Protective Effects of Kaempferitrin on Advanced Glycation End Products Induce Mesangial Cell Apoptosis and Oxidative Stress

 ,
,
Abstract
:1. Introduction
2. Results
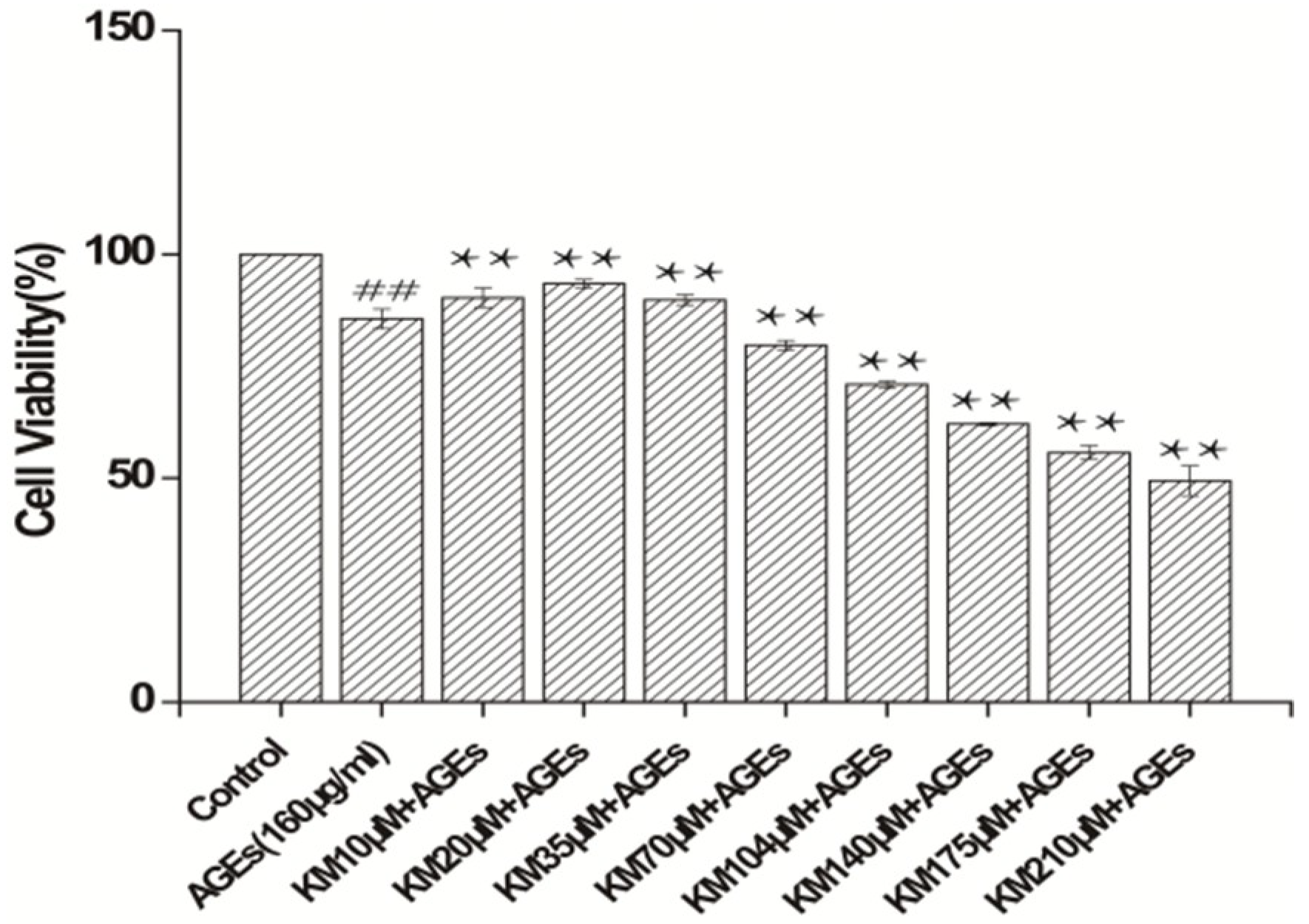
2.1. KM Increases the Viability of AGE-Treated GMCs
2.2. KM Treatment Protects against AGE-Induced GMC Injury
2.3. KM Treatment Reduced the Production of ROS in AGE-Treated GMCs
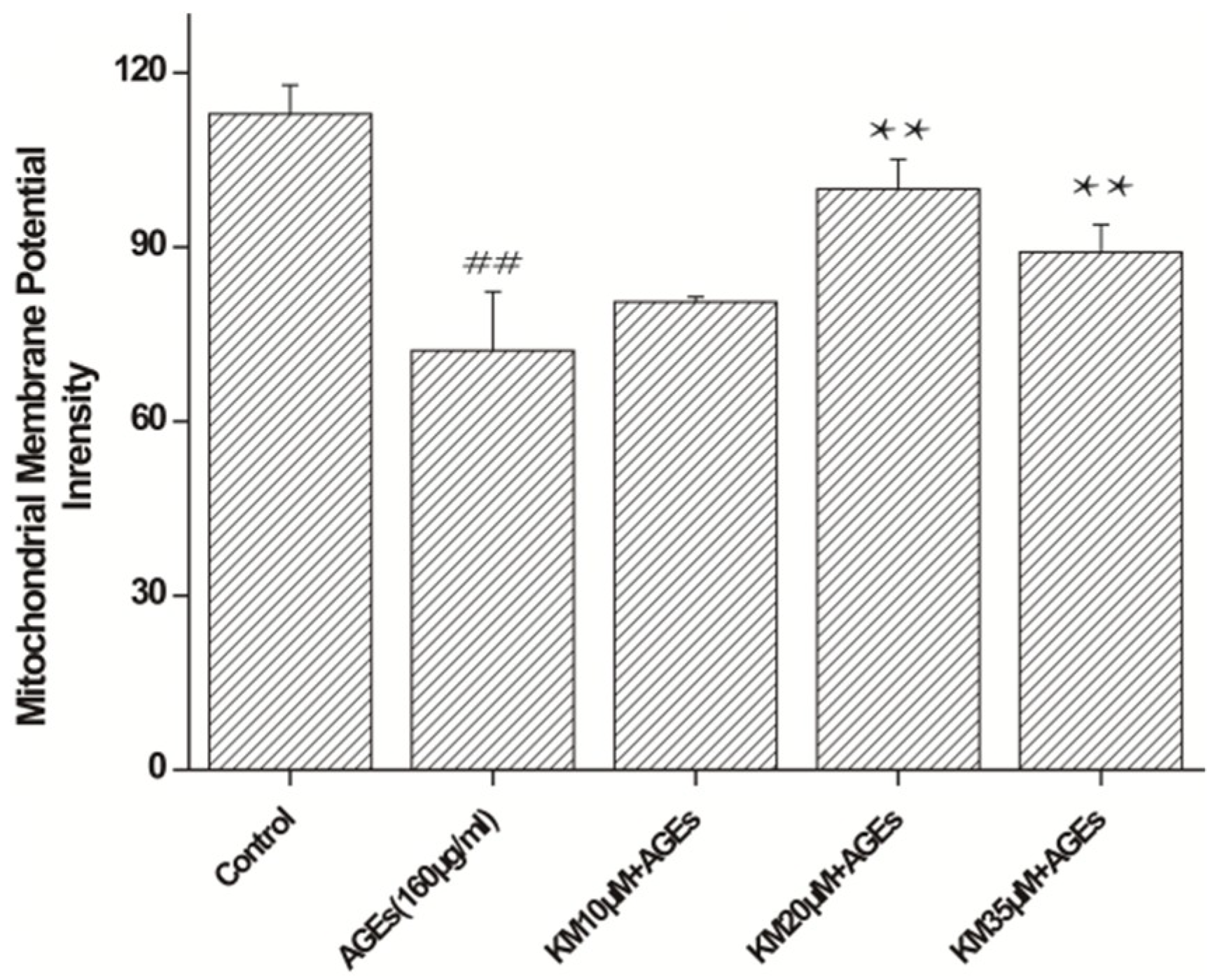
2.4. KM Treatment Improves Mitochondrial Membrane Potential Recovery in AGE-Treated GMCs
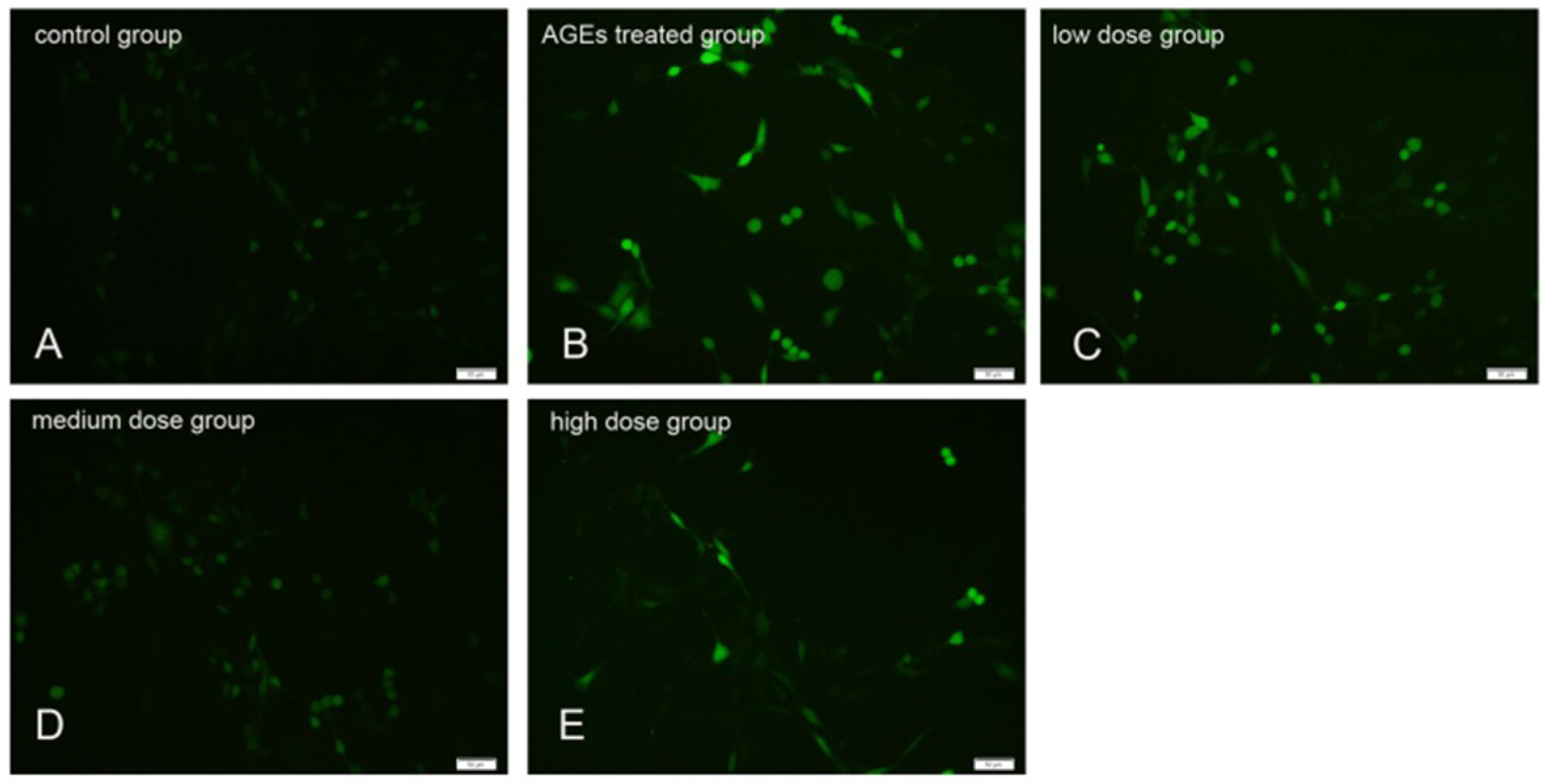
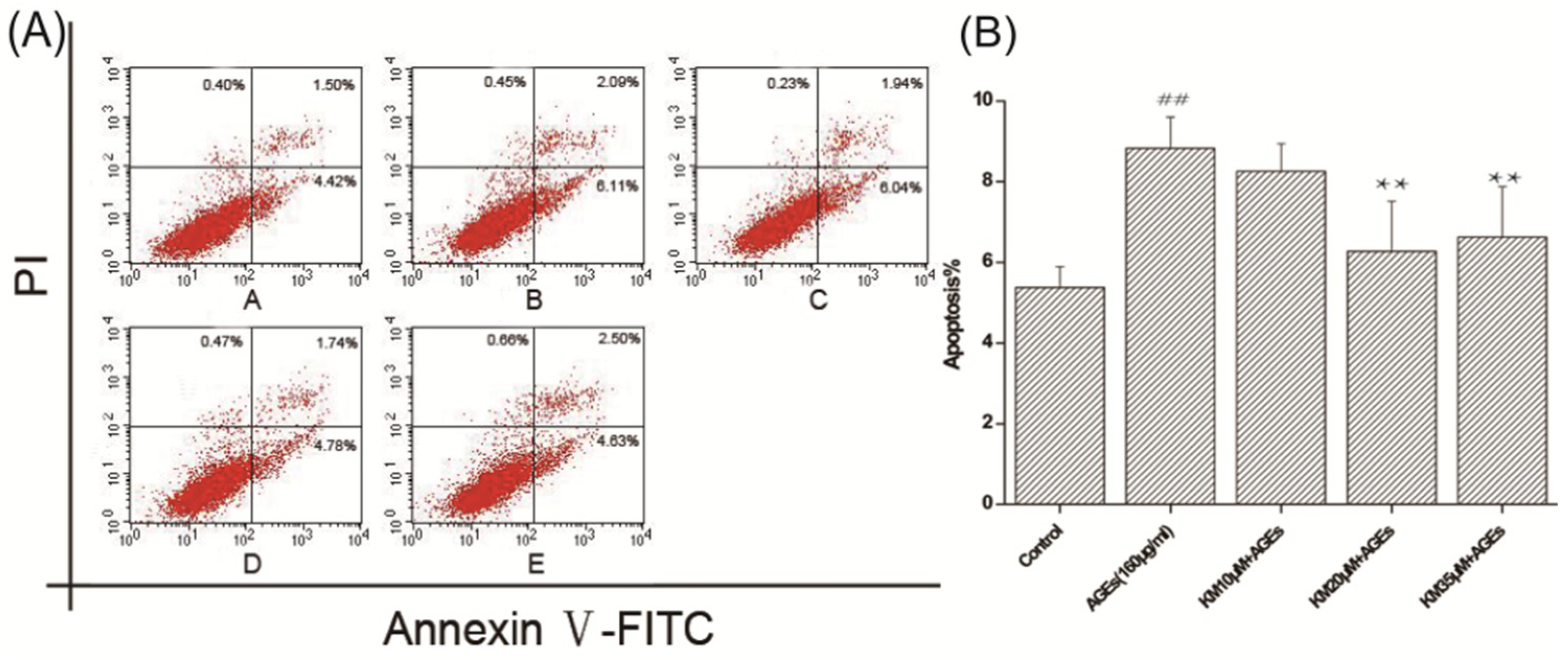
2.5. KM Treatment Abates the Morphological Changes Induced by Apoptosis and Reduces the Rate of Apoptosis in AGE-Treated GMCs
2.6. KM Modulates the Expression of Both Pro- and Anti-Apoptotic Proteins in GMCs Treated with AGEs
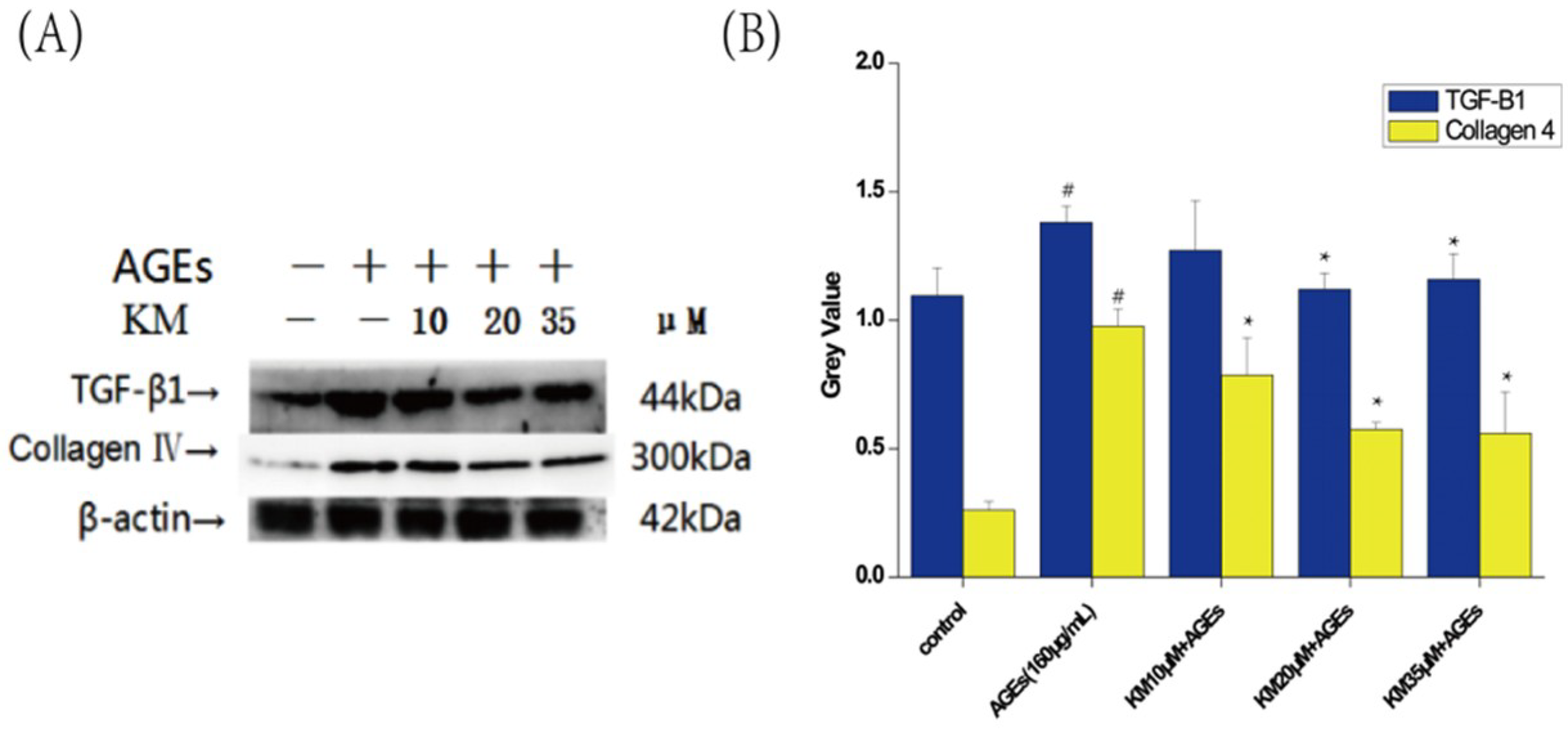
2.7. KM Treatment Enhances Antioxidation and Reduces the Proliferation of the Extracellular Matrix in AGE-Treated GMCs
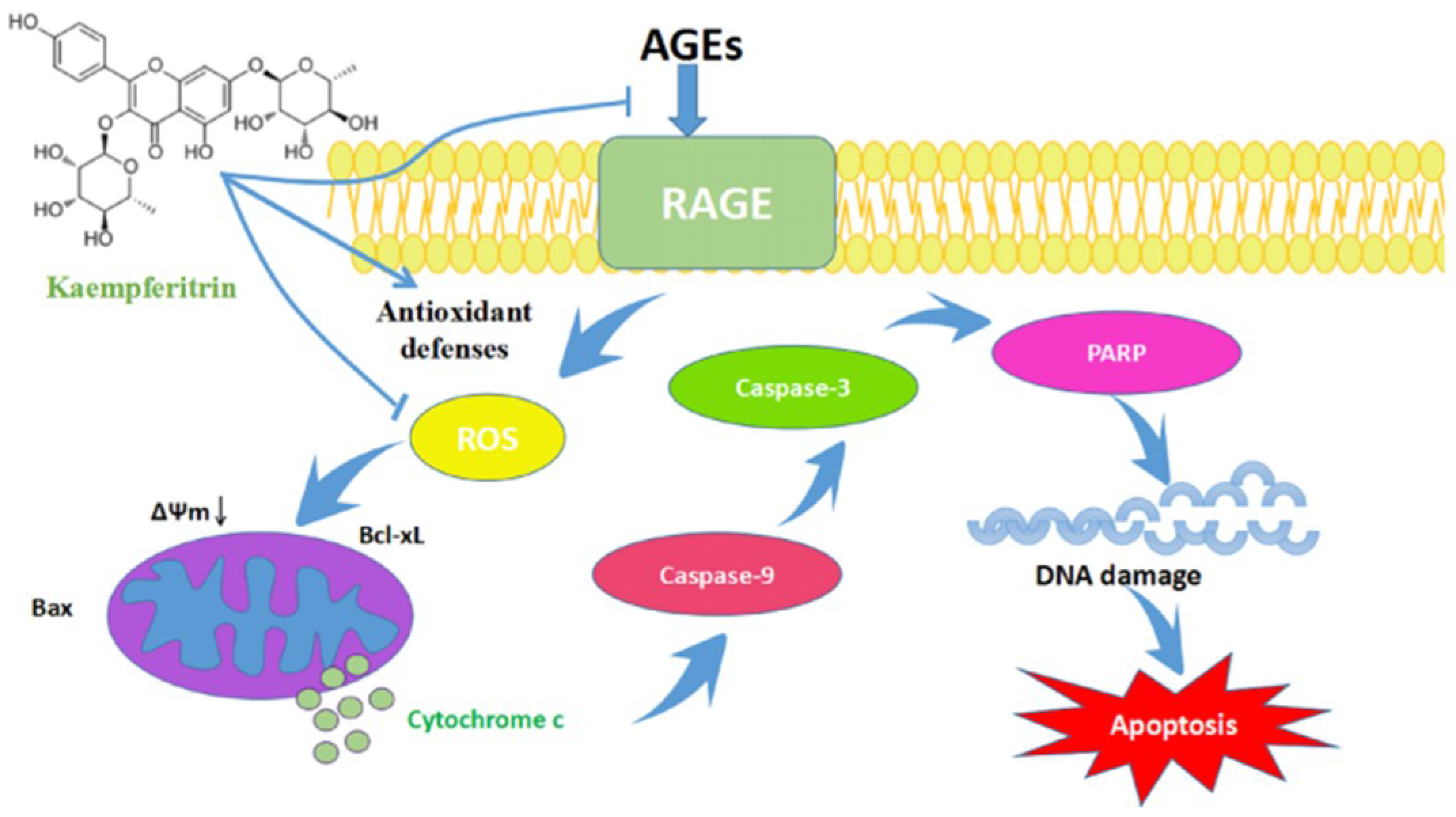
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. AGEs Preparation
4.3. Cell Culture
4.4. MTT Assay
4.5. Cellular Morphology Analysis
4.6. Determination of SOD and MDA Content
4.7. Detection of Intracellular ROS Accumulation
4.8. Assessment of Mitochondrial Membrane Potential
4.9. Hoechst 33258 Staining
4.10. Annexin V and PI Double Staining
4.11. Subcellular Fractionation for the Detection of Cytochrome C Release
4.12. Western Blotting
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Husseny, M.W.; Mamdouh, M.; Shaban, S. Adipokines: Potential therapeutic targets for vascular dysfunction in type II diabetes mellitus and obesity. J. Diabetes Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. Diabetic nephropathy: Heparanase mediates renal injury. Nat. Rev. Nephrol. 2014, 10, 483. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P. Diabetic nephropathy: Worldwide epidemic and effects of current treatment on natural history. Curr. Diabetes Rep. 2006, 6, 479–483. [Google Scholar] [CrossRef]
- Chu, Y.W.; Lin, H.M.; Wang, J.J. Epidemiology and outcomes of hypoglycemia in patients with advanced diabetic kidney disease on dialysis: A national cohort study. PLoS ONE 2017, 12, e0174601. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Palace, M.R. Diabetes and advanced glycation end products. J. Intern. Med. 2010, 251, 87–101. [Google Scholar] [CrossRef]
- Reiniger, N.; Kai, L.; Mccalla, D. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.T. Regulation of advanced glycation end product (AGE)-receptor (RAGE) system by PPAR-gamma agonists and its implication in cardiovascular disease. Pharmacol. Res. 2009, 60, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Diabetic stress: New triumphs and challenges to maintain vascular longevity. Expert Rev. Cardiovasc. Ther. 2008, 6, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Matavelli, L.C.; Siragy, H.M. Renal (pro)renin receptor contributes to development of diabetic kidney disease through TGFβ1-CTGF signaling cascade. Clin. Exp. Pharmacol. Physiol. 2011, 38, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Martínezpalacián, A.; Castillo, G.D.; Suárezcausado, A. Mouse Hepatic Oval Cells Require Met-Dependent PI3K to Impair TGF-β-Induced Oxidative Stress and Apoptosis. PLoS ONE 2013, 8, e53108. [Google Scholar] [Green Version]
- Fukami, K.; Yamagishi, S.; Ueda, S. Role of AGEs in diabetic nephropathy. Curr. Pharm. Des. 2008, 14, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, J.A.; Shankland, S.J.; Pichler, R.H. Proteinuria in diabetic kidney disease: A mechanistic viewpoint. Kidney Int. 2008, 74, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W. AGEs and diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.P.; Horst, H.; De, S.E. Insulinomimetic effects of kaempferitrin on glycaemia and on 14C-glucose uptake in rat soleus muscle. Chem. Biol. Interact. 2004, 149, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jiang, Z.T.; Ma, L. Investigation of antioxidant activities and free radical scavenging of laurel essential oil. China Food Additives 2012, 2, 69–74. [Google Scholar]
- Lin, L.; Ni, B.; Lin, H. Traditional usages, botany, phytochemistry, pharmacology and toxicology of Polygonum multiflorum Thunb: A. review. J. Ethnopharmacol. 2015, 159, 158–183. [Google Scholar] [CrossRef] [PubMed]
- Prakash, D.; Sudhandiran, G. Dietary flavonoid fisetin regulates aluminium chloride-induced neuronal apoptosis in cortex and hippocampus of mice brain. J. Nutr. Biochem. 2015, 26, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Menezes Sousaminto, F.D.; Mattosruela, A.B. Hypoglycemic activity of two Brazilian Bauhinia species: Bauhinia forficata L. and Bauhinia monandra Kurz. Rev. Bras. Farmacogn. 2007, 17, 8–13. [Google Scholar] [CrossRef]
- Lino, C.S.; Diógenes, J.P.; Pereira, B.A. Antidiabetic activity of Bauhinia forficata extracts in alloxan-diabetic rats. Biol. Pharm. Bull 2004, 27, 125–127. [Google Scholar] [CrossRef]
- Ashafaq, M.; Raza, S.S.; Khan, M.M. Catechin hydrate ameliorates redox imbalance and limits inflammatory response in focal cerebral ischemia. Neurochem. Res. 2012, 37, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.M.; Chen, K.; Rao, Y.K. Kaempferitrin activates the insulin signaling pathway and stimulates secretion of adiponectin in 3T3-L1 adipocytes. Eur. J. Pharmacol. 2009, 607, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghani, N.T.; Shoukry, A.F.; EI Nashar, R.M. Flow injection potentiometric determination of pipazethate hydrochloride. Analyst 2004, 78, 107–113. [Google Scholar] [CrossRef]
- Alonso-Castro, A.J.; Ortiz-Sánchez, E.; García-Regalado, A. Kaempferitrin induces apoptosis via intrinsic pathway in HeLa cells and exerts antitumor effects. J. Ethnopharmacol. 2013, 145, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Vellosa, J.C.; Regasini, L.O.; Belló, C. Preliminary in vitro and ex vivo evaluation of afzelin, kaempferitrin and pterogynoside action over free radicals and reactive oxygen species. Arch. Pharm. Res. 2015, 38, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Vishnu-Prasad, C.N.; Suma, M.S.; Banerji, A. Kaempferitrin inhibits GLUT4 translocation and glucose uptake in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2009, 380, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Cazarolli, L.H.; Pereira, D.F.; Kappel, V.D. Insulin signaling: A potential signaling pathway for the stimulatory effect of kaempferitrin on glucose uptake in skeletal muscle. Eur. J. Pharmacol. 2013, 712, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Manach, C.; Morand, C. Dietary polyphenols and the prevention of diseases. Crit. Rev. Food Sci. Nutr. 2005, 45, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; He, B.; Zheng, Z.G. Inhibitory effects of two major isoflavonoids in Radix Astragali on high glucose-induced mesangial cells proliferation and AGEs-induced endothelial cells apoptosis. Planta Med. 2011, 77, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Rao, Y.K.; Chen, K. Andrographolide and 14-deoxy-11,12-didehydroandrographolide from Andrographis paniculata attenuate high glucose-induced fibrosis and apoptosis in murine renal mesangeal cell lines. J. Ethnopharmacol. 2010, 132, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.H.; Shao, Z.M.; Tang, D.Q. Preventive effects of rutin on the development of experimental diabetic nephropathy in rats. Life Sci. 2012, 91, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhao, L.; Chen, Y. Advanced glycation end products (AGEs) increase human mesangial foam cell formation by increasing Golgi SCAP glycosylation in vitro. Am. J. Physiol. Renal Physiol. 2011, 301, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, Y.S.; Wada, J.; Sun, L. Diabetic nephropathy: Mechanisms of renal disease progression. Exp. Biol Med. 2008, 233, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Sun, L.; Xiao, L. Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr. Med. Chem. 2015, 22, 2858–2870. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Ueda, S.; Yamagishi, S. AGEs activate mesangial TGF-β-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004, 66, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Navarrogonzález, J.F.; Morafernández, C.; Fuentes, M.M.D. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Giribabu, N.; Karim, K.; Kilari, E.K. Phyllanthus niruri leaves aqueous extract improves kidney functions, ameliorates kidney oxidative stress, inflammation, fibrosis and apoptosis and enhances kidney cell proliferation in adult male rats with diabetes mellitus. J. Ethnopharmacol. 2017, 205, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Qian, Z.Y.; Zhou, C.H. Crocetin inhibits leukocyte adherence to vascular endothelial cells induced by AGEs. J. Ethnopharmacol. 2006, 107, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Poupel, F.; Aghaei, M.; Movahedian, A. Dihydroartemisinin induces apoptosis in human bladder cancer cell lines through reactive oxygen species, mitochondrial membrane potential, and cytochrome c pathway. Int. J. Prev. Med. 2017, 8, 78. [Google Scholar] [PubMed]
- Lu, M.; Sun, L.; Zhou, J. Dihydroartemisinin induces apoptosis in colorectal cancer cells through the mitochondria-dependent pathway. Tumor Biol. 2014, 35, 5307–5314. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Hail, N.; Lotan, R. Apoptosis as a Novel Target for Cancer Chemoprevention. J. Natl. Cancer Inst. 2004, 96, 662–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.F.; Lo, Y.H.; Hsieh, M.C. Cytotoxicities, cell cycle and caspase evaluations of 1,6-diaryl-3(Z.)-hexen-1,5-diynes, 2-(6-aryl-3(Z.)-hexen-1,5-diynyl) anilines and their derivatives. Bioorg. Med. Chem. 2005, 13, 3565–3575. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hou, Y.; Dong, M. PA-MSHA inhibits proliferation and induces apoptosis through the up-regulation and activation of caspases in the human breast cancer cell lines. J. Cell Biochem. 2010, 108, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Granadoserrano, A.B.; Martín, M.A.; Bravo, L. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J. Nutr. 2006, 136, 2715–2721. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, B.; Lin, S. Astaxanthin inhibits apoptosis in alveolar epithelial cells type II in vivo and in vitro through the ROS-dependent mitochondrial signalling pathway. J. Cell Mol. Med. 2015, 18, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Osen-Sand, A.; Nichols, A. Bid-Induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999, 144, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization. Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Wang, I.K.; Lin-Shiau, S.Y.; Lin, J.K. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukaemia HL-60 cells. Eur. J. Cancer 1999, 35, 1517–1525. [Google Scholar] [CrossRef]
- Chen, Y.; Du, X.; Zhou, Y. Paeoniflorin protects HUVECs from AGE-BSA-induced injury via an autophagic pathway by acting on the RAGE. Int. J. Clin. Exp. Pathol. 2015, 8, 53–62. [Google Scholar] [PubMed]
- Zhang, M.; Feng, L.; Gu, J. The attenuation of moutan cortex on oxidative stress for renal injury in AGEs-induced mesangial cell dysfunction and streptozotocin-induced diabetic nephropathy rats. Oxid. Med. Cell Longev. 2015, 2014, 463815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Meng, X.; Li, D. Angiotensin(1–7) attenuates the progression of streptozotocin-induced diabetic renal injury better than angiotensin receptor blockade. Kidney Int. 2015, 87, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, J.; Zhang, Y. Intermittent high glucose-induced oxidative stress modulates retinal pigmented epithelial cell autophagy and promotes cell survival via increased HMGB1. BMC Ophthalmol. 2018, 18, 192. [Google Scholar] [CrossRef] [PubMed]
- Sureda, F.X.; Escubedo, E.; Gabriel, C. Mitochondrial membrane potential measurement in rat cerebellar neurons by flow cytometry. Cytometry 2015, 28, 74–80. [Google Scholar] [CrossRef]
- Li, W.; Yan, M.H.; Liu, Y.; Liu, Z.; Wang, Z.; Chen, C.; Zhang, J.; Sun, Y.S. Ginsenoside Rg5 ameliorates cisplatin-induced nephrotoxicity in mice through inhibition of inflammation, oxidative stress, and apoptosis. Nutrients 2016, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Singh, R.L.; Kakkar, P. Modulation of Bax/Bcl-2 and caspases by probiotics during acetaminophen induced apoptosis in primary hepatocytes. Food Chem. Toxicol. 2011, 49, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Wu, Y.C.; Chia, Y.C. Gallic acid, a major component of Toona sinensis leaf extracts, contains a ROS-mediated anti-cancer activity in human prostate cancer cells. Cancer Lett. 2009, 286, 161–171. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | SOD (U/mL) | MDA (n mol/mL) |
|---|---|---|
| Control | 1.29 ± 0.067 | 0.51 ± 0.006 |
| AGEs group | 0.65 ± 0.006 # | 0.89 ± 0.009 # |
| KM low-dose group | 0.81 ± 0.007 * | 0.78 ± 0.020 * |
| KM medium-dose group | 0.93 ± 0.021 * | 0.58 ± 0.012 * |
| KM high-dose group | 0.76 ± 0.005 * | 0.69 ± 0.007 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Wang, R.; Liu, D.; Zuo, M.; Zhao, C.; Zhang, T.; Li, W. Protective Effects of Kaempferitrin on Advanced Glycation End Products Induce Mesangial Cell Apoptosis and Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 3334. https://doi.org/10.3390/ijms19113334
Jiang W, Wang R, Liu D, Zuo M, Zhao C, Zhang T, Li W. Protective Effects of Kaempferitrin on Advanced Glycation End Products Induce Mesangial Cell Apoptosis and Oxidative Stress. International Journal of Molecular Sciences. 2018; 19(11):3334. https://doi.org/10.3390/ijms19113334
Chicago/Turabian StyleJiang, Wenxian, Rongshen Wang, Di Liu, Min Zuo, Chunzhen Zhao, Tianliang Zhang, and Wanzhong Li. 2018. "Protective Effects of Kaempferitrin on Advanced Glycation End Products Induce Mesangial Cell Apoptosis and Oxidative Stress" International Journal of Molecular Sciences 19, no. 11: 3334. https://doi.org/10.3390/ijms19113334