Complete Genome Sequence and Comparative Analysis of Synechococcus sp. CS-601 (SynAce01), a Cold-Adapted Cyanobacterium from an Oligotrophic Antarctic Habitat




Abstract
:
1. Introduction
2. Results and Discussion
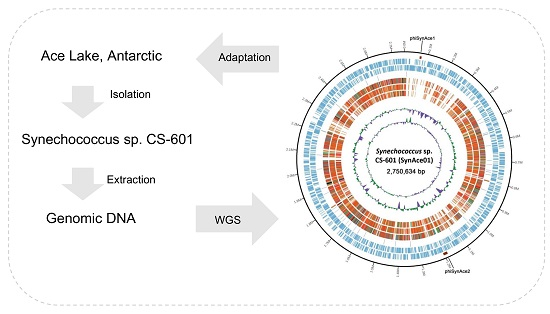
2.1. General Features of Synechococcus sp. CS-601 (SynAce01) Genome
2.2. Mobile Genetic Elements
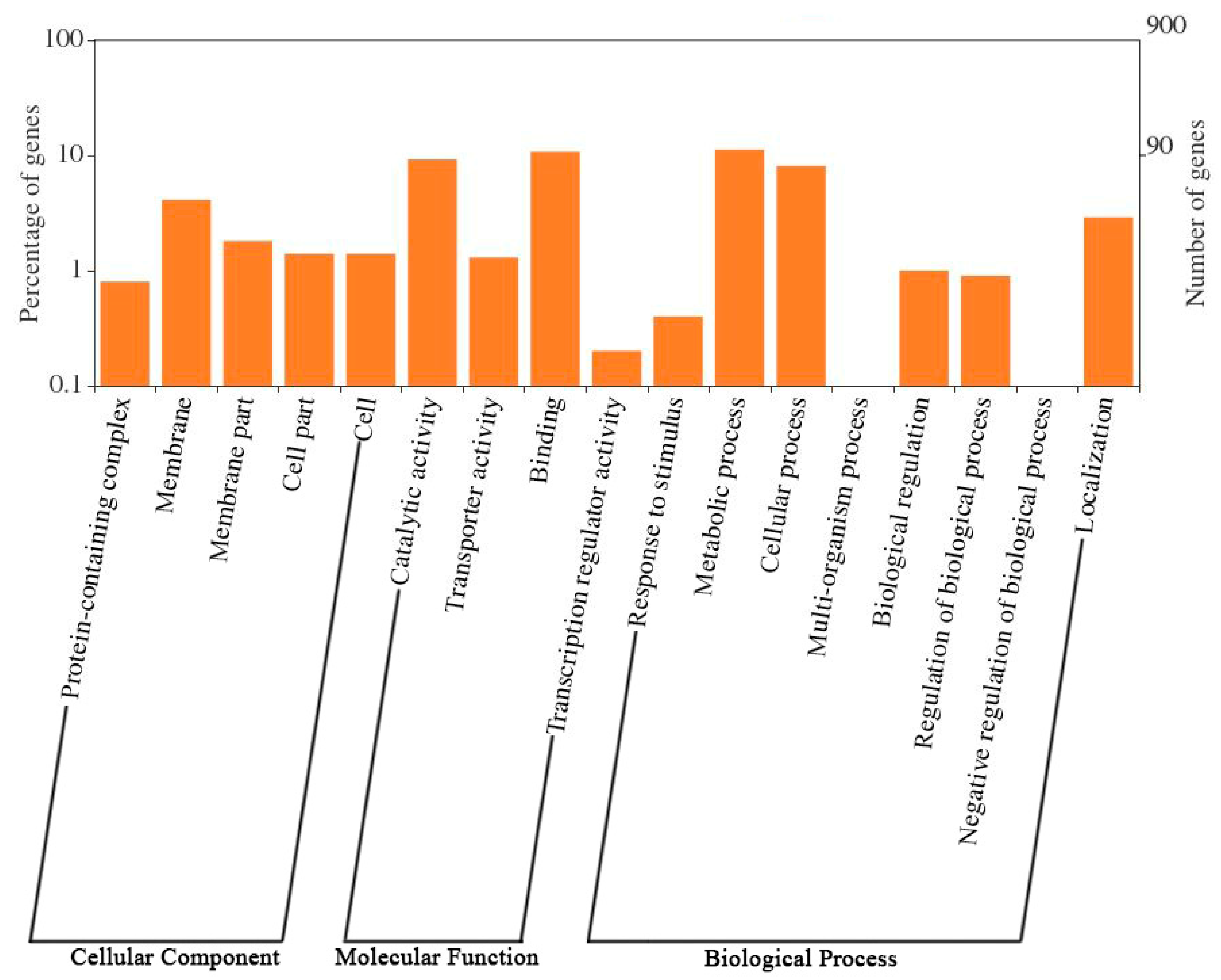
2.3. Gene Contents in Synechococcus sp. CS-601 (SynAce01)
2.4. Cold Adaptation Strategy
2.5. Strategy for Living in Oligotrophic Environment
2.6. Salinity Adaptation Strategy
2.7. Phylogenetic Analysis
3. Conclusion
4. Materials and Methods
4.1. Bacterial Strain and DNA Extraction
4.2. Genome Sequencing and De Novo Assembly
4.3. Genome Annotation
4.4. Comparative Genome Analysis
4.5. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pittera, J.; Humily, F.; Thorel, M.; Grulois, D.; Garczarek, L.; Six, C. Connecting thermal physiology and latitudinal niche partitioning in marine Synechococcus. ISME J. 2014, 8, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Buitenhuis, E.T.; Li, W.K.W.; Vaulot, D.; Lomas, M.W.; Landry, M.R.; Partensky, F.; Karl, D.M.; Ulloa, O.; Campbell, L.; Jacquet, S. Picophytoplankton biomass distribution in the global ocean. Earth Syst. Sci. Data 2012, 5, 301–315. [Google Scholar] [CrossRef]
- Pittera, J.; Partensky, F.; Six, C. Adaptive thermostability of light-harvesting complexes in marine picocyanobacteria. ISME J. 2016, 11, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.M.; Castenholz, R.W. Cyanobacteria in geothermal habitats. In The Ecology of Cyanobacteria; Springer: Dordrecht, The Netherlands, 2000; pp. 37–59. [Google Scholar]
- Tang, J.; Jiang, D.; Luo, Y.; Liang, Y.; Li, L.; Shah, M.M.R.; Daroch, M. Potential new genera of cyanobacterial strains isolated from thermal springs of western Sichuan, China. Algal Res. 2018, 31, 14–20. [Google Scholar] [CrossRef]
- Powell, L. Ecology of a novel Synechococcus clade occurring in dense populations in saline Antarctic lakes. Mar. Ecol. Prog. 2005, 291, 65–80. [Google Scholar] [CrossRef]
- Vincent, W.F.; Bowman, J.P.; Rankin, L.M.; McMeekin, T.A. Phylogenetic diversity of picocyanobacteria in Arctic and Antarctic ecosystems. In Microbial Biosystems: New Frontiers, Proceedings of the 8th International Symposium on Microbial Ecology; Atlantic Canada Society for Microbial Ecology: Halifax, NS, Canada, 2000; pp. 317–322. [Google Scholar]
- Chrismas, N.; Anesio, A.M.; Sánchezbaracaldo, P. The future of genomics in polar and alpine cyanobacteria. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrismas, N.A.M.; Barker, G.; Anesio, A.M.; Sánchezbaracaldo, P. Genomic mechanisms for cold tolerance and production of exopolysaccharides in the Arctic cyanobacterium Phormidesmis priestleyi BC1401. BMC Genom. 2016, 17, 533. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.W.; Middendorf, L.R.; Benner, S.A.; Church, G.M.; Harris, T.; Huang, X.; Jovanovich, S.B.; Nelson, J.R.; Schloss, J.A.; Schwartz, D.C. The challenges of sequencing by synthesis. Nat. Biotechnol. 2009, 27, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B. Real-time DNA sequencing from single polymerase molecules. Methods Enzymol. 2010, 472, 431–455. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.S.; Iliopoulos, D. Origins of the E. coli strain causing an outbreak of hemolytic–uremic syndrome in Germany. N. Engl. J. Med. 2011, 187, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Sorenson, J.; Harris, J.B.; Robins, W.P.; Charles, R.C.; Jeancharles, R.R.; Bullard, J.; Webster, D.R.; Kasarskis, A.; Peluso, P. The origin of the Haitian Cholera outbreak strain. N. Engl. J. Med. 2011, 364, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Klammer, A.A.; Robins, W.P.; Chin, C.S.; Webster, D.; Paxinos, E.; Hsu, D.; Ashby, M.; Wang, S.; Peluso, P. A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 2012, 30, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoniewski, M.J.; Meienberg, J.; Patrignani, A.; Szabelska, A.; Matyas, G.; Schlapbach, R. Precise breakpoint localization of large genomic deletions using PacBio and Illumina next-generation sequencers. BioTechniques 2013, 54, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Gui, S.; Quan, Z.; Pan, L.; Wang, S.; Ke, W.; Liang, D.; Ding, Y. A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: Insight into the plastid evolution of basal eudicots. BMC Plant Biol. 2014, 14, 289. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, L.E.; Karbysheva, E.A.; Shestakov, S.V. The role of mobile genetic elements in the evolution of cyanobacteria. Russ. J. Genet. 2013, 3, 91–101. [Google Scholar] [CrossRef]
- Palenik, B.; Brahamsha, B.; Larimer, F.W.; Land, M.; Hauser, L.; Chain, P.; Lamerdin, J.; Regala, W.; Allen, E.E.; Mccarren, J. The genome of a motile marine Synechococcus. Nature 2003, 424, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 2008, 322, 1843–1845. [Google Scholar] [CrossRef]
- Blindauer, C.A. Zinc-handling in cyanobacteria: An update. Chem. Biodivers. 2008, 5, 1990–2013. [Google Scholar]
- Saita, E.; Albanesi, D.; De, M.D. Sensing membrane thickness: Lessons learned from cold stress. Biochim. Biophys. Acta 2016, 1861, 837. [Google Scholar] [CrossRef] [PubMed]
- Barria, C.; Malecki, M.; Arraiano, C.M. Bacterial adaptation to cold. Microbiology 2013, 159, 2437–2443. [Google Scholar] [CrossRef] [Green Version]
- Los, D.A.; Murata, N. Responses to cold shock in cyanobacteria. J. Mol. Microbiol. Biotechnol. 1999, 1, 221–230. [Google Scholar] [PubMed]
- Sinetova, M.A.; Los, D.A. New insights in cyanobacterial cold stress responses: Genes, sensors, and molecular triggers. Biochim. Biophys. Acta 2016, 1860, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Kappell, A.D.; van Waasbergen, L.G. The response regulator RpaB binds the high light regulatory 1 sequence upstream of the high-light-inducible hliB gene from the cyanobacterium Synechocystis PCC 6803. Arch. Microbiol. 2007, 187, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Hantke, K. Is the bacterial ferrous iron transporter FeoB a living fossil? Trends Microbiol. 2003, 11, 192–195. [Google Scholar] [CrossRef]
- Pereira, S.B.; Mota, R.; Vieira, C.P.; Vieira, J.; Tamagnini, P. Phylum-wide analysis of genes/proteins related to the last steps of assembly and export of extracellular polymeric substances (EPS) in cyanobacteria. Sci. Rep. 2015, 5, 14835. [Google Scholar] [CrossRef] [Green Version]
- Tuominen, I.; Tyystjärvi, E.; Tyystjärvi, T. Expression of primary sigma factor (PSF) and PSF-like sigma factors in the cyanobacterium Synechocystis sp. strain PCC 6803. J. Bacteriol. 2003, 185, 1116–1119. [Google Scholar] [CrossRef]
- Peters, J.M.; Mooney, R.A.; Grass, J.A.; Jessen, E.D.; Tran, F.; Landick, R. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 2012, 26, 2621–2633. [Google Scholar] [CrossRef]
- Sato, N.; Tachikawa, T.; Wada, A.; Tanaka, A. Temperature-dependent regulation of the ribosomal small-subunit protein S21 in the cyanobacterium Anabaena variabilis M3. J. Bacteriol. 1997, 179, 7063–7071. [Google Scholar] [CrossRef]
- Karzai, A.W.; Susskind, M.M.; Sauer, R.T. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA). Embo J. 1999, 18, 3793–3799. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa, Y.; Tohno, M.; Kaminuma, E.; Nakamura, Y.; Arita, M. Complete genome sequence and analysis of Lactobacillus hokkaidonensis LOOC260T, a psychrotrophic lactic acid bacterium isolated from silage. BMC Genom. 2015, 16, 240. [Google Scholar] [CrossRef] [PubMed]
- Price, G.D.; Badger, M.R.; Woodger, F.J.; Long, B.M. Advances in understanding the cyanobacterial CO2-concentrating-mechanism (CCM): Functional components, Ci transporters, diversity, genetic regulation and prospects for engineering into plants. J. Exp. Bot. 2008, 59, 1441–1461. [Google Scholar] [CrossRef] [PubMed]
- Price, G.D.; Sültemeyer, D.; Klughammer, B.; Ludwig, M.; Badger, M.R. The functioning of the CO2 concentrating mechanism in several cyanobacterial strains: A review of general physiological characteristics, genes, proteins, and recent advances. Can. J. Bot. 1998, 76, 973–1002. [Google Scholar] [CrossRef]
- Maeda, S.I.; Badger, M.R.; Price, G.D. Novel gene products associated with NdhD3/D4-containing NDH-1 complexes are involved in photosynthetic CO2 hydration in the cyanobacterium, Synechococcus sp. PCC7942. Mol. Mircrobiol. 2010, 43, 425–435. [Google Scholar] [CrossRef]
- Shibata, M.; Ohkawa, H.; Kaneko, T.; Fukuzawa, H.; Tabata, S.; Kaplan, A.; Ogawa, T. Distinct constitutive and low-CO2-induced CO2 uptake systems in cyanobacteria: Genes involved and their phylogenetic relationship with homologous genes in other organisms. Proc. Natl. Acad. Sci. USA 2001, 98, 11789–11794. [Google Scholar] [CrossRef] [PubMed]
- Badger, M.R.; Price, G.D. CO2 concentrating mechanisms in cyanobacteria: Molecular components, their diversity and evolution. J. Exp. Bot. 2003, 54, 609–622. [Google Scholar] [CrossRef]
- Price, G.D.; Woodger, F.J.; Badger, M.R.; Howitt, S.M.; Tucker, L. Identification of a SulP-type bicarbonate transporter in marine cyanobacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 18228–18233. [Google Scholar] [CrossRef] [Green Version]
- Omata, T.; Price, G.D.; Badger, M.R.; Okamura, M.; Gohta, S.; Ogawa, T. Identification of an ATP-binding cassette transporter involved in bicarbonate uptake in the Cyanobacterium Synechococcus sp. strain PCC7942. Proc. Natl. Acad. Sci. USA 1999, 96, 13571–13576. [Google Scholar] [CrossRef]
- Bryant, D.A. The Beauty in Small Things Revealed. Proc. Natl. Acad. Sci. USA 2003, 100, 9647–9649. [Google Scholar] [CrossRef]
- Shibata, M.; Katoh, H.; Sonoda, M.; Ohkawa, H.; Shimoyama, M.; Fukuzawa, H.; Kaplan, A.; Ogawa, T. Genes essential to sodium-dependent bicarbonate transport in cyanobacteria: Function and phylogenetic analysis. J. Biol. Chem. 2002, 277, 18658–18664. [Google Scholar] [CrossRef] [PubMed]
- Bonfil, D.J.; Ronen-Tarazi, M.; Sültemeyer, D.; Lieman-Hurwitz, J.; Schatz, D.; Kaplan, A. A putative HCO3− transporter in the cyanobacterium Synechococcus sp. strain PCC7942. FEBS Lett. 1998, 430, 236–240. [Google Scholar] [CrossRef]
- Platt, T.; Li, W.K.W. Photosynthetic Picoplankton; Canadian Bulletin of Fisheries and Aquatic Sciences: Ottawa, ON, Canada, 1986; Volume 214, pp. 71–120. [Google Scholar]
- Dolganov, N.; Grossman, A.R. A polypeptide with similarity to phycocyanin α-subunit phycocyanobilin lyase involved in degradation of phycobilisomes. J. Bacteriol. 1999, 181, 610–617. [Google Scholar] [PubMed]
- Fukaya, F.; Promden, W.; Hibino, T.; Tanaka, Y.; Nakamura, T.; Takabe, T. An mrp-Like cluster in the halotolerant cyanobacterium Aphanothece halophytica functions as a Na+/H+ antiporter. Appl. Environ. Microbiol. 2009, 75, 6626–6629. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, M.; Ghai, R.; Leon, M.J.; Rodríguez-Olmos, Á.; Copa-Patiño, J.L.; Soliveri, J.; Sanchez-Porro, C.; Ventosa, A.; Rodriguez-Valera, F. Genomes of Spiribacter, a streamlined, successful halophilic bacterium. BMC Genom. 2013, 14, 787. [Google Scholar] [CrossRef] [PubMed]
- Mongodin, E.F.; Nelson, K.E.; Daugherty, S.; DeBoy, R.T.; Wister, J.; Khouri, H.; Weidman, J.; Walsh, D.A.; Papke, R.T.; Sanchez Perez, G.; et al. The genome of Salinibacter ruber: Convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc. Natl. Acad. Sci. USA 2005, 102, 18147–18152. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Ishitani, M.; Takabe, T.; Rai, A.K. Synechococcus sp. PCC7942 transformed with Escherichia coli bet genes produces glycine betaine from choline and acquires resistance to salt stress. Plant Physiol. 1995, 107, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Bardavid, R.E.; Khristo, P.; Oren, A. Interrelationships between Dunaliella and halophilic prokaryotes in saltern crystallizer ponds. Extremophiles 2008, 12, 5–14. [Google Scholar] [CrossRef]
- Blank, C.E.; Sánchez-Baracaldo, P. Timing of morphological and ecological innovations in the cyanobacteria—A key to understanding the rise in atmospheric oxygen. Geobiology 2010, 8, 1–23. [Google Scholar] [CrossRef]
- Scanlan, D.J.; Ostrowski, M.; Mazard, S.; Dufresne, A.; Garczarek, L.; Hess, W.R.; Post, A.F.; Hagemann, M.; Paulsen, I.; Partensky, F. Ecological genomics of marine picocyanobacteria. Microbiol. Mol. Biol. Rev. 2009, 73, 249–299. [Google Scholar] [CrossRef]
- Cabelloyeves, P.J.; Haromoreno, J.M.; Martincuadrado, A.B.; Ghai, R.; Picazo, A.; Camacho, A.; Rodriguezvalera, F. Novel Synechococcus genomes reconstructed from freshwater reservoirs. Front. Microbiol. 2017, 8, 1151. [Google Scholar] [CrossRef]
- Haverkamp, T.H.A.; Schouten, D.; Doeleman, M.; Wollenzien, U.; Huisman, J.; Stal, L.J. Colorful microdiversity of Synechococcus strains (picocyanobacteria) isolated from the Baltic Sea. ISME J. 2009, 3, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Callieri, C.; Coci, M.; Corno, G.; Macek, M.; Modenutti, B.; Balseiro, E.; Bertoni, R. Phylogenetic diversity of nonmarine picocyanobacteria. FEMS Microbiol. Ecol. 2013, 85, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosbie, N.D.; Pöckl, M.; Weisse, T. Dispersal and phylogenetic diversity of nonmarine picocyanobacteria, inferred from 16S rRNA gene and cpcBA-intergenic spacer sequence analyses. Appl. Environ. Microbiol. 2003, 69, 5716–5721. [Google Scholar] [CrossRef]
- Vézina, S.; Vincent, W.F. Arctic cyanobacteria and limnological properties of their environment: Bylot Island, Northwest Territories, Canada (73° N, 80° W). Polar Biol. 1997, 17, 523–534. [Google Scholar] [CrossRef]
- Ernst, A.; Becker, S.; Wollenzien, U.I.A.; Postius, C. Ecosystem-dependent adaptive radiations of picocyanobacteria inferred from 16S rRNA and ITS-1 sequence analysis. Microbiology 2003, 149, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iii, A.R.L.; Smith, V.E. Chloroplast pigments of the marine dinoflagellate Gyrodinium resplendens. Lipids 1968, 3, 5–13. [Google Scholar]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Qi, P.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Fang, X.; Yang, H.; Wang, J.; Kristiansen, K.; Wang, J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009, 19, 545–552. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Klimke, W.; Maglott, D.R. NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res. 2009, 37, D32–D36. [Google Scholar] [CrossRef]
- Varani, A.M.; Siguier, P.; Gourbeyre, E.; Charneau, V.; Chandler, M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011, 12, R30. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, 52–57. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; Garcíagómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; Wang, J. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.I.; Schein, J.E.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synechococcus sp. CS-601 (SynAce01) | Synechococcus sp. WH8102 | P. marinus MED4 | S. elongatus PCC7942 | |
|---|---|---|---|---|
| Isolation source | Ace Lake, Antarctic | Sargasso Sea 22.495° N 65.6° W | Eastern Mediterranean Sea 35° N 20°E (approx.) | California USA |
| Habitat | Saline lake | Tropical Seawater | Shallow seawater (depth 5M) high light adapted strain | Freshwater |
| Temperature range | −17–29.5 | 12–30 | 17–27 | 25–45 |
| Genome status | Complete | Complete | Complete | Complete |
| Genome size (bp) | 2,750,634 | 2,434,428 | 1,657,990 | 2.74227 |
| GC content (%) | 63.92 | 59.40 | 30.80 | 55.46 |
| CDSs | 2984 | 2513 | 1790 | 2685 |
| rRNA operons | 2 | 2 | 1 | 2 |
| tRNAs | 44 | 43 | 37 | 44 |
| Other RNA | 4 | 4 | 4 | 4 |
| Accession no. | CP018091 | NC_005070 | NC_005072 | NC_007604 |
| Gene 1 | Description | Number of BLASTP Hits | |||
|---|---|---|---|---|---|
| Synechococcus sp. SynAce01 | Synechococcus sp. WH8102 | P. marinus MED4 | S. elongatus PCC 7942 | ||
| aceF | Pyruvate dehydrogenase E2 component | 1 | 1 | 1 | 1 |
| csp-family | Cold shock protein/cold-inducible RNA chaperone | 2 | 3 | 2 | 3 |
| deaD | DEAD-like RNA helicase | 2 | 2 | 1 | 0 |
| desA | Fatty acid desaturase | 2 | 2 | 2 | 0 |
| dnaA | Replication initiation protein | 1 | 1 | 1 | 1 |
| dnaJ | Molecular chaperone | 8 | 7 | 5 | 5 |
| gyrA | DNA gyrase subunit A | 1 | 2 | 2 | 2 |
| infA | Translation initiation factor IF-1 | 1 | 1 | 1 | 1 |
| infB | Translation initiation factor IF-2 | 1 | 1 | 1 | 1 |
| infC | Translation initiation factor IF-3 | 1 | 1 | 1 | 1 |
| mtnA | Translation initiation factor IF-2B subunit alpha | 1 | 1 | 0 | 1 |
| nusA | Transcription termination factor | 1 | 1 | 1 | 1 |
| pdhA | Pyruvate dehydrogenase E1 subunit alpha | 1 | 1 | 2 | 1 |
| pdhB | Pyruvate dehydrogenase E1 subunit beta | 1 | 1 | 1 | 1 |
| rbfA | Ribosome-binding factor A | 1 | 1 | 1 | 1 |
| recA | Recombination and DNA repair | 1 | 1 | 1 | 1 |
| tig | Protein-folding chaperone | 1 | 1 | 1 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Du, L.-M.; Liang, Y.-M.; Daroch, M. Complete Genome Sequence and Comparative Analysis of Synechococcus sp. CS-601 (SynAce01), a Cold-Adapted Cyanobacterium from an Oligotrophic Antarctic Habitat. Int. J. Mol. Sci. 2019, 20, 152. https://doi.org/10.3390/ijms20010152
Tang J, Du L-M, Liang Y-M, Daroch M. Complete Genome Sequence and Comparative Analysis of Synechococcus sp. CS-601 (SynAce01), a Cold-Adapted Cyanobacterium from an Oligotrophic Antarctic Habitat. International Journal of Molecular Sciences. 2019; 20(1):152. https://doi.org/10.3390/ijms20010152
Chicago/Turabian StyleTang, Jie, Lian-Ming Du, Yuan-Mei Liang, and Maurycy Daroch. 2019. "Complete Genome Sequence and Comparative Analysis of Synechococcus sp. CS-601 (SynAce01), a Cold-Adapted Cyanobacterium from an Oligotrophic Antarctic Habitat" International Journal of Molecular Sciences 20, no. 1: 152. https://doi.org/10.3390/ijms20010152