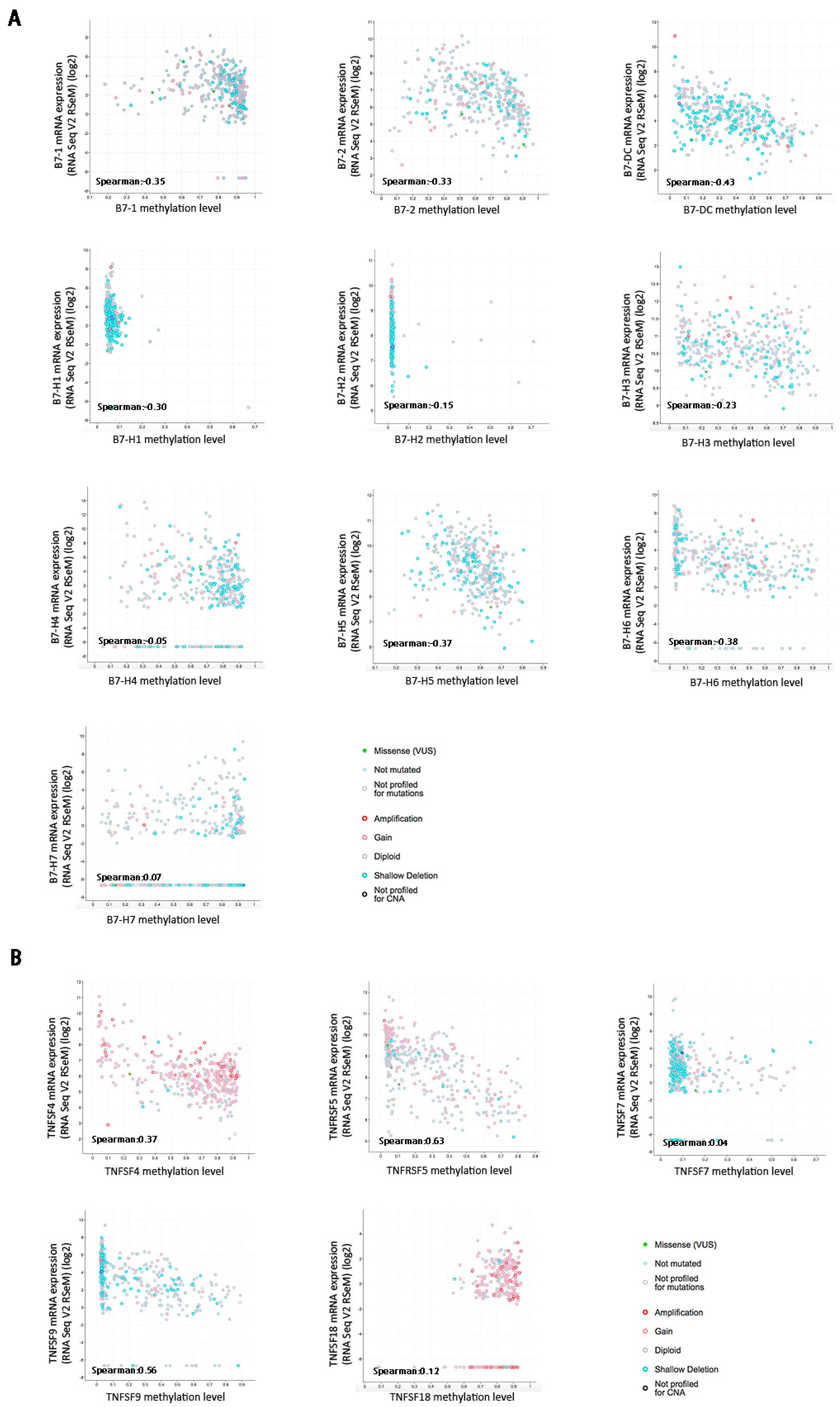
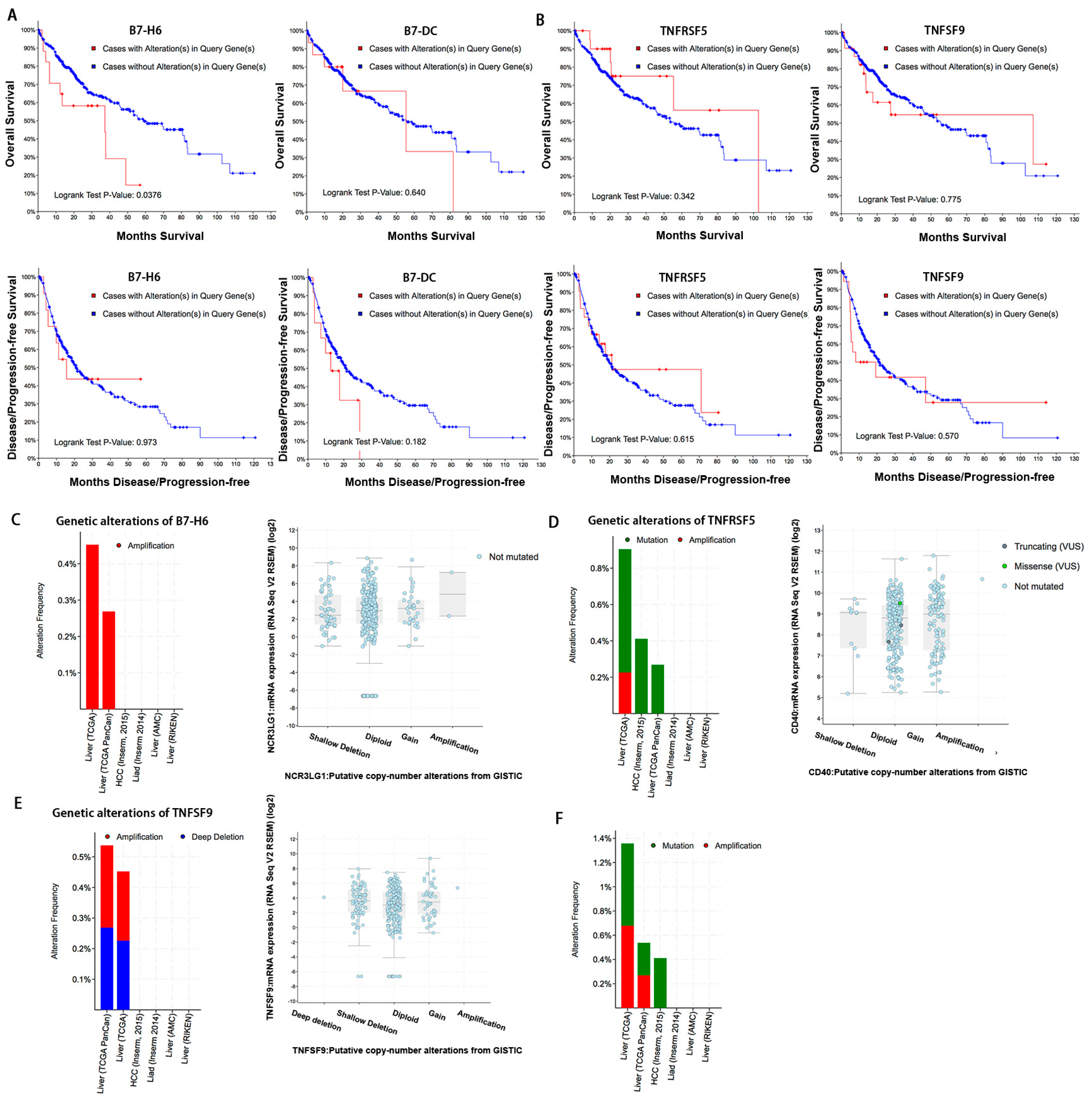
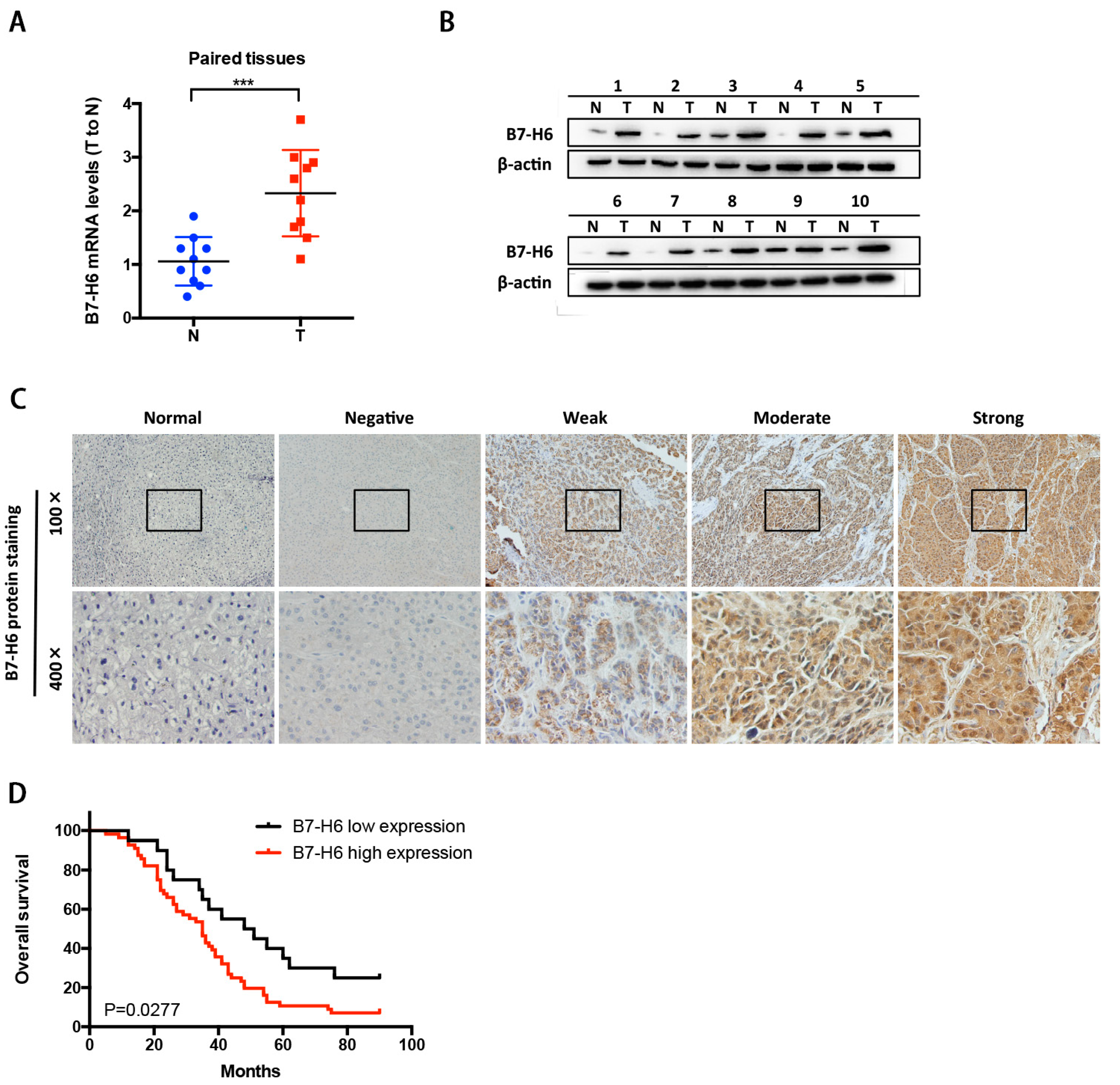
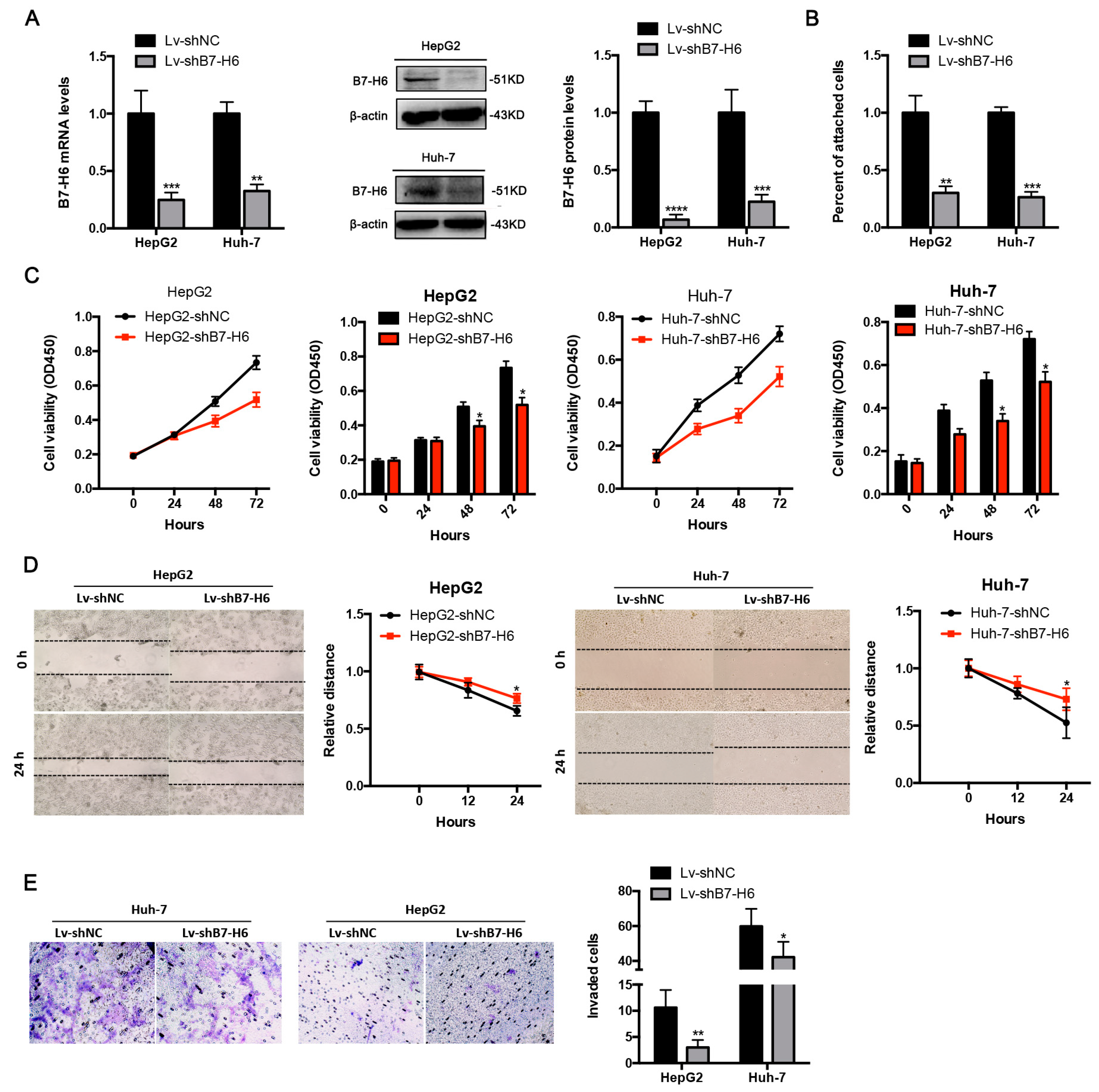
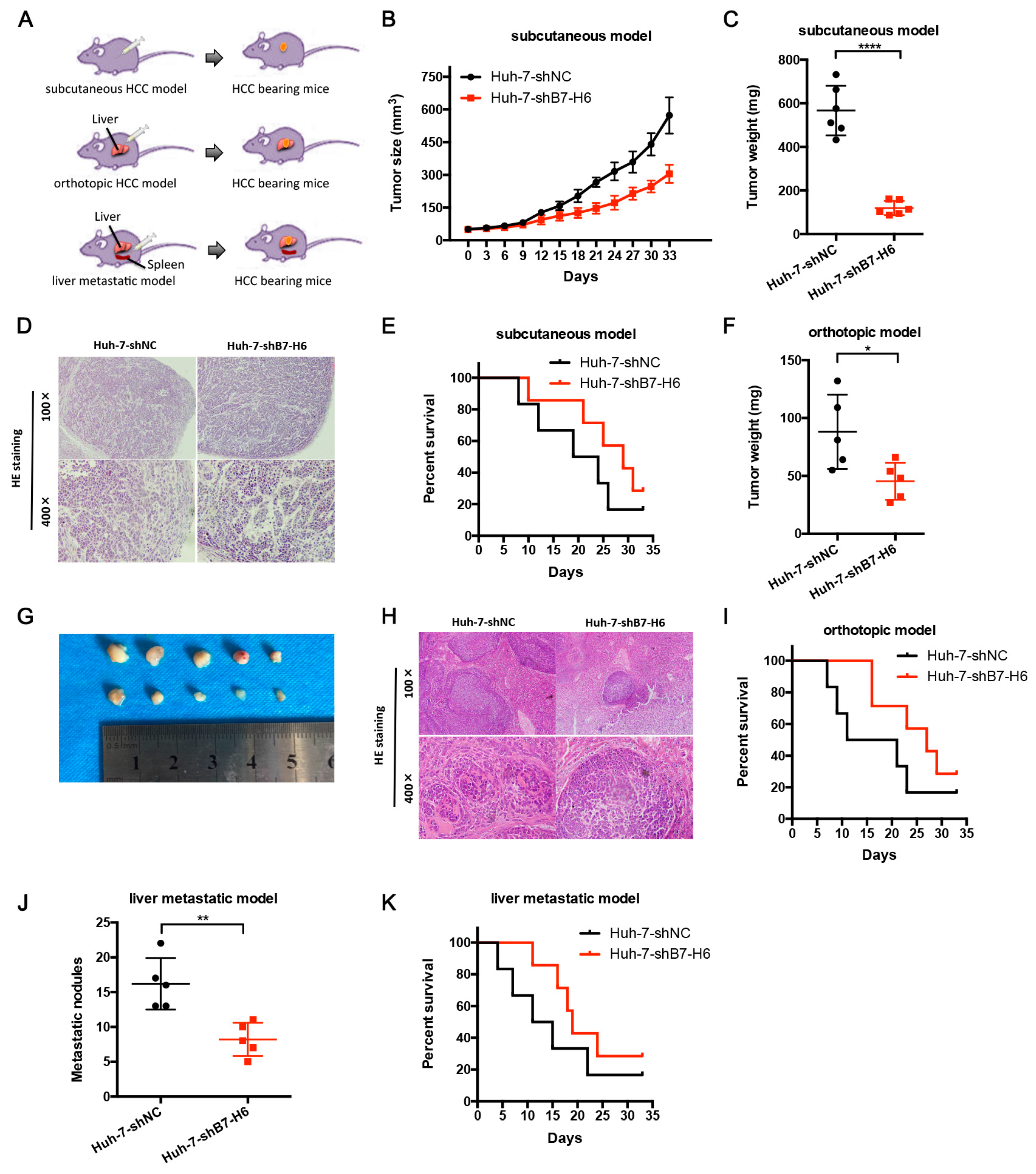
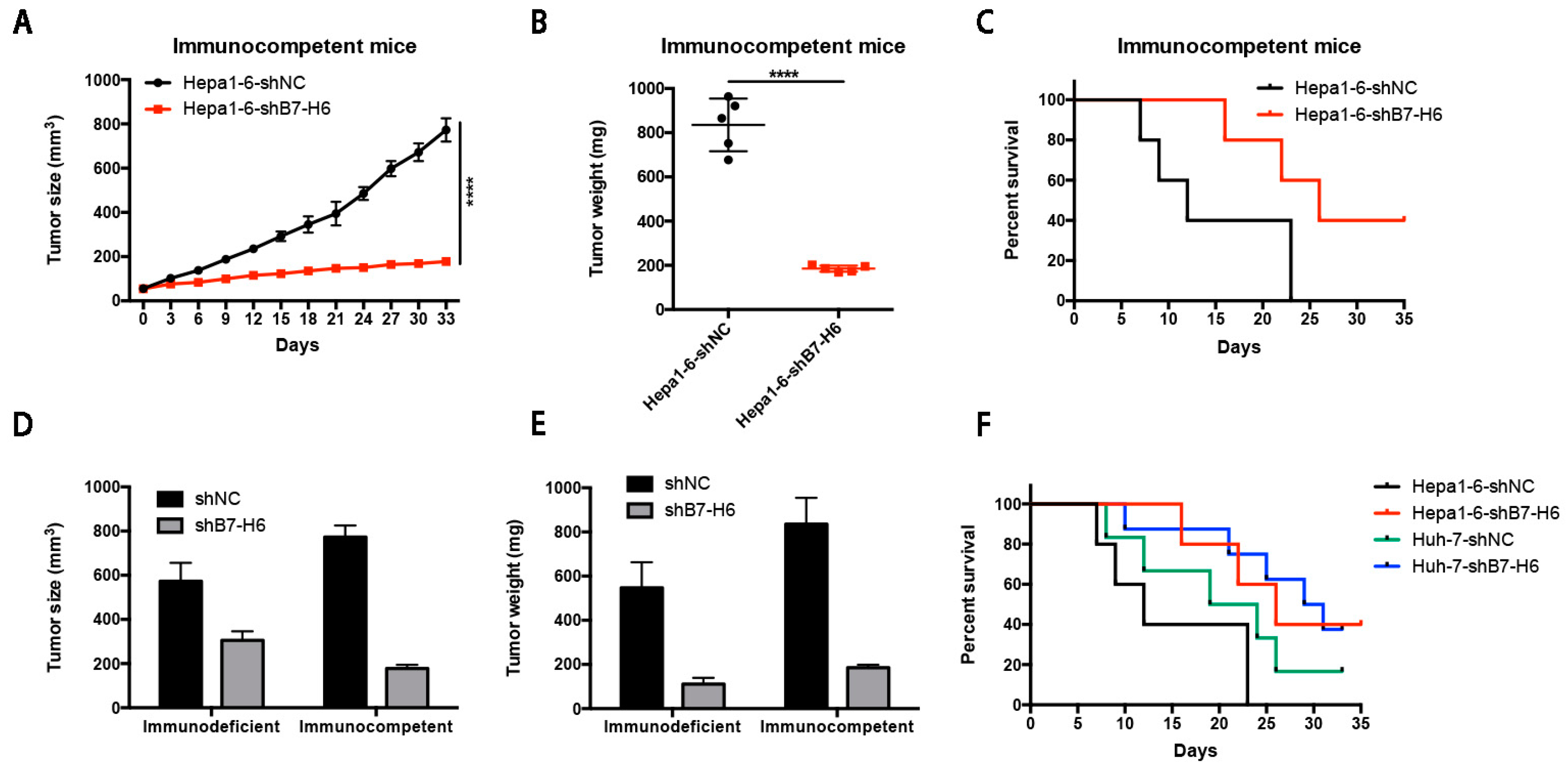
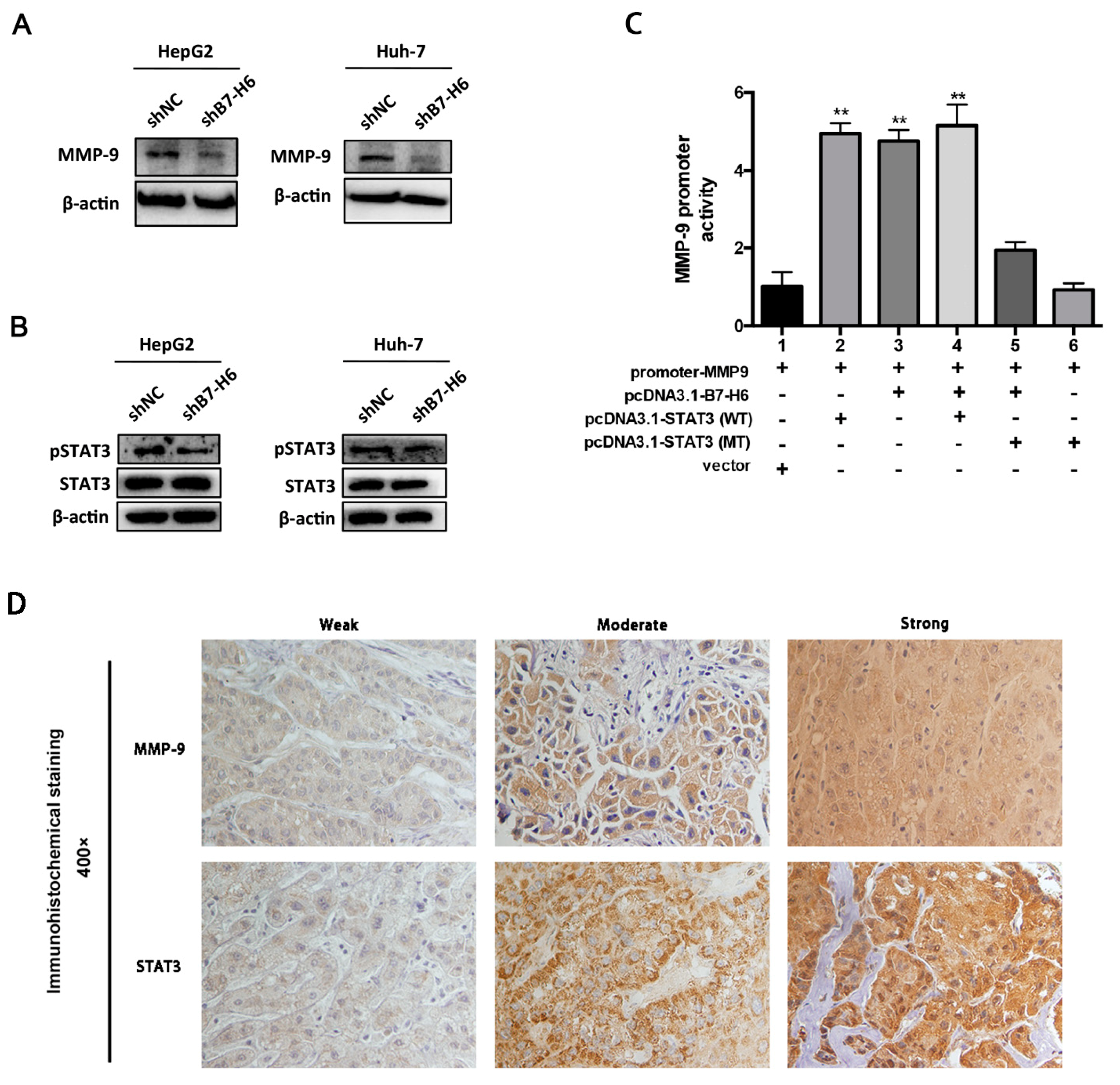
In cancer, dysregulation of co-inhibitory B7 and TNFR family molecules is associated with attenuated anti-tumor immunity and enhanced immune evasion. In this study, we portrayed the comprehensive perspectives and clinical relevance of immune co-stimulator B7 and TNSF family genes in HCC. Given the promising results from clinical trials assessing the immunotherapy of advanced or treatment-refractory cancer with immune checkpoint inhibitors such as anti-PD-1/PD-L1, the B7 and TNFR family members may be closely evaluated as potential immunotherapeutic targets in HCC. Here, we provide a systematic overview of 10 B7 family members and 6 TNFR family members respectively, under a unified nomenclature according to previous studies. Both mutations and CNAs were observed in B7 and TNFR family genes. Intriguingly, we found that their mRNA dysregulation was more common and associated with promoter DNA methylation. Furthermore, the alterations of B7-H6 were correlated with unfavorable overall survival, which may serve as a promising candidate for HCC immunotherapy. We found that B7-H6 overexpression was associated with aggressive clinicopathologic features and poor prognosis in HCC. Downregulation of B7-H6 in HCC cells significantly inhibited cell adhesion, proliferation, migration, and invasion. Knockdown of B7-H6 in HCC cells inhibited tumor growth and metastasis in vivo. Furthermore, B7-H6 promtes MMP-9 expression and STAT3 activation in HCC cells. B7-H6 and STAT3 perform functional overlapping roles on enhancing the MMP-9 promoter activity in HCC. Hence, B7-H6 might be a potential target for HCC treatment.
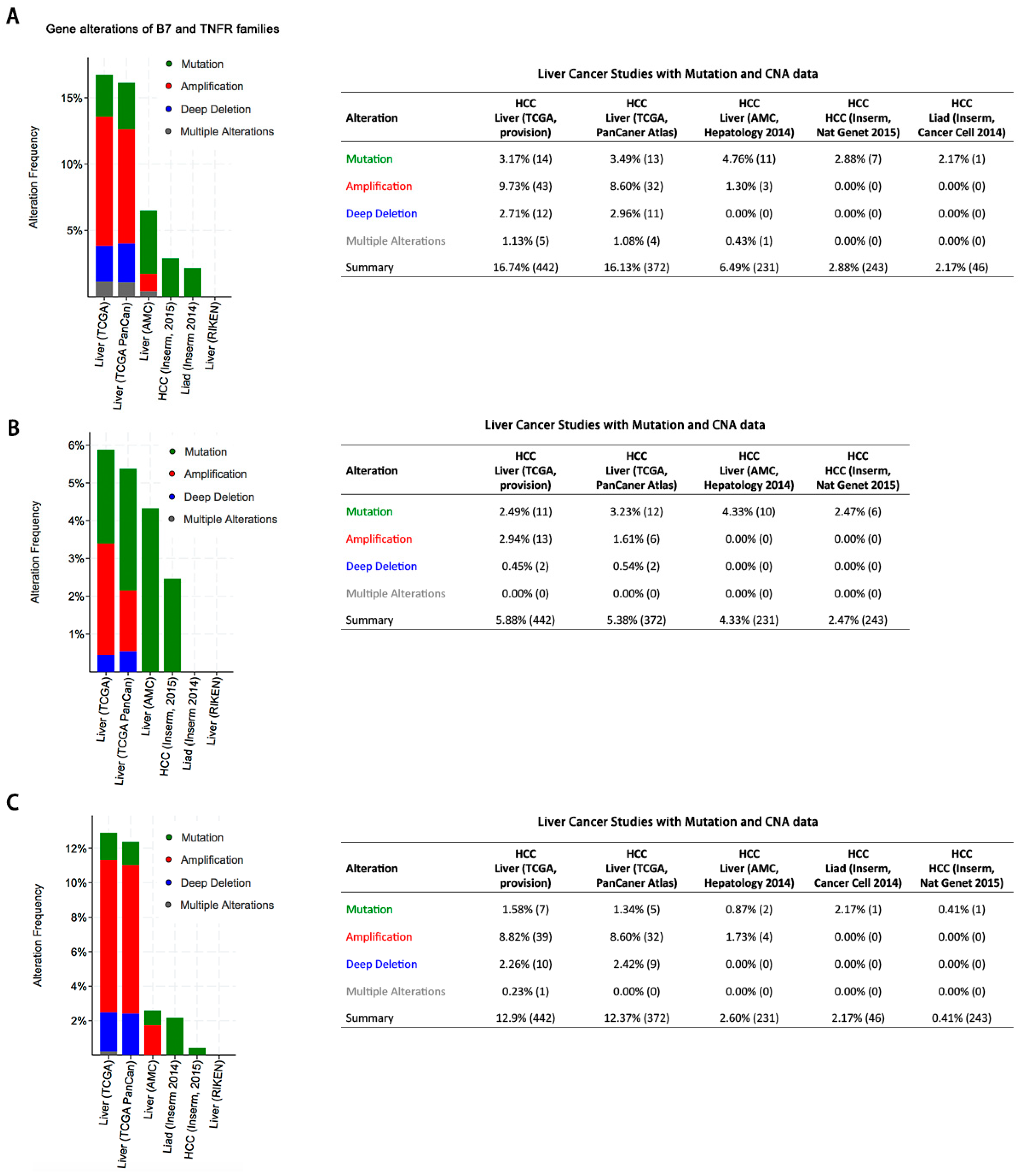
Despite that checkpoint blockade has provided prolonged clinical benefits, but this benefit remains limited to only a small subset of patients [
14,
15,
16]. Therefore, it has been much warranted to explore novel and effective candidates for cancer immunotherapy. In this study, we performed a comprehensive genetic analysis of B7 and TNFR family genes and found that both of the family genes exhibited mutation, amplification or deletion in HCC, which is consistent with previous studies in breast cancer, colorectal cancer, and head and neck cancer [
17,
18,
19]. Overall, both B7 and TNFR gene alterations were observed in about 16% of patients and might contribute to activating or inhibiting immune response in HCC, although the mutation and CNA rates were relatively low. Besides, we found that the B7-H6 alterations were associated with poor overall survival, demonstrating its potential role as a predictive biomarker of immunotherapy regimens. Taken together, our findings suggested that targeting B7-H6 might represent an attractive and promsing therapeutic approach against HCC.
The B7 family and their cognate receptors are the major drivers of T cell co-stimulated activation and co-inhibited inactivation. In addition to the B7 family, the TNFR superfamily members possess the indispensible properties for immune regulation in cancer. Based on the fundamental mechanisms of the TNFR family members, the translational investigation has brought about the research and development of new therapeutic immune agents besides CTLA-4 and PD-L1/PD-1 antibodies, such as dacetuzumab (humanized TNFRSF5 monoclonal antibody) and urelumab (fully human TNFRSF9 monoclonal antibody) [
8]. Various clinical trials have demonstrated that blockade of TNFRSF9 altered the tumor microenvironment and minimized systemic exposure [
20]. However, we found that alterations in the six TNFR family members were relatively less common and were not significantly correlated with prognosis. We speculated that the role of the TNFR family in anti-tumor immunity might not be as important as that of the B7 family in HCC. Further basic studies are warranted to explore the therapeutic value of the TNFR family members.
The limitation of this study should be acknowledged. First, although the the data from cBioPortal for Cancer Genomics enrolled large-scale cancer genomic projects, more specialized and representative cohorts should be conducted to illustrate the landscape of the B7 and TNFR families in HCC. Second, with the exception of B7-H6, the protein expression of other B7 and TNFR family genes was not validated by immunohistochemistry in HCC specimens. Moreover, more large-scale prospective studies and multicenter clinical trials are much warranted to further validate our findings. Nevertheless, this study steps forward to establishing the comprehensive genetic and immune landscape of the B7 and TNFR families in HCC, and these findings could help provide potential candidates to stratify patients based on response to immune checkpoint therapy.
In conclusion, our study provided a systematic analysis of B7 and TNFR family gene alterations in HCC. Notably, B7-H6 alteration is significantly associated with poor overall survival, and its mRNA dysregulation plausibly correlates to both gene amplification and DNA methylation. B7-H6 overexpression was associated with aggressive clinicopathologic features and poor prognosis in HCC. Downregulation of B7-H6 in HCC cells significantly inhibited cell adhesion, proliferation, migration, and invasion. Knockdown of B7-H6 in HCC cells inhibited tumor growth and metastasis in vivo. To our knowledge, this is the first study on the immune molecular landscape of the pivotal B7 and TNFR families in HCC, thereby facilitating the development of novel promising immunotherapy against HCC.
4. Materials and Methods
4.1. Cell Culture and Gene Silencing
HepG2, Huh-7, Hepa1-6, and HEK-293 cells were purchased from Institute of Cell Biology (Shanghai, China). Cells were cultured in RPMI 1640 medium (11875-093, Gibco, Waltham, MA, USA), containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and 2% L-glutamine at 37 °C in a humidified atmosphere of 5% CO2. Lentivirus was used to establish stable B7-H6 knockdown HCC cells, namely shNC and shB7-H6 cells, respectively. The shNC cells were used as control mock.
4.2. Patients and Specimens
A total of 74 cases of human HCC tissuses from patients who underwent surgical dissections were enrolled in this study, and were all diagnosed with HCC. The surgical samples were supplied by the Xijing Hospital at the Fourth Military Medical University. Histopathological sections of each patient by HE staining were reviewed by two independent pathologists to evaluate the grades and TNM stages. The TNM stages were assessed based on the American Joint Committee on Cancer (AJCC) TNM classification for HCC. Tumor differentiation grading was classified by the Edmondson grading system. Eligible patients in this study were those with histopathological confirmation, sufficient samples for IHC staining and qRT-PCR, and complete clinical and outcome statistics. Through screening electronic database by follow-up, we retrospectively collected those patients with adequate follow-up information. Overall survival (OS) rates were calculated from the date of surgery to death or until the last follow-up (censored).
4.3. Integrative Alteration Analysis of B7 and TNFR Family Members
Integrative alteration analysis of B7 and TNFR family members on HCC was conducted using the cBioPortal for Cancer Genomics database and TCGA with more than 1000 patients’ details [
21]. Six large-scale studies were enrolled to assess the frequency of B7 and TNFR gene alterations including mutations, deletions, and amplifications. Mutations included missense mutations and truncating mutations. Whole-exome sequencing was utilized to identify gene mutations. Mutations were validated by targeted re-sequencing and interrogation of RNA for expression of the mutated alleles, which included truncating, inframe, and missense mutations. Missense mutations are point mutations changing a single nucleotide that results in the substitution of a different amino acid and a nonfunctional protein. Truncating mutations are point mutations in the genetic code that generate one stop codon and interrupt protein translation. Any insertion or deletion that is evenly divisible by three is termed an inframe mutation, which would not disrupt the reading frame, given the triplet nature of gene expression by codons. Furthermore, we evaluated the genomic alterations including mRNA dysregulation and promoter methylation of both families across 371 sequenced HCC samples with complete TCGA data, as queried with cBioPortal. All mRNA data were assayed by mRNA-seq and gene expression values were represented as RNA-Seq by expectation maximization (RSEM) data normalized within each sample to the upper quartile of total reads [
22].
4.4. Association of B7 and TNFR Families with Prognosis in HCC Patients
The clinicopathological information and survival data were obtained from the cBioPortal for Cancer Genomics database and TCGA. B7 and TNFR family members’ mRNA dysregulations were assessed to correlate with the prognosis of patients with HCC. We performed receiver operating characteristic (ROC) curve analysis to determine cut-off value of mRNA.
4.5. Immunohistochemistry (IHC)
4µm thick sections were deparaffinized, followed by antigen retrieval with 10 mmol/L citrate buffer (ZLI-9605, Zhongshan Jinqiao Co., Beijing, China) at pH6.0. Endogenous peroxidase activity was blocked with methanol containing 3% hydrogen peroxide for 15 min. After serum block, sections were incubated with anti-B7-H6 (ab121794, Abcam, Cambridge, MA, USA), anti-MMP9 (ab38898, Abcam, Cambridge, MA, USA), anti-STAT3 (ab119352, Abcam, Cambridge, MA, USA) at 4 °C overnight. Following incubation, immunoperoxidase staining was carried out by a streptavidin-peroxidase-3,3′-diaminobenzidine kit (SPN-9001, Zhongshan Jinqiao Co., Beijing, China) to visualize the target proteins. Hematoxylin was utilized to counterstain the nuclei. Isotype control and negative control were used to evaluate specificity of all antibodies. Two independent pathologists who were blinded to the experiment evaluated the sections. Major evaluation parameters include the intensity and density of positive cells.
4.6. Real-Time Quantitative PCR and Data Analysis
RT-PCR was conducted to confirm the knockdown of B7-H6 expression at the mRNA level in HepG2 and Huh-7 cells. Total RNA was extracted by utilization of Total RNA Kit II (R6934-01, Omega Bio-tek, Norcross, GA, USA) following manufacturer’s instructions, and then reversely transcribed to cDNA by use of the PrimeScript RT Reagent Kit (TaKaRa). The SYBR Premix Ex Taq II Kit (RR037A, TaKaRa) was used in qRT-PCR. Human GAPDH mRNA was used as an internal control. PCR Primers were synthesized as follows: GAPDH forward primer, 5′- CTGGGCTACACTGAGCACC-3′, and reverse primer, 5′-AAGTGGTCGTTGAGGGCAATG-3′; B7-H6 forward primer, 5′-CTCCTGATTCTGCT GTGGGC-3′, and reverse primer, 5′-GTCGGAATGCCTCTTGGTGA -3′. Real-time quantitative PCR was performed with a Stratagene M × 3005P Multiplex Quantitative PCR System (Agilent Technologies, Santa Clara, CA, USA).
4.7. Western Blot Analysis
Cells were harvested and then treated with RIPA lysis buffer containing phenylmethylsulfonyl floride (KGP610, KeyGen BioTech, Nanjing, China), protease inhibitors (KGP603, KeyGen BioTech, Nanjing, China) and phosphatase inhibitors (KGP602, KeyGen BioTech, Nanjing, China). Lysates were incubated on ice for 30 min, centrifuged (14,000 rpm, 20 min, 4 °C) and were quantified by the BCA Protein Assay Kit (23225, Thermo Fisher Scientific, Waltham, MA, USA). The supernatants were subjected to the subsequent SDS-PAGE. Protein samples were boiled for 5 min and then loaded to 12% SDS-PAGE gel by equal amounts. Gels were transferred to PVDF membranes (IPVH00010, Millipore, Temecula, CA, USA). After blockade in 5% non-fat milk in TBST (50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20) for 1 h, the membrane was incubated with the designated primary antibodies (anti-B7-H6 ab121794, anti-MMP9 ab38898, anti-STAT3 ab119352, anti-pSTAT3 ab76315, Abcam; anti-β-actin, M1210-2, HuaBio, Hangzhou, China) at 4 °C overnight. The images were developed after incubation with the secondary antibodies at room temperature for 1 h. Bands were visualized by Chemidoc Touch (Bio-Rad, Hercules, CA, USA).
4.8. Cell Adhesion Assay
96-well plates were coated 100 μL Matrigel (354234, BD Biosciences, San Jose, CA, US) per well at 4 °C overnight. The plates were washed twice with PBS and blocked with 2% BSA for 30 min at 37 °C. HCC cells transfected with shB7-H6 or shNC lentivirus were collected and added into each gel-coated well and incubated for 1 h at 37 °C in 5% CO2. Unattached cells were gently washed away with PBS. The attached cells were fixed with 4% formaldehyde, stained with 0.2% crystal violet for 20 min, and destained with distilled water. After exposure in air for 24 h, solubilized in lysis buffer for 30 min and quantified by the microplate reader (BioTek, Winooski, VT, USA).
4.9. Cell Proliferation Assay
HCC cells transfected with shB7-H6 or shNC lentivirus were seeded into 96-well plates at a density of 4 × 104 cells per well. Cell Counting Kit-8 (CCK-8, C0038, Beyotime, Beijing, China) was used to determine the OD value at a wavelength of 450 nm after 1 h according to the manufacturer’s instructions. Cell proliferation was examined every 24 h over a period of three days. All experiments were performed in triplicate, and the mean proliferation rate was adopted.
4.10. Wound Healing Assay
HCC cells transfected with shB7-H6 or shNC lentivirus were cultured in 6-well plates. A straight wound area was created using a 100-μL pipette tip when cells reached a confluence of 90–100%. Then the scraped cells were washed with PBS twice and cultured in fresh serum-free RPMI 1640 medium (Gibco, Waltham, MA, USA), at 37 °C in a humidified atmosphere of 5% CO2. Images were acquired at 0, 12 and 24 h for at least five non-overlapping fields. All experiments were performed in triplicate, and the mean proliferation rate was adopted.
4.11. Transwell Invasion Assay
The upper chamber of Transwell inserts (8 μm) was coated with 100 μL of Matrigel (diluted 1:4 in serum-free RPMI 1640 medium) and was incubated at 37 °C for 2 h. For invasion assay, 3 × 105 cells transfected with shB7-H6 or shNC in 200 μL serum-free RPMI 1640 medium were added to the upper chamber of the coated inserts (Merck Millipore, Temecula, CA, USA) of the 24-well plates with 500 μL of RPMI 1640 medium containing 10% FBS in the bottom chamber. After incubation at 37 °C for 24 h, the non-invading cells were removed slightly from the upper chamber with a cotton-tipped swab. The inserts were rinsed with PBS and the migrated cells were fixed with 95% ethanol with 0.2% crystal violet for 30 min. The invaded cells were counted in five randomly selected fields. All experiments were performed in triplicate, and the mean proliferation rate was adopted.
4.12. Subcutaneous HCC Model in Nude Mice
For subcutaneous HCC model, 5 × 106 Huh-7-shB7-H6 or Huh-7-shNC cells were injected into the left flanks of BALB/C nude mice. Tumors were measured with a caliper and calculated with the formula length × width × depth × 0.52 (cubic millimeters). The volume of the tumor was measured every third day for 33 days. The survival time was also calculated for each mouse. Then mice were sacrificed, and their tumors were harvested and weighed. Livers and tumors were dissected, fixed with 10% formalin and washed with 75% ethanol. Then the tissues were prepared for standard histological examination. Animal studies were approved by the Laboratory Animal Research Center of the Fourth Military Medical University.
4.13. Orthotopic HCC Model in Nude Mice
For orthotopic implantation, 5 × 106 Huh-7-shB7-H6 or Huh-7-shNC cells were injected into the left lobe of the livers of BALB/c nude mice via a microsyringe through an 8-mm transverse incision under anesthesia. The survival time was calculated for 33 consecutive days. Then mice were sacrificed, and their livers and tumors were dissected, fixed with 10% formalin and washed with 75% ethanol. Then the tissues were prepared for standard histological examination.
4.14. Metastatic HCC Model in Nude Mice
For establishement of metastatic HCC model, 5 × 106 Huh-7-shB7-H6 or Huh-7-shNC cells were injected into the spleens of BALB/c nude mice via a microsyringe through an 8-mm transverse incision under anesthesia. The survival time was calculated for 33 consecutive days. Then mice were sacrificed, and their livers were dissected, fixed with 10% formalin and washed with 75% ethanol. Then the tissues were prepared for standard histological examination.
4.15. Subcutaneous HCC Model in Immune Competent Mice
For subcutaneous HCC model in immune competent mice, 2 × 106 Hepa1-6-shB7-H6 or Hepa1-6-shNC cells were injected into the left flanks of C57BL/6 mice. Tumors were measured with a caliper and calculated with the formula length × width × depth × 0.52 (cubic millimeters). The volume of the tumor was measured every third day for 33 days. The survival time was also calculated for each mouse. Then mice were sacrificed, and their tumors were harvested and weighed. Livers and tumors were dissected, fixed with 10% formalin, and washed with 75% ethanol. Then the tissues were prepared for standard histological examination.
4.17. Dual-Luciferase Reporter Assay
Huh-7 and 293T cells were transfected with firefly luciferase reporter gene constructs and hRluc-CMV vector (Promega, Madison, WI, USA). The MMP-9 promoter region was cloned by gene synthesis in according with coding sequence and was inserted into pGL4.10 luciferase vector (Promega). The MMP-9 promoter-reporter construct was then transfected into Huh-7 and 293T cells alone or in combination with the following vectors by using Lipofectamine 2000 (11668019, Invitrogen) as recommended by the manufacturer: B7-H6-pcDNA3.1, STAT3-pcDNA3.1 (Wide Type, WT), STAT3-pcDNA3.1 (Mutation Type, MT), and Vector-pcDNA3.1. In each transfection system, an empty vector was added to the reaction mixture to normalize the amount of total DNA. At 48 h post-transfection, Firefly and Renilla luciferase intensities were measured using the Dual-Luciferase Reporter Assay System (E1980, Promega) according to the manufacturer.
4.18. Statistics
The statistical analyses were performed by SPSS 23.0 (SPSS, Chicago, IL, USA) and GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). Student’s t-test (two-tailed) was used to compare the means between two groups. The Pearson chi-square test was used to compare categorical variables, and Fisher’s exact test was used if indicated. The Spearman correlation test was conducted to assess the relationship between promoter methylation and mRNA expression. Overall survival was calculated using Kaplan–Meier analysis and compared by the log-rank test. The mRNA expression data were presented as mean ± SD. p values less than 0.05 were considered statistically significant (two-tailed).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}