Genome-Wide Identification, Molecular Evolution, and Expression Profiling Analysis of Pectin Methylesterase Inhibitor Genes in Brassica campestris ssp. chinensis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification and Phylogenetic Analysis of PMEI Gene Family in B. campestris
2.2. Characterization of Gene Structure, Conserved Motif, and Physicochemical Properties
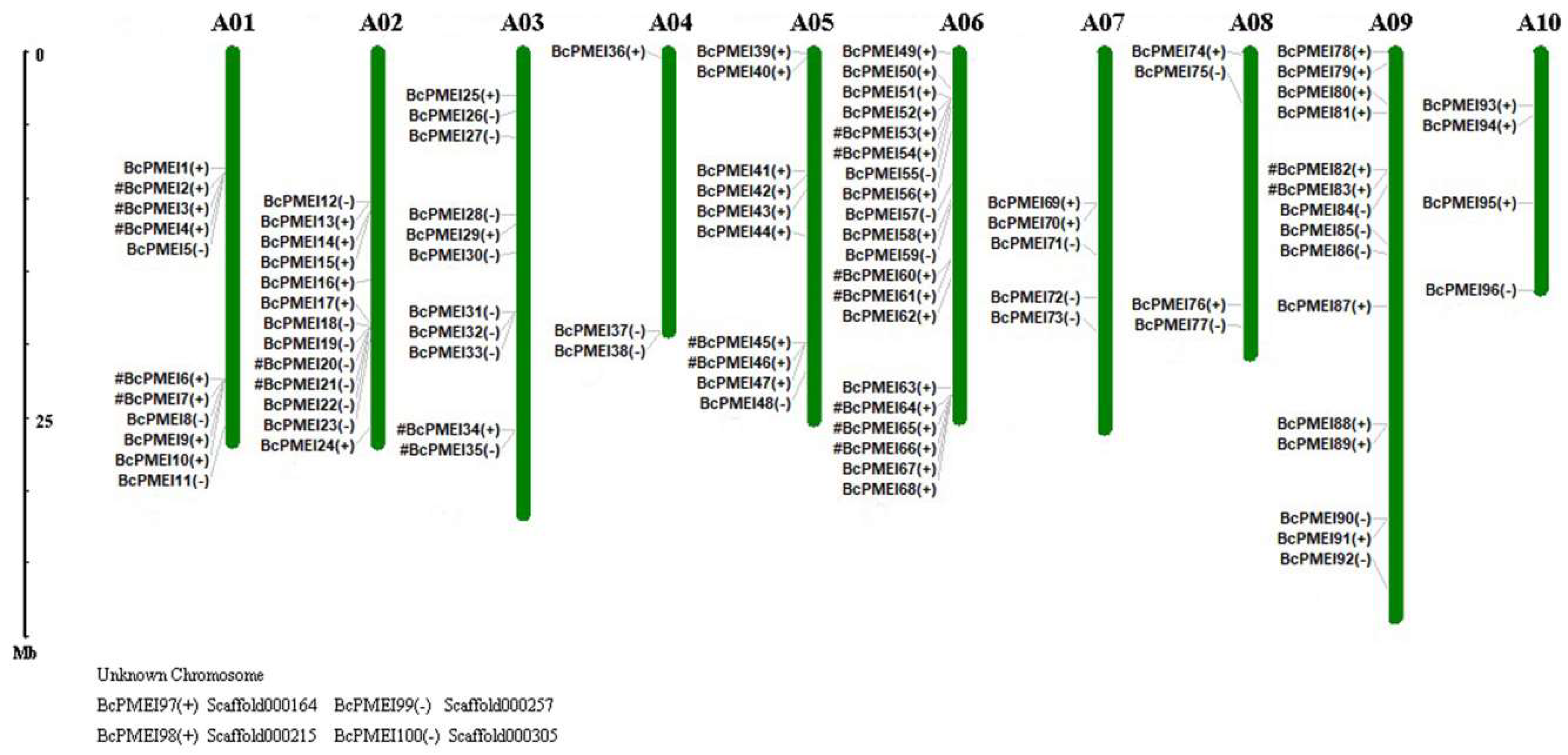
2.3. Chromosomal Distribution of PMEIs in B. campestris
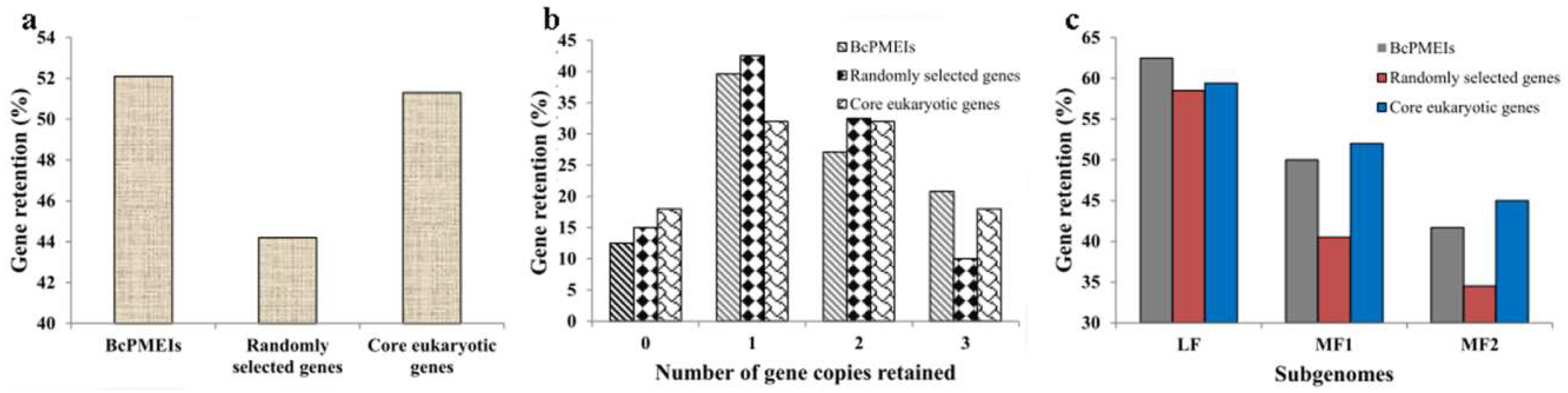
2.4. Synteny and Retained Proportion Analysis
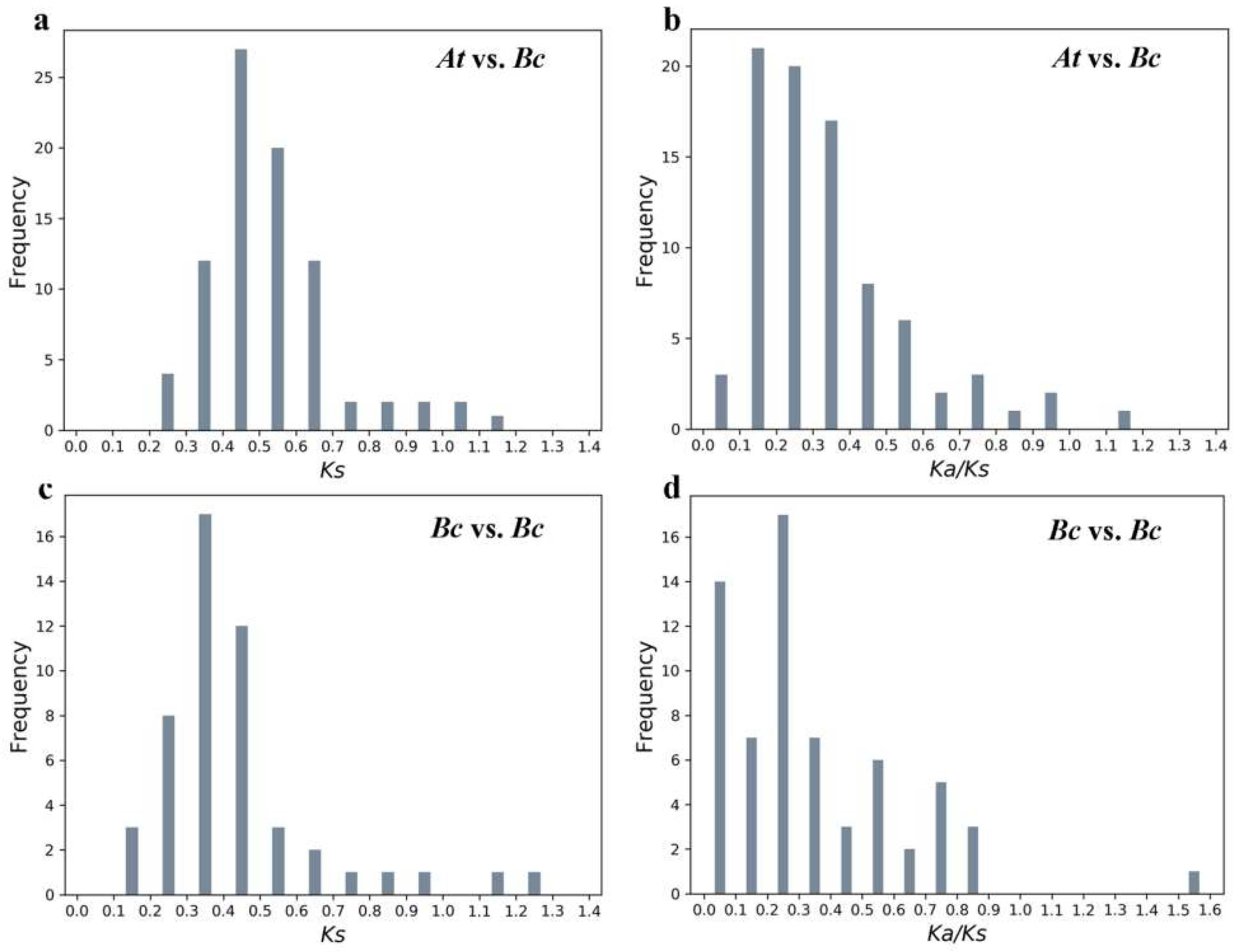
2.5. Evolutionary Analysis of BcPMEIs
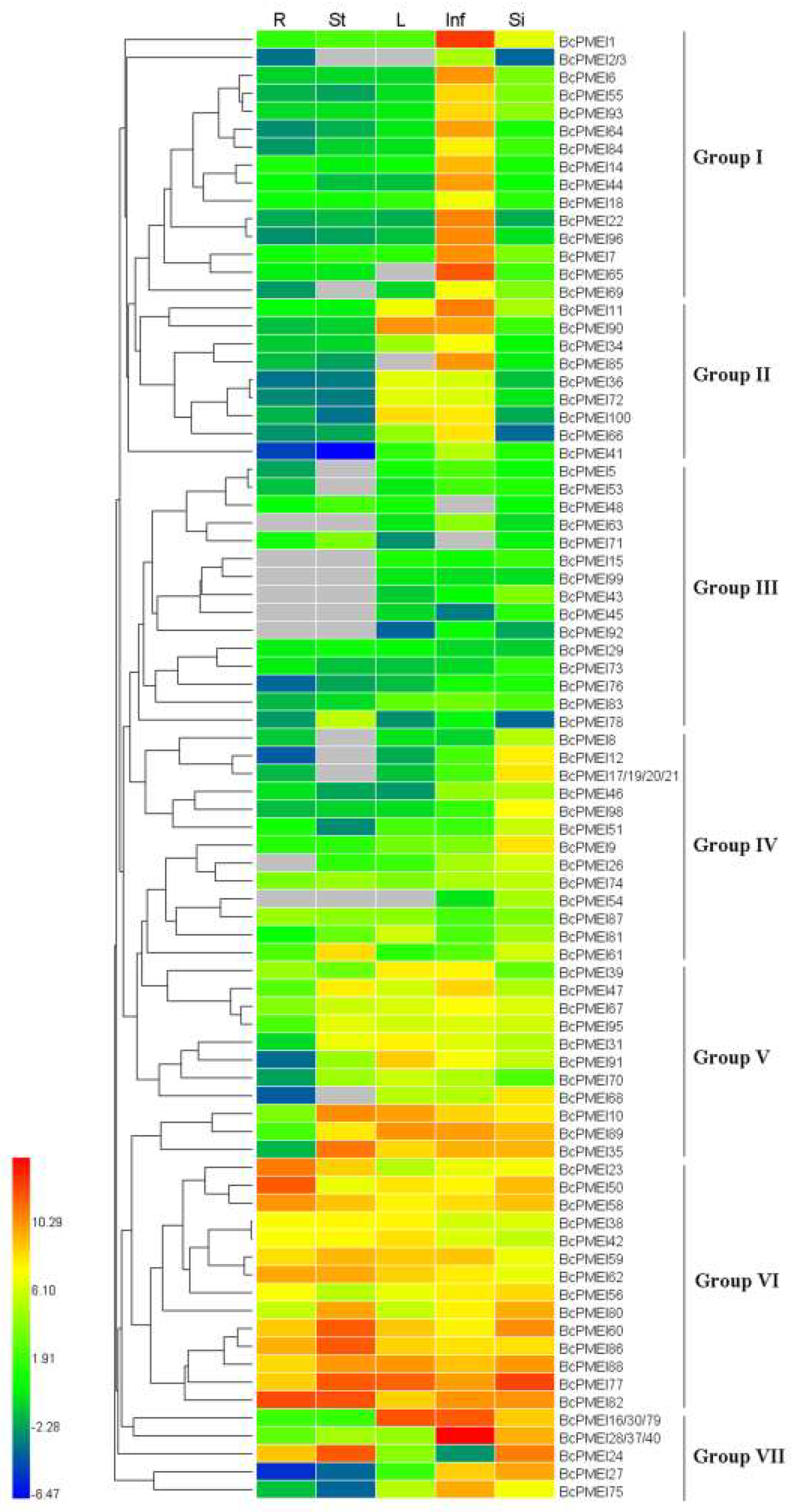
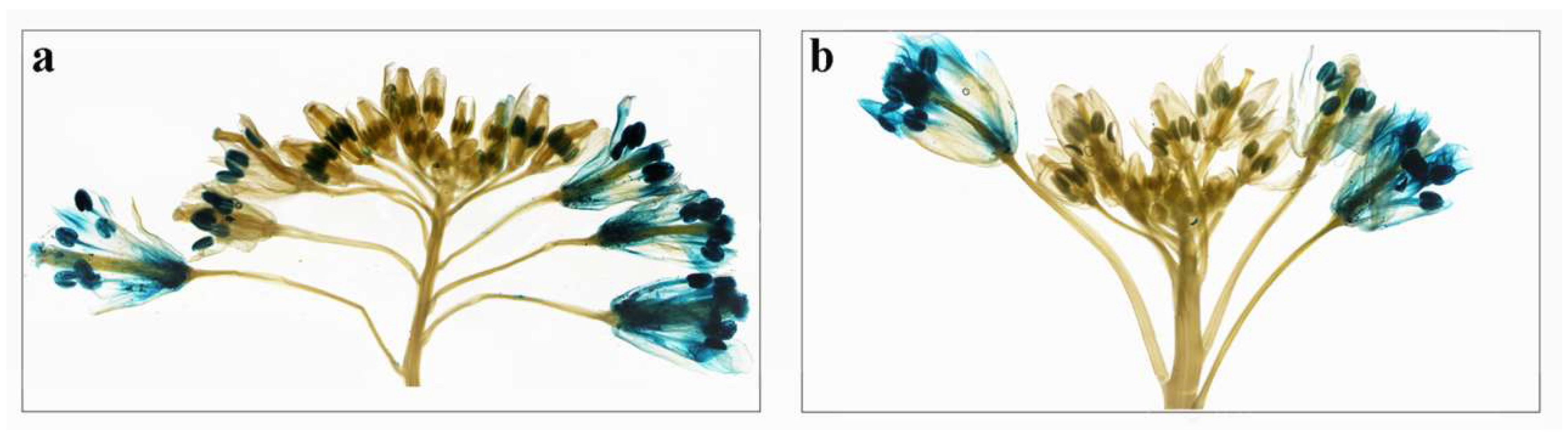
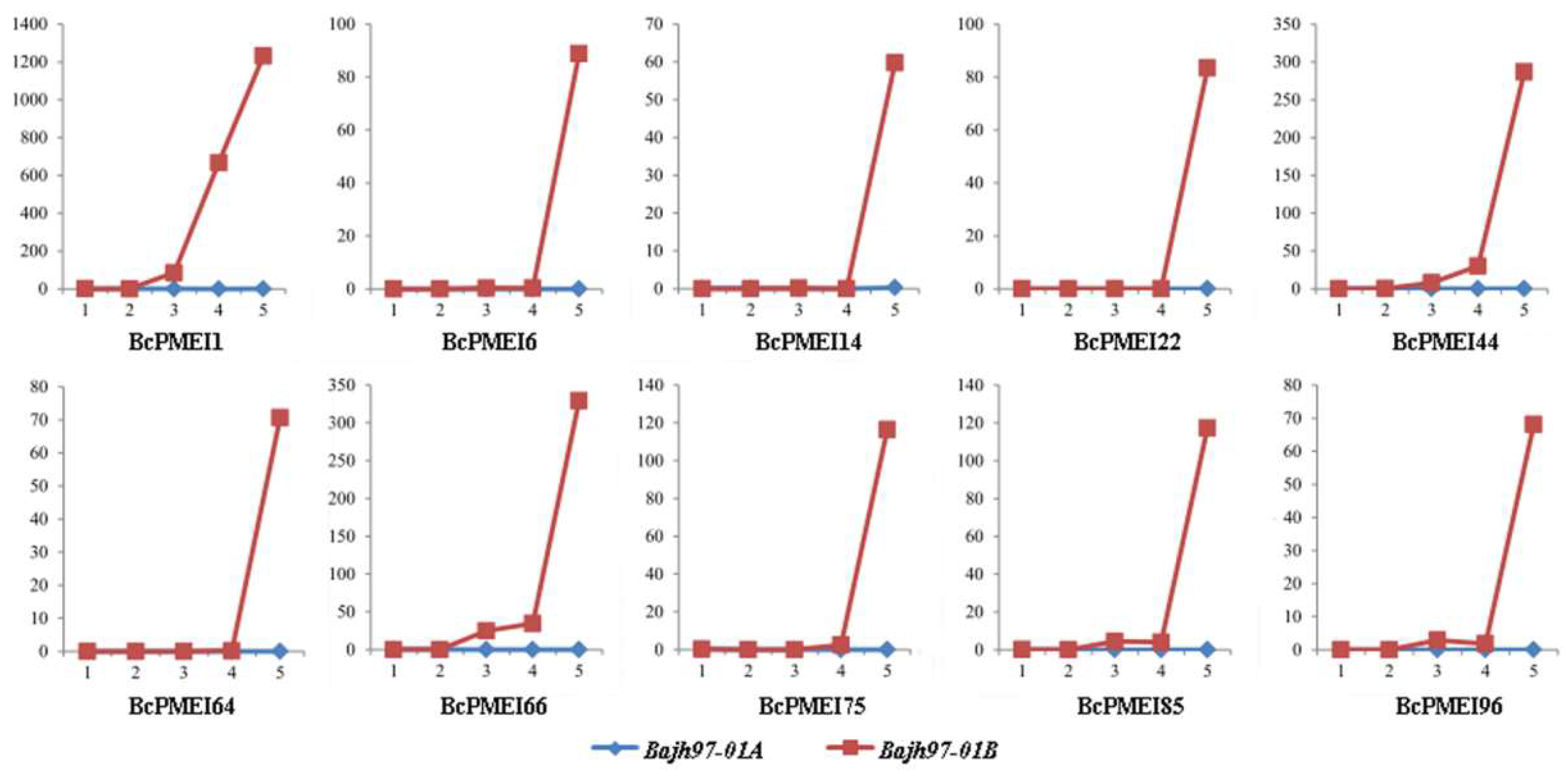
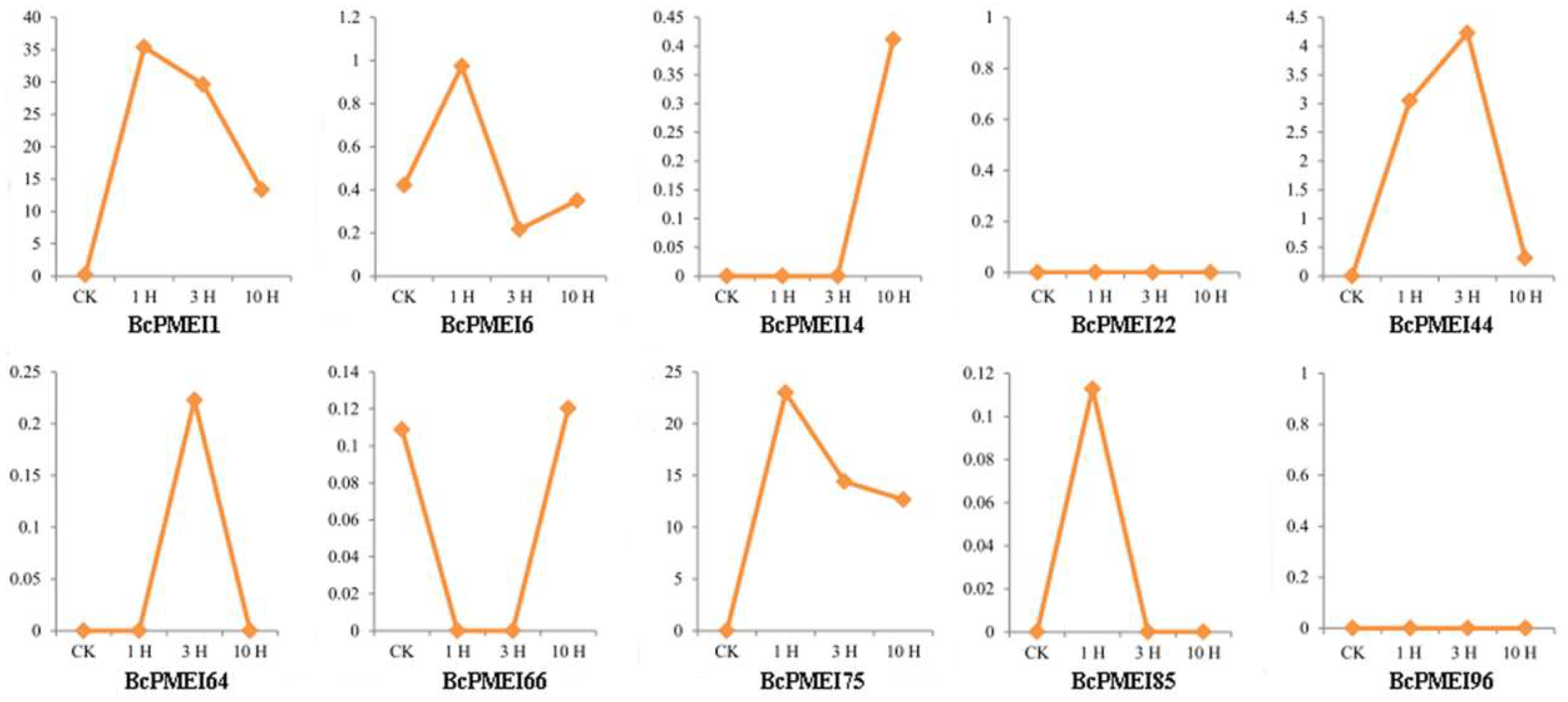
2.6. Expression Analysis of BcPMEIs in Different Tissues and Promoter::GUS Fusions
2.7. Roles of BcPMEIs in Pollen Development
2.8. Cis-Elements in the Putative Promoter Regions of BcPMEIs
2.9. The Microarray Data Analysis of PMEIs in Arabidopsis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Identification of PMEIs in B. campestris
4.3. Phylogenetic, Gene Structural, and Physicochemical Properties Analysis
4.4. Motif Recognition, Putative Promoter Region Analysis, and Promoter::GUS Fusion Construction
4.5. Chromosome Location, Synteny, Retained Rate, and Evolutionary Analysis
4.6. RNA Isolation and Expression Profile Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AtPMEIs | Arabidopsis thaliana PMEI genes |
| BcPMEIs | Brassica campestris PMEI genes |
| BRAD | brassica database |
| GMS | genic male sterile |
| HAP | hour after pollination |
| HAT | hour after treatment |
| HMM | hidden Markov model |
| HG | homogalacturonan |
| Ka | nonsynonymous substitution rate |
| Ks | synonymous substitution rate |
| LF | least fractionated subgenome |
| MEME | multiple em for motif elicitation |
| MF1 | medium fractionated subgenome |
| MF2 | most fractionated subgenome |
| MYA | million years ago |
| NJ | neighbor-joining |
| PGs | polygalacturonases |
| PLs | pectate lyases |
| PMEs | pectin methylesterases |
| PMEIs | pectin methylesterase inhibitors |
| qRT-PCR | quantitative real-time PCR |
| RGI | rhamnogalacturonan I |
| RGII | rhamnogalacturonan II |
| TAIR | the arabidopsis information resource |
| TD | tandem duplication |
| WGT | whole genome triplication |
| WGD | whole genome duplication |
| XGA | xylogalacturonan |
References
- Varner, J.E.; Lin, L.S. Plant cell wall architecture. Cell 1989, 56, 231–239. [Google Scholar] [CrossRef]
- Micheli, F. Pectin methylesterases: Cell wall enzymes with important roles in plant physiology. Trends Plant Sci. 2001, 6, 414–419. [Google Scholar] [CrossRef]
- Caffall, K.H.; Mohnen, D. The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr. Res. 2009, 344, 1879–1900. [Google Scholar] [CrossRef] [PubMed]
- Popper, A.; Michel, G.; Domozych, D.S.; Tuohy, M.G.; Kloareg, B.; Stengel, D.B. Evolution and diversity of plant cell walls: From algae to flowering plants. Annu. Rev. Plant Biol. 2011, 62, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Cervone, F.; Bellincampi, D. Methyl esterification of pectin plays a role during plant–pathogen interactions and affects plant resistance to diseases. J. Plant Physiol. 2012, 169, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Bellincampi, D.; Cervone, F.; Lionetti, V. Plant cell wall dynamics and wall-related susceptibility in plant–pathogen interactions. Front. Plant Sci. 2014, 5, 228. [Google Scholar] [CrossRef] [PubMed]
- Ridley, B.L.; Oneill, M.A.; Mohnen, D. Pectins: Structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 2001, 57, 929–967. [Google Scholar] [CrossRef]
- Giovane, A.; Servillo, L.; Balestrieri, C.; Raiola, A.; D’Avino, R.; Tamburrini, M.; Ciardiello, M.A.; Camardella, L. Pectin methylesterase inhibitor. Biochim. Biophys. Acta 2004, 1696, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Y.; Feng, J.; Wu, J.; Wang, X.W. BoPMEI1, a pollen-specific pectin methylesterase inhibitor, has an essential role in pollen tube growth. Planta 2010, 231, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, S.; Van Orden, J.; Wolf, S.; Vissenberg, K.; Delacourt, J.; Ndong, Y.A.; Pelloux, J.; Bischoff, V.; Urbain, A.; Mouille, G.; et al. A role for pectin de-methylesterification in a developmentally regulated growth acceleration in dark-grown Arabidopsis hypocotyls. New Phytol. 2010, 188, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Reca, I.B.; Lionetti, V.; Camardella, L.; D’Avino, R.; Giardina, T.; Cervone, F.; Bellincampi, D. A functional pectin methylesterase inhibitor protein (SolyPMEI) is expressed during tomato fruit ripening and interacts with PME-1. Plant Mol. Biol. 2012, 79, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Levesque-Tremblay, G.; Bartels, S.; Weitbrecht, K.; Wormit, A.; Usadel, B.; Haughn, G.; Kermode, A.R. Demethylesterification of cell wall pectins in Arabidopsis plays a role in seed germination. Plant Physiol. 2013, 161, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Fabri, E.; De Caroli, M.; Hansen, A.R.; Willats, W.G.T.; Piro, G.; Bellincampi, D. Three pectin methylesterase inhibitors protect cell wall integrity for immunity to Botrytis. Plant Physiol. 2017, 173, 1844–1863. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Liu, Z.; Huang, S.; Li, C.; Ren, J.; Tang, X.; Liu, W.; Peng, S.; Feng, H. Pectin methylesterase inhibitor (PMEI) family can be related to male sterility in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol. Genet. Genom. 2017, 293, 243–357. [Google Scholar] [CrossRef] [PubMed]
- Peaucelle, A.; Louvet, R.; Johansen, J.N.; Hofte, H.; Laufs, P.; Pelloux, J.; Mouille, G. Arabidopsis phyllotaxis is controlled by the methyl-esterification status of cell-wall pectins. Curr. Biol. 2008, 18, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Saez-Aguayo, S.; Ralet, M.C.; Berger, A.; Botran, L.; Ropartz, D.; Marion-Poll, A.; North, H.M. Pectin methylesterase inhibitor6 promotes Arabidopsis mucilage release by limiting methylesterification of homogalacturonan in seed coat epidermal cells. Plant Cell 2013, 25, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Jeong, H.Y.; Jeon, S.H.; Kim, D.; Lee, C. Rice pectin methylesterase inhibitor28 (OsPMEI28) encodes a functional PMEI and its overexpression results in a dwarf phenotype through increased pectin methylesterification levels. J. Plant Physiol. 2017, 208, 17–25. [Google Scholar] [CrossRef] [PubMed]
- An, S.H.; Sohn, K.H.; Choi, H.W.; Hwang, I.S.; Lee, S.C.; Hwang, B.K. Pepper pectin methylesterase inhibitor protein CaPMEI1 is required for antifungal activity, basal disease resistance and abiotic stress tolerance. Planta 2008, 228, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Tang, H.; Tan, X.; Ficklin, S.P.; Feltus, F.A.; Paterson, A.H. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS ONE 2011, 6, e28150. [Google Scholar] [CrossRef] [PubMed]
- Edger, P.P.; Pires, J.C. Gene and genome duplications: The impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009, 17, 699–717. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Duan, W.; Lyu, S.; Li, Y.; Hou, X. Genome-wide identification, evolution, and expression analysis of the ATP-binding cassette transporter gene family in Brassica rapa. Front. Plant Sci. 2017, 8, 349. [Google Scholar] [CrossRef] [PubMed]
- Warwick, S.I.; Francis, A.; Alshehbaz, I.A. Brassicaceae: Species checklist and database on CD-Rom. Plant Syst. Evol. 2006, 259, 249–258. [Google Scholar] [CrossRef]
- Bailey, C.D.; Koch, M.A.; Mayer, M.S.; Mummenhoff, K.; Okane, S.L.; Warwick, S.I.; Windham, M.D.; Alshehbaz, I.A. Toward a global phylogeny of the Brassicaceae. Mol. Biol. Evol. 2006, 23, 2142–2160. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Town, C.D.; Cheung, F.; Maiti, R.; Crabtree, J.; Haas, B.J.; Wortman, J.R.; Hine, E.E.; Althoff, R.; Arbogast, T.S.; Tallon, L.J.; et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 2006, 18, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.W.; Lai, K.N.; Tai, P.Y.; Li, W.H. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J. Mol. Evol. 1999, 48, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Jeong, H.Y.; Kim, H.; Kim, Y.C.; Lee, C. Molecular and biochemical characterization of rice pectin methylesterase inhibitors (OsPMEIs). Plant Physiol. Biochem. 2016, 101, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Pinzón-Latorre, D.; Deyholos, M.K. Characterization and transcript profiling of the pectin methylesterase (PME) and pectin methylesterase inhibitor (PMEI) gene families in flax (Linum usitatissimum). BMC Genom. 2013, 14, 742. [Google Scholar] [CrossRef] [PubMed]
- Scheler, C.; Weitbrecht, K.; Pearce, S.P.; Hampstead, A.; Büttner-Mainik, A.; Lee, K.J.D.; Voegele, A.; Oracz, K.; Dekkers, B.J.W.; Wang, X.; et al. Promotion of testa rupture during garden cress germination involves seed compartment-specific expression and activity of pectin methylesterases. Plant Physiol. 2015, 167, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yuan, D.; Gao, W.; Li, Y.; Tan, J.; Zhang, X. A comparative genome analysis of PME and PMEI families reveals the evolution of pectin metabolism in plant cell walls. PLoS ONE 2013, 8, e72082. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wu, J.; Fang, L.; Wang, X. Syntenic gene analysis between Brassica rapa and other Brassicaceae species. Front. Plant Sci. 2012, 3, 198. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Dong, H.; Cui, J.; Li, M.; Lin, S.; Cao, J.; Huang, L. Genomic, molecular evolution, and expression analysis of genes encoding putative classical AGPs, lysine-rich AGPs, and AG peptides in Brassica rapa. Front. Plant Sci. 2017, 8, 397. [Google Scholar] [CrossRef] [PubMed]
- Lou, P.; Wu, J.; Cheng, F.; Cressman, L.G.; Wang, X.; McClung, C.R. Preferential retention of circadian clock genes during diploidization following whole genome triplication in Brassica rapa. Plant Cell 2012, 24, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wu, J.; Wang, X. Genome triplication drove the diversification of Brassica plants. Hortic. Res. 2014, 1, 14024. [Google Scholar] [CrossRef] [PubMed]
- Toufighi, K.; Brady, S.M.; Austin, R.; Ly, E.; Provart, N.J. The botany array resource: E-northerns, expression angling, and promoter analyses. Plant J. 2005, 43, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Juge, N. Plant protein inhibitors of cell wall degrading enzymes. Trends Plant Sci. 2006, 11, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, C.; Castaldo, D.; Giovane, A.; Quagliuolo, L.; Servillo, L. A glycoprotein inhibitor of pectin methylesterase in kiwi fruit (Actinidia chinensis). FEBS J. 1990, 193, 183–187. [Google Scholar] [CrossRef]
- Woriedh, M.; Wolf, S.; Marton, M.L.; Hinze, A.; Gahrtz, M.; Becker, D.; Dresselhaus, T. External application of gametophyte-specific ZmPMEI1 induces pollen tube burst in maize. Plant Reprod. 2013, 26, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Grsic-Rausch, S.; Rausch, T.; Greiner, S. Identification of pollen-expressed pectin methylesterase inhibitors in Arabidopsis. FEBS Lett. 2003, 555, 551–555. [Google Scholar] [CrossRef]
- Lionetti, V.; Raiola, A.; Mattei, B.; Bellincampi, D. The grapevine VvPMEI1 gene encodes a novel functional pectin methylesterase inhibitor associated to grape berry development. PLoS ONE 2015, 10, e0133810. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.G.; Harris, P.J. The polysaccharide composition of Poales cell walls: Poaceae cell walls are not unique. Biochem. Syst. Ecol. 1999, 27, 33–53. [Google Scholar] [CrossRef]
- Pelloux, J.; Rusterucci, C.; Mellerowicz, E.J. New insights into pectin methylesterase structure and function. Trends Plant Sci. 2007, 12, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Cheng, F.; Wu, J.; Wang, X. The impact of genome triplication on tandem gene evolution in Brassica rapa. Front. Plant Sci. 2012, 3, 261. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, G.L. Variation and Evolution in Plants; Columbia University Press: New York, NY, USA, 1950. [Google Scholar]
- Sankoff, D.; Zheng, C.; Zhu, Q. The collapse of gene complement following whole genome duplication. BMC Genom. 2010, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Messing, J.; Bharti, A.K.; Karlowski, W.M.; Gundlach, H.; Kim, H.R.; Yu, Y.; Wei, F.; Fuks, G.; Soderlund, C.A.; Mayer, K.F.; et al. Sequence composition and genome organization of maize. Proc. Natl. Acad. Sci. USA 2004, 101, 14349–14354. [Google Scholar] [CrossRef] [PubMed]
- Schnable, J.C.; Springer, N.M.; Freeling, M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA 2011, 108, 4069–4074. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.C.; Pedersen, B.; Freeling, M. Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 2006, 16, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Veitia, R.A. The gene balance hypothesis: From classical genetics to modern genomics. Plant Cell 2007, 19, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Senechal, F.; Wattier, C.; Rusterucci, C.; Pelloux, J. Homogalacturonan-modifying enzymes: Structure, expression, and roles in plants. J. Exp. Bot. 2014, 65, 5125–5160. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Lin, K.; Bonnema, G.; Wang, X. Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS ONE 2012, 7, e36442. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, Y.; Shang, C.; Li, J.; Wang, J.; Wu, Z.; Ma, L.; Qi, T.; Fu, C.; Bai, Z. Genome-wide characterization of GRAS family genes in Medicago truncatula reveals their evolutionary dynamics and functional diversification. PLoS ONE 2017, 12, e0185439. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, X.; Ye, L.; Pan, C.; Chen, L.; Zou, T.; Lu, G. Genome-wide identification and expression analysis of two-component system genes in tomato. Int. J. Mol. Sci. 2016, 17, 1204. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Z.; Xiao, Y.; Wang, P.; Wang, Y.; Ge, X.; Zhang, C.; Zhang, X.; Li, F. Genome-wide analysis of the NF-YB gene family in Gossypium hirsutum L. and characterization of the role of GhDNF-YB22 in embryogenesis. Int. J. Mol. Sci. 2018, 19, 483. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.; Kozlovsky, S.V.; Tian, G.W.; Chen, M.H.; Zaltsman, A.; Citovsky, V. How pollen tubes grow. Dev. Biol. 2007, 303, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Rockel, N.; Wolf, S.; Kost, B.; Rausch, T.; Greiner, S. Elaborate spatial patterning of cell-wall PME and PMEI at the pollen tube tip involves PMEI endocytosis, and reflects the distribution of esterified and de-esterified pectins. Plant J. 2008, 53, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Jithesh, M.N.; Wally, O.S.; Manfield, I.; Critchley, A.T.; Hiltz, D.; Prithiviraj, B. Analysis of seaweed extract-induced transcriptome leads to identification of a negative regulator of salt tolerance in Arabidopsis. HortScience 2012, 47, 704–709. [Google Scholar]
- Huang, L.; Cao, J.; Ye, W.; Liu, T.; Jiang, L.; Ye, Y. Transcriptional differences between the male-sterile mutant bcms and wild-type Brassica campestris ssp. chinensis reveal genes related to pollen development. Plant Biol. 2008, 10, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ye, W.Z.; Liu, T.T.; Cao, J.S. Characterization of the male-sterile line Bcajh97-01A/B and identification of candidate genes for genic male sterility in Chinese cabbage-pak-choi. J. Am. Soc. Hortic. Sci. 2009, 134, 632–640. [Google Scholar]
- Huang, L.; Zhao, X.; Liu, T.; Dong, H.; Cao, J. Developmental characteristics of floral organs and pollen of Chinese cabbage (Brassica campestris L. ssp. chinensis). Plant Syst. Evol. 2010, 286, 103–115. [Google Scholar] [CrossRef]
- Jiang, J.; Jiang, J.; Qiu, L.; Miao, Y.; Yao, L.; Cao, J. Identification of gene expression profile during fertilization in Brassica campestris subsp. chinensis. Genome 2013, 56, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Liu, S.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Li, P.; Hua, W.; Wang, X. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; La Bastide, M.D.; Hamilton, J.P.; Kanamori, H.; Mccombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S. Improvement of the Oryza sativa nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012, 40, D302–D305. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 2010, 5, e11335. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, R.A.; Kavanagh, T.A.; Bevan, M.W. GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987, 6, 3901–3907. [Google Scholar] [PubMed]
- Sanders, P.M.; Bui, A.Q.; Weterings, K.; McIntire, K.; Hsu, Y.C.; Lee, P.Y.; Truong, M.T.; Beals, T.; Goldberg, R. Anther developmental defects in Arabidopsis thaliana male-sterile mutants. Sex. Plant Reprod. 1999, 11, 297–322. [Google Scholar] [CrossRef]
- Cheng, F.; Mandakova, T.; Wu, J.; Xie, Q.; Lysak, M.A.; Wang, X. Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. Plant Cell 2013, 25, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shao, Z.Q.; Wu, X.Z.; Wang, Q.; Wang, B.; Chen, J.Q.; Hang, Y.Y.; Xue, J.Y. Loss/retention and evolution of NBS-encoding genes upon whole genome triplication of Brassica rapa. Gene 2014, 540, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A toolkit for illustrating heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Yu, H.; Xiong, X.; Yue, X.; Yu, Y.; Huang, L.; Cao, J. Genome-Wide Identification, Molecular Evolution, and Expression Profiling Analysis of Pectin Methylesterase Inhibitor Genes in Brassica campestris ssp. chinensis. Int. J. Mol. Sci. 2018, 19, 1338. https://doi.org/10.3390/ijms19051338
Liu T, Yu H, Xiong X, Yue X, Yu Y, Huang L, Cao J. Genome-Wide Identification, Molecular Evolution, and Expression Profiling Analysis of Pectin Methylesterase Inhibitor Genes in Brassica campestris ssp. chinensis. International Journal of Molecular Sciences. 2018; 19(5):1338. https://doi.org/10.3390/ijms19051338
Chicago/Turabian StyleLiu, Tingting, Hui Yu, Xingpeng Xiong, Xiaoyan Yue, Youjian Yu, Li Huang, and Jiashu Cao. 2018. "Genome-Wide Identification, Molecular Evolution, and Expression Profiling Analysis of Pectin Methylesterase Inhibitor Genes in Brassica campestris ssp. chinensis" International Journal of Molecular Sciences 19, no. 5: 1338. https://doi.org/10.3390/ijms19051338