1. Introduction
Lead (Pb) is a potent neurotoxin and has been a global public health concern for many years [
1]. High-level acute exposure can result in damage to the central nervous system, cause a person to enter into a coma, or even result in death, while low level chronic exposure can damage brain development, the central nervous system, the heart, lungs, and kidneys. Chronic Pb poisoning has been associated with a loss of cognition, shortening of attention span, alteration of behaviour, dyslexia, attention deficit disorder, hypertension, renal impairment, kidney damage, immunotoxicity, reproductive toxicity, and increased risk of cardiovascular death [
1,
2]. Due to the known detrimental health outcomes at any level of Pb, there is currently no level of exposure that is considered to be safe [
2].
Children are considered to be at a particularly high risk of the adverse health effects of Pb, especially those under 5 years of age, for several reasons: (i) per unit of body mass, children inhale and ingest more food, water, and dust than adults, so their relative intake of Pb by these routes is therefore greater; (ii) once in the body, children are up to five times more effective at absorbing Pb than adults [
2,
3]; (iii) hand-to-mouth characteristics are much more prevalent in young children and consequently their ingestion of Pb from sources such as soil, dust, and flaking paint is likely to be considerably greater [
3]; (iv) the brains of young children are rapidly developing, but the presence of Pb can interfere with this development and result in long-term irreversible neurological damage [
4,
5]. Although the negative impacts of Pb poisoning are known, it is estimated to affect hundreds of millions of children globally [
1].
Lead exposure is typically assessed by measuring blood lead concentrations (BLCs). As Pb has a half-life of approximately 1–2 months in blood, repeated measurements can be used to monitor changes in exposure with time [
6]. Despite there being no safe level of exposure, many public health agencies have blood lead reference values (BLRV) for children. The current BLRV for England and Georgia is 5 µg dL
−1, while in the U.S., this level has recently been reduced to 3.5 µg dL
−1 [
6,
7]. The BLRV is not intended to define an acceptable BLC but rather to identify children of particular concern where public health intervention measures may be implemented and where follow-up monitoring may be required. Indeed, studies have found neurological effects at these levels; for example, at a population level, children with a BLC of 5 µg dL
−1 have been shown to score several IQ points lower than unaffected peers [
8,
9].
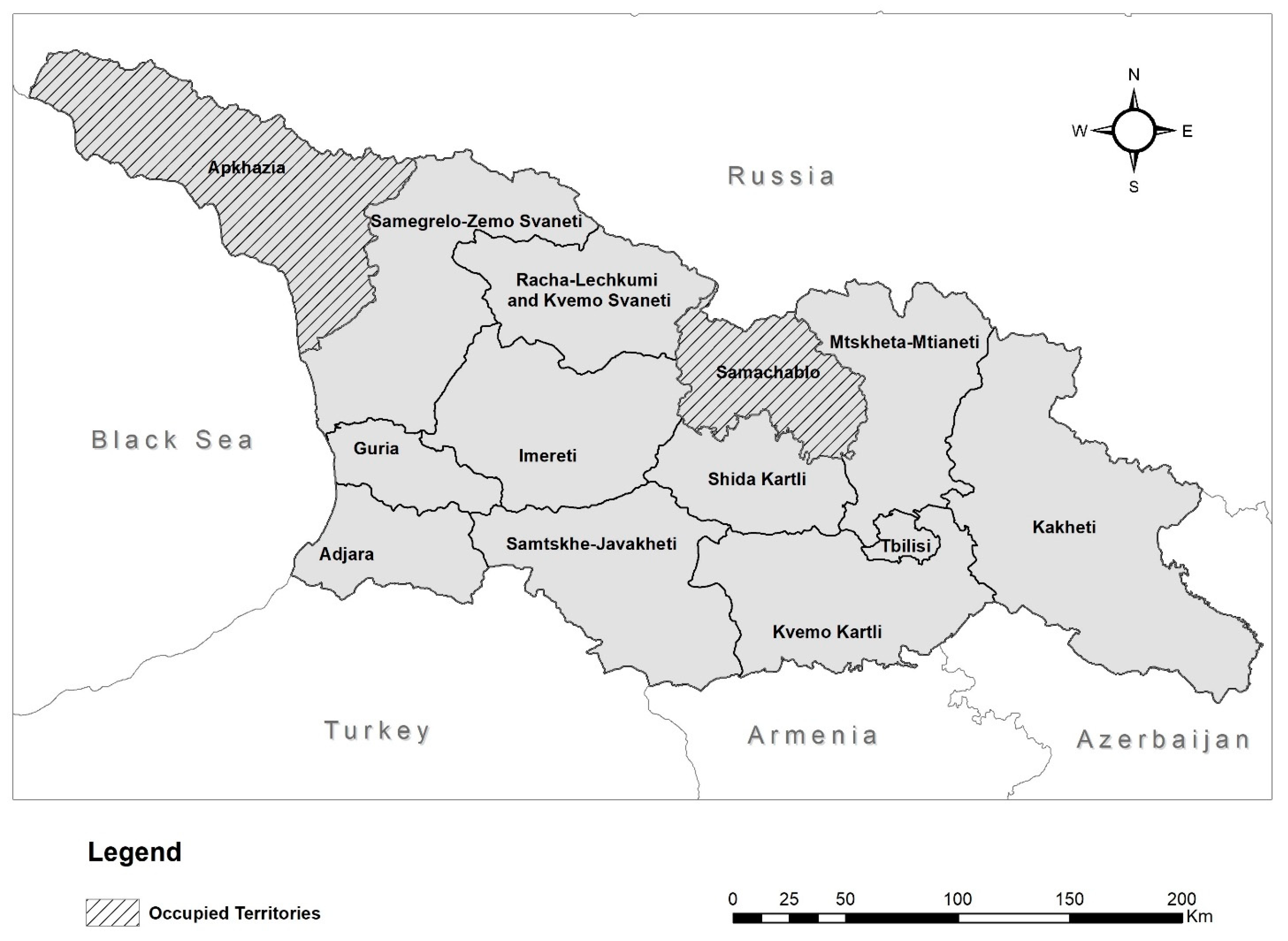
Concerns about the severity of Pb exposure in Georgia were raised in late 2015 after a collaborative study of 254 children aged 2–5 years at the Lasvili Children’s Hospital in the Georgian capital Tbilisi, performed by Georgia’s National Center for Disease Control and Public Health (NCDC) and U.S. Centers for Disease Control and Prevention [
10]. The study found that a third of participants had BLCs above 5 µg dL
−1. At the same time, the NCDC also conducted a similar study with a cohort of 46 children aged 4–6 years from 10 villages in the Bolnisi and Dmanisi districts, with 14 of the participants (30.4%) having BLCs above 5 µg dL
−1. The BLC of these participants was retested in 2017–18, with 37% still having BLCs above 5 µg dL
−1. This led to the UNICEF-funded national Multiple Indicator Cluster Survey (MICS) of Georgia, conducted in 2018 and including a BLC sampling component, where a nationally representative sample of 1578 children aged 2–7 years was tested. It found that, nationally, 41% of participants had BLCs above the BLRV of 5 µg dL
−1. However, it also demonstrated that there was a significant regional variation, with regions in the west of the country having a higher number of participants with BLCs above the BLRV than those in the east. For example, the region with the lowest proportion of participants having BLCs above the BLRV was Kvemo Kartli (18%), while the highest was Adjara (85%) (
Figure 1) [
11].
In response, the Georgian authorities instigated ‘The State Program for Disease Early Diagnosis and Screening’ (Ministerial decree No. 240, 23 May 2019, Governmental decree #693, 31 December 2018) in an attempt to mediate the elevated BLCs [
12]. This programme included raising public awareness of Pb toxicity and advice on reducing the Pb exposure together with BLC monitoring for MICS participants and their family members under the age of 18. Additionally, children were assessed for physical and mental development and nutritional status. Where BLCs were found to exceed 5 µg dL
−1, nutritional and behavioural advice was given to the child and their parent/guardian, as well as nutritional supplements of iron, calcium, and multivitamins being prescribed, during a consultation with a doctor. To investigate the impact of these mitigation measures and in an attempt to identify additional potential sources of Pb exposure in these children, a follow-up study was designed by the NCDC, the UK Health Security Agency (UKHSA) (formally Public Health England), and the British Geological Survey (BGS). This study recruited 36 households from across Georgia that had previously participated in the MICS survey and where BLC were previously determined as >5 µg dL
−1. In addition to the children’s blood, a questionnaire relating to potential Pb exposure and children’s behaviour was completed by the parents/guardians. Based upon the survey responses, a range of samples from the child’s environment were taken where available. The environmental samples included soil, household dust, paint, drinking water, milk, spices, flour, tea, and toys. All samples collected were first analysed to determine the total Pb amount. Samples from three households were also analysed for their Pb isotope composition. Used together, this information can aid in the identification or dismissal of environmental sources of Pb exposure for an individual [
13].
The isotope composition of blood Pb is inherited from its source(s); source apportionment can therefore be achieved by comparing Pb isotope ratios in blood with likely sources of exposure. The concept of Pb isotope ratios for environmental tracing has been well established [
14,
15,
16]; however, there are limited applications at a population level. One example of such work was presented by Glorennec et al. [
17] and Oulhote et al. [
18], where blood samples from 125 French children, along with samples of dust, paint, water, and soil from the child’s household, were analysed for their total Pb concentrations and Pb isotope ratio. In those studies, the observed variability in the isotope ratios and their analytical uncertainty was used to identify that
207Pb/
206Pb and
206Pb/
204Pb were the most appropriate ratios for discrimination in that case. Glorennec et al. [
17] and Oulhote et al. [
18] reported that 57% of the children had a BLC > 2.5 µg dL
−1 and of these, a suspected source of Pb exposure was identified in 32% of cases by Pb isotope analysis. In a recent study, similar to the one presented here, by Becker et al. [
19], Pb isotope ratios were used to determine that the environmental sources of Pb in five children from Kansas City with BLCs > 5 µg dL
−1 included household dust, soil, turmeric, and paint. Other studies by Gulson et al. [
20] and Takagi et al. [
21,
22] that used Pb isotope ratios to identify the most likely dominant sources of Pb in the blood of children in Australia and Japan, respectively, also found a strong link with household dust. In a study by Forsyth et al. [
23], the blood of 45 pregnant women from a rural area of Bangladesh was analysed and found to be most isotopically similar to that of turmeric, which had elevated levels of Pb and Cr, and indicated that the dominant source of Pb found in blood was from the adulterant spices with lead chromate. Fillion et al. [
24] present data from a study of 156 participants from an Inuit population living in Nunavut, Canada, where household members had previously been identified as having a BLC > 10 µg dL
−1. This study concluded that household dust and ammunition were the likely major sources of Pb exposure. However, as indicated by Gulson et al. [
25], some of the ratios presented by Fillion et al. [
24] are impossible values and therefore the reliability of the presented data is questionable. Such work highlights the need for robust and reliable methods for data analysis and treatment with the appropriate quality controls and validation.
We aimed to assess the feasibility of applying a Pb isotope ratio tracing approach to the linking of environmental samples as potential sources of Pb exposure in selected children in Georgia, with blood Pb seen as the exposure biomarker and numerous candidate environmental media seen as environmental exposure indicators. We focused on a few of the most exposed children based on total Pb concentration in their blood, but intended to provide an approach for the evaluation of competing environmental sources of exposure in the context of a nationally representative sample. In the following sections, we outline the procedures used for sample preparation and analysis, demonstrate that results are comparable between laboratories, and present preliminary data based on the analyses of samples from three households.
2. Materials and Methods
2.1. Reagents
Ultrapure water (UPW) from a Milli-Q Reference Water Purification System with a quality of 18.2 MΩ-cm was used throughout. Trace metal grade nitric acid (16 M) and 30% hydrogen peroxide solution for trace analysis were purchased from Fisher Scientific and Sigma-Aldrich, respectively. For total Pb analysis, the following reference materials were included to cover a range of sample types and Pb concentrations: ‘lead in powdered paint’ (Sigma-Aldrich, product ID SQC074), Seronorm Trace Elements in Whole Blood L-3 (SERO), GBW08505 tea leaves, SRM2711a Montana II Soil (NIST) BGS Guidance Material 102 Ironstone Soil (BGS), SRM2584 Trace Elements in Indoor Dust (NIST), and BCR-2 Basalt Columbia River (USGS). Of these reference materials, BCR-2 is the only one with Pb isotope ratio values included on the certificate of analysis. The natural Pb isotope standard SRM981 (NIST) was used as the Pb isotope reference material.
2.2. Sample Collection, Transport, and Storage
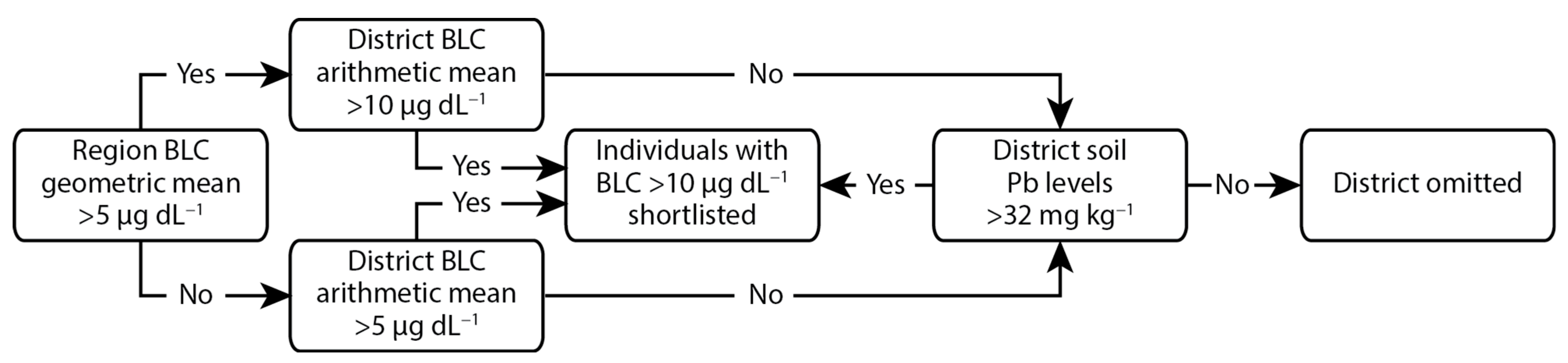
To focus on participants with the highest levels of Pb exposure, only children identified in the MICS as having BLCs > 10 µg dL
−1 were considered as potential candidates for this study. This covered 11 of the 13 regions of Georgia (2 regions occupied by Russia were not included), with each region being subdivided into districts. The criteria used to determine if potential candidates should be shortlisted from a given district is outlined in
Figure 2. Briefly, where the geometric mean of the BLC for a region was >5 µg dL
−1, only districts where the arithmetic mean of the BLC was >10 µg dL
−1 were included. This was the case for 5 of the regions, all in the west of the country. In the 6 regions where the geometric mean of the BLC was <5 µg dL
−1, only districts where the arithmetic mean of the BLC was >5 µg dL
−1 were included. The geometric mean was used for regional selection due to the BLC data from the MICS being skewed, with higher incidences of elevated BLCs and the highest BLCs being seen in the west of the country [
11]. Any districts that were not selected through the above criteria were still included where soil Pb concentrations, identified through routine state monitoring, exceeded the Georgian reference value of 32 mg kg
−1 [
26]. Prior to the final selection of a household, ethical approval was obtained from the NCDC institutional review board and the parents/guardians were contacted, informed of the aims of the study, invited to be included in the cohort, and provided signed written consent. Thirty-six households were recruited from the shortlisted participants, with a heavier weighting towards the regions and districts with the highest mean BLCs.
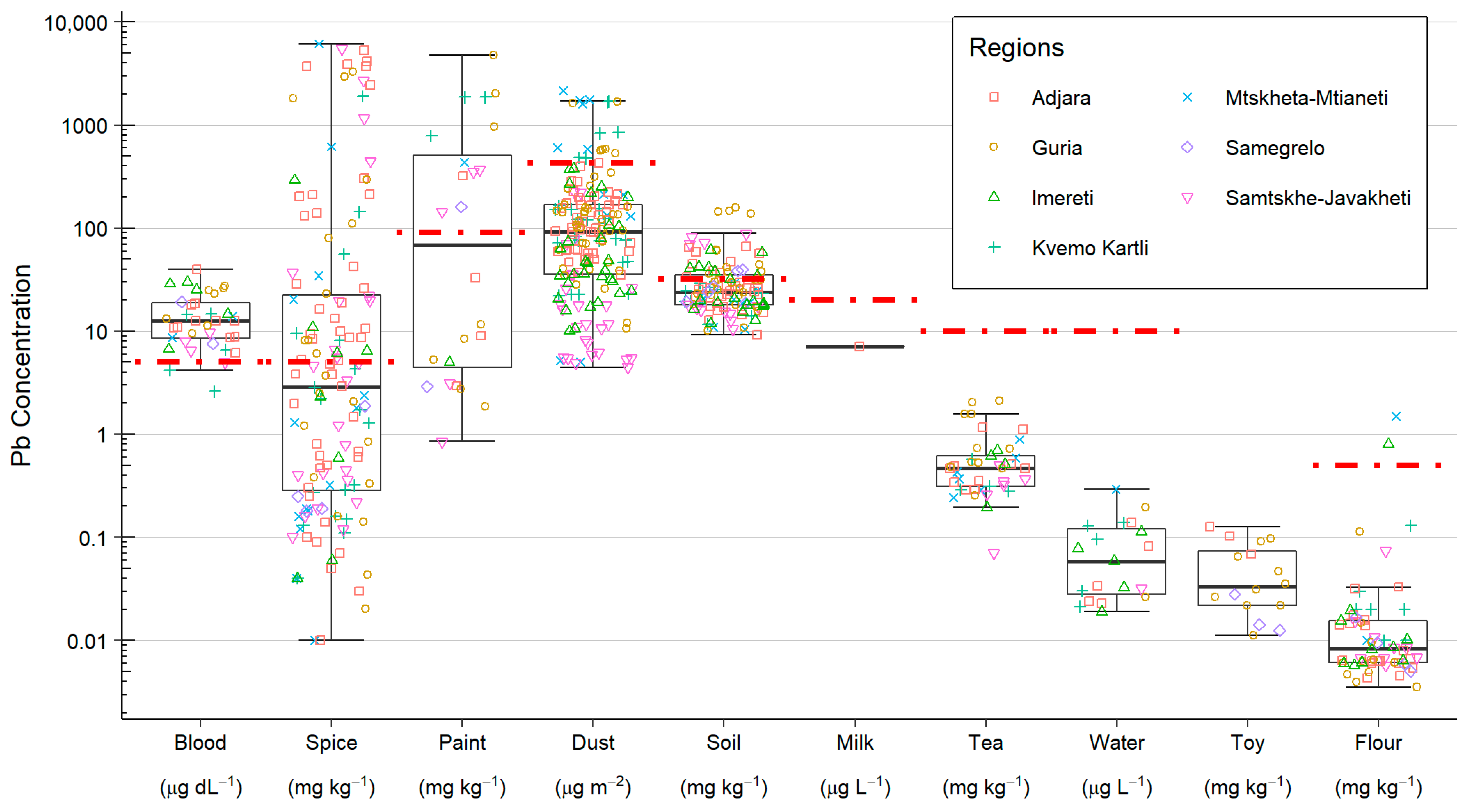
For sample collection, a phlebotomist and a trained NCDC scientist visited each household to collect samples and complete a questionnaire with the parents/guardians. In addition to blood samples from the child being taken during the visit, environmental samples of soil, spices, milk, water, household dust, flour (maize and wheat), paint, and toys were taken where available, being potential sources of Pb exposure for the child, and the parent/guardian gave informed consent. The types of environmental samples that were included in this study were selected based on them being reasonable potential sources of Pb, either from the published literature and/or informed by data and information from the Georgian National Food Agency and the Georgian Environment Agency [
21,
27,
28,
29].
A single blood sample was taken from the child in all 36 households. The sample was taken from the same child that gave a blood sample for the MICS in all but one instance, where a sibling provided the blood sample. Blood samples of 1–3 mL were collected by a phlebotomist in a vacutainer tube and frozen at −20 °C for storage.
A total of 60 soil and 2 construction sand samples were collected from gardens and yards that the children had access to, and where any vegetables or fruits were grown for household consumption. Approximately 200 g of sample, comprising 10–20 subsamples taken from different locations and a depth of 1–15 cm, was collected. These samples were stored at room temperature.
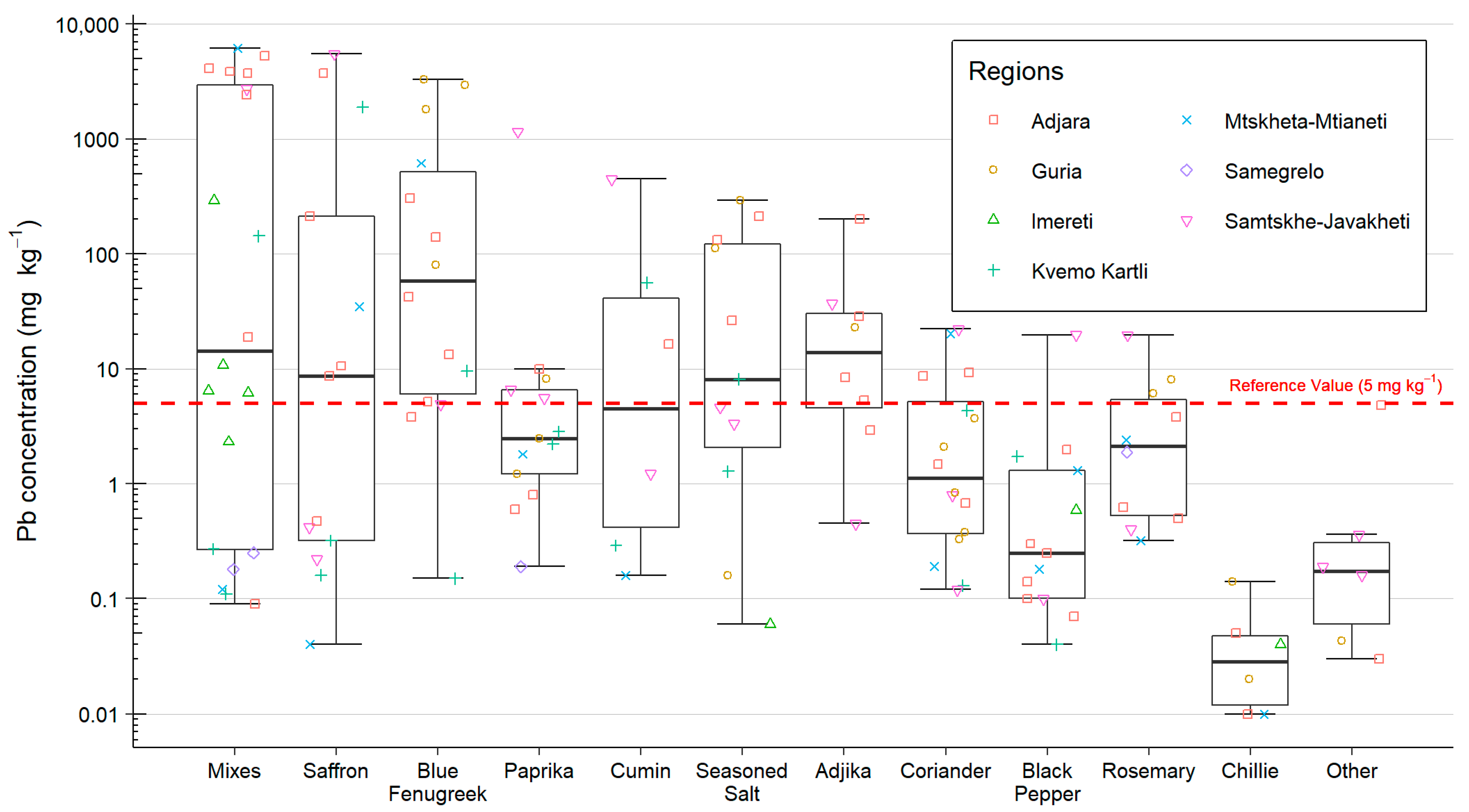
Aliquots of 2–3 g of the most frequently used spices in the household were sampled, up to a maximum of nine spice types per household. Spices were collected in individual zip lock bags, each labelled with the household reference and spice type. Spices from each household were grouped into plastic zip lock wallets and stored at room temperature.
Approximately 10–20 g of wheat and maize flour used in the household was taken and transferred to labelled zip lock bags and stored in the zip lock wallets with the spices. All households gave at least 1 flour sample, with 61 samples collected in total, including 2 samples of dried corn.
Water and milk samples of between 10 and 50 mL were collected in 50 mL polypropylene tubes and stored at −20 °C. In total, 15 households provided a milk sample sourced from their own domestic supply. All 36 households provided a sample of their main residential drinking water supply, with 17 of these taken from a mains residential water supply and 19 from a private or residential supply from a well or spring. A third (12) of the households also provided a sample from a second source where the child spent a substantial amount of time (e.g., school or family member’s house).
The sampling of household dust followed the procedure outlined by Middleton et al. [
30]. Briefly, samples were taken from appropriate raised, flat surfaces where dust is likely to collect, e.g., windowsills, shelves, and tops of wardrobes and cupboards. A plastic template was used to mark out an area 10 cm × 10 cm, then, using Ghost Wipes (Environmental Express, Charleston, SC, USA), the area was sampled and any dust was collected before being transferred to a labelled polypropylene tube. A total of 92 dust samples were collected from 30 households.
Paint samples were collected from 18 households, with 22 samples being collected in total. Samples were taken from both indoor and outdoor locations where painted surfaces were observed as flaking or chipping away. Paint flakes, chips, or dust were collected in labelled zip lock bags.
Where appropriate, toy samples were taken, with a focus on those where the child was known to put the toy in their mouth. In total, 16 toy samples were collected; of these, 12 were plastic toys (e.g., building blocks, animal figures, etc.), 2 were painted metal toys, and 2 were malleable toys (Play-Doh and kinetic sand). These toys were collected in labelled zip lock bags.
Tea samples were gathered from 25 households, with a total of 31 samples being collected, ranging from supermarket black tea bags, herbal teas, and loose tea leaves purchased from local bazaars. A single tea bag or 2–3 g of loose tea leaves was transferred to labelled zip lock bags and stored at room temperature.
2.3. Sample Preparation
Blood (1 mL), flour (0.5 g), spice (0.5 g), tea (0.5 g), paint (<0.25 g), and milk (1.5 mL) samples were prepared by microwave-assisted acid digestion. Apart from milk, these samples were prepared in UKHSA laboratories using an Anton Paar Multiwave Go microwave digestion system. Samples were weighed directly into digestion vessels followed by the digestion solution. To the blood samples, 1 mL HNO
3, 1 mL UPW, and 1 mL H
2O
2 were added. To flour, spice, and tea samples, 2 mL HNO
3, 2 mL UPW, and 1 mL H
2O
2 were added. To paint samples, 6 mL HNO
3 was added. For the blood, flour, spice, and tea sample digests, the temperature was ramped to 180 °C over 10 min and held for 20 min, while for paint samples, the temperature was ramped to 175 °C over 15 min and held for 15 min. Due to restrictions on the importation of dairy products, milk samples were prepared in Georgia by Laboratory Multitest, with 0.5 mL HNO
3 added and allowed to stand at room temperature for 15 min before the addition of 3 mL UPW. Microwave-assisted digestion was then performed using a Berghof Speedwave Two system, with temperatures ramped to 180 °C over 10 min and held for 20 min. A summary of sample types, amounts, reagents, and digestion protocol used are provided in
Table 1. Digested samples were transferred to 15 mL metal-free centrifuge tubes (VWR) and any samples containing undigested material were centrifuged and the supernatant removed. Samples were stored at room temperature prior to analysis.
Water samples were prepared in UKHSA laboratories and 3 mL aliquots were acidified to 5% HNO3 and analysed directly. To improve the limits of detection, the remaining samples were preconcentrated using a Genevac EZ-2 Evaporator to reduce sample volumes of up to 50 mL to a final volume of 5 mL in 5% HNO3.
To assess the availability of Pb from toy samples, a metal migration protocol was performed as outlined in BSI standard BS EN 71-3:2019 [
31]. The toy material was placed into a 0.07 mol L
−1 HCl solution at a mass ratio of 1:50. The mixture was agitated for 1 h and left to stand for an additional hour. The solid samples were then removed and the solutions acidified to 5% HNO
3.
Soil samples were prepared in BGS laboratories, with 0.25 g powdered soil weighed into PFA vials. A pre-digest was made by adding 8 M HNO3 to the soil and slowly drying down overnight to decompose any organic component. A mixed acid attack consisting of 2 mL 16 M HNO3, 2.5 mL HF, and 1 mL 9.2 M HClO4 was performed by stepped heating using a programmable heat block. The final heating stage was used to dry down the sample. Once cool, the samples were taken up in 2.5 mL 8 M HNO3, 2.5 mL H2O2, and 20 mL UPW, and transferred to LDPE bottles.
Dust wipe samples were also prepared in BGS laboratories, where they were carefully placed in a disposable DigiTUBE (SCP Science) tube using plastic tweezers. A 10 mL aliquot of 8 M HNO3 was added to the sample and left for a minimum of 10 min to allow any initial reaction to die down. A further 4 mL 16 M HNO3, 2.5 mL HCl, and 1 mL HF were added before covering with a plastic watch glass and heating overnight at 70 °C on a temperature-controlled hot block. The watch glass was removed and the samples further heated to 110 °C before cooling and the further addition of 4 mL 16 M HNO3 and 1 mL H2O2. A final drying at 90 °C was made until samples achieved a gel-like consistency. The samples were reconstituted with 1 mL 8M HNO3 followed by the addition of 9 mL UPW.
2.4. Total Pb Concentration Determination
All samples were analysed for total Pb in UKHSA laboratories, where the samples were diluted to 5% HNO
3 with UPW and any additional dilution was performed with a stock solution of 5% HNO
3 as required. Analysis was performed using a Thermo iCAP-Q ICP-MS instrument operating in helium collision cell mode and equipped with an ASX-520 autosampler. Calibrations were prepared using standard solutions with Pb concentrations of 0, 0.05, 0.1, 0.5, 1, 5, 10, 50, and 100 µg L
−1, with a mixing tee used for online internal standard addition of a solution containing 1 µg L
−1 of Rh. Quality control solutions with Pb of known concentrations were analysed periodically between samples. Quality control solutions of the reference materials SRM2711a, BCR-2, BGS 102, SQC074, and Seronorm Trace Elements in Whole Blood L-3 were analysed between samples, with a summary of this data provided in
Table S1.
All soil and dust samples, and 10 of the tea samples, were also analysed in BGS laboratories, where the sample digests were diluted by a factor of 40 using a mixture of 1%
v/
v 16 M nitric acid and 0.5%
v/
v 12 M hydrochloric acid. Analysis was performed using an Agilent 8900 ICP-MS instrument operating in helium collision cell mode and equipped with an SPS4 auto-sampler and an ISIS flow injection system. The instrument was calibrated for Pb using 1, 5, 10, 25, 50, and 100 µg L
−1 dilutions of a custom multi-element mixture from QMX (UK). A quality control solution of SRM2711a, SRM2584, BCR-2, BGS 102, and GBW08505 with a Pb concentration of 25 µg L
−1 was analysed between samples, with a summary of this data provided in
Table S1.
2.5. Pb Isotope Ratio Analysis
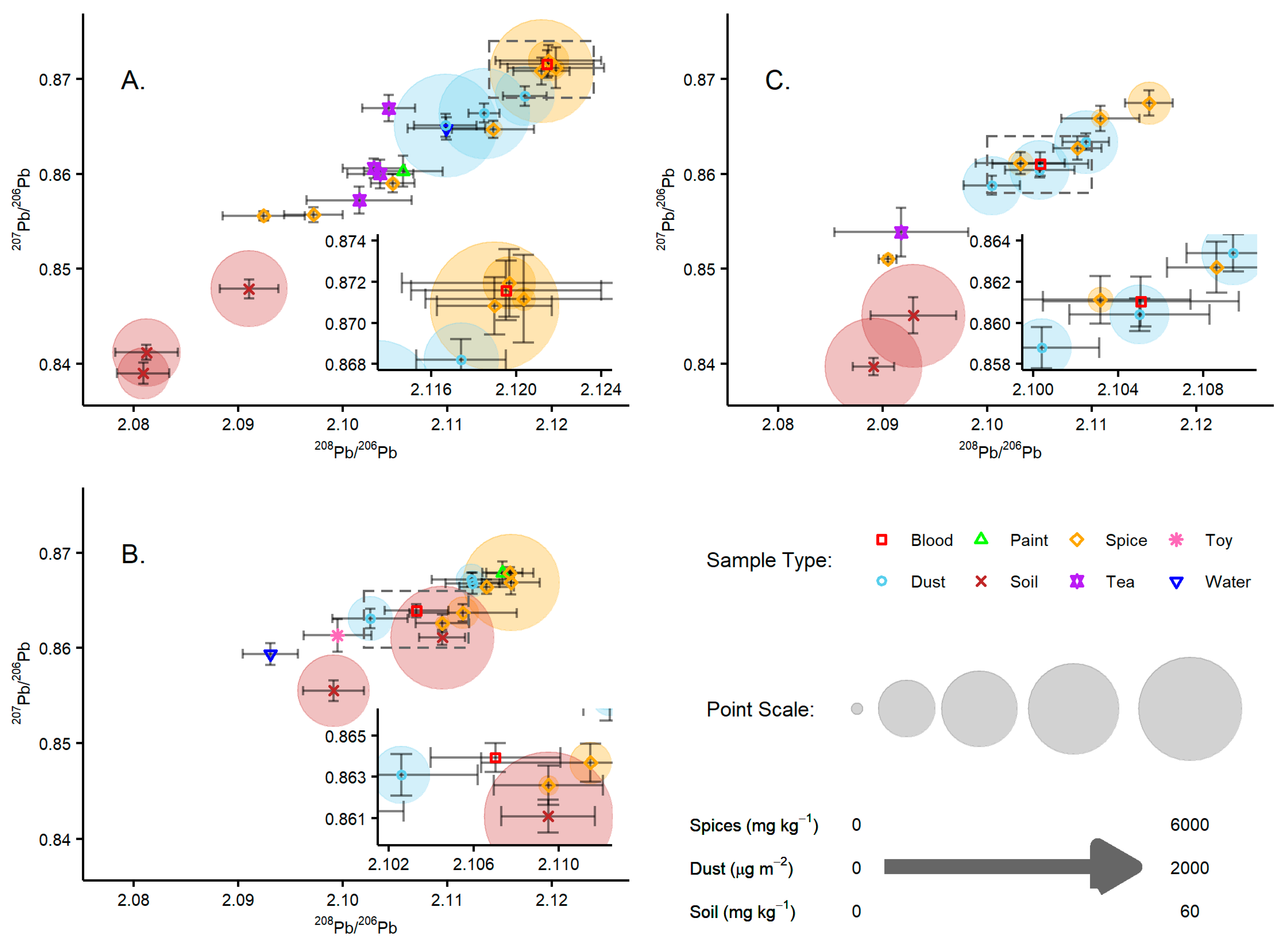
Of the 36 households included in this study, the blood and environmental samples from 3 households, hereby referred to as A, B, and C, were selected for Pb isotope ratio analysis. These 3 households were selected based on the types of environmental samples provided and their total Pb concentrations, such that all sample types were included in the isotope analysis and these covered a range of the observed total Pb concentrations.
The sample solutions of blood, paint, spices, tea, flour wheat, and toys were analysed for their Pb isotope composition in UKHSA laboratories. The sample solutions of soil and dust were analysed in BGS laboratories. In addition, four each of the soil and dust samples were analysed in UKHSA laboratories to demonstrate comparability.
The sample analysis and raw data processing performed in UKHSA laboratories were based on the optimised approach developed by Usman et al. [
32]. A Thermo Scientific iCAP Q ICP-MS was used for all analyses, measurements were performed in helium collision mode, and the following masses were monitored:
200Hg,
203Tl,
204Pb/Hg,
205Tl,
206Pb,
207Pb, and
208Pb. Dwell times of 10 ms were used for each mass, 1000 sweeps per scan were performed, and each analysis consisted of six scans. This resulted in an acquisition time of approximately 7 min, including time for sample uptake and a post-analysis rinse cycle. To ensure that precise and accurate isotope ratios can be determined, it is essential that all masses are monitored in pulse detection mode. When signal intensities reach a certain threshold, the detection mode will switch from pulse to analogue mode; this transition is at approximately 2 × 10
6 cps. The optimal concentration is therefore low enough for the major
208Pb isotope to be monitored without switching to the analogue detection mode while providing a reasonable signal intensity to monitor the minor
204Pb isotope.
The deadtime correction factor was established at the start of each measurement session [
33]. This was achieved by preparing SRM981 standard solutions at concentrations of 0, 1, 3, 5, 7, and 9 µg L
−1. With the instrumental deadtime set to zero, these solutions were analysed as above. If the detector switched to analogue mode while monitoring
208Pb, the data were omitted. A blank correction was applied to the Pb isotope intensities by subtracting the baseline values from the measured signal intensities (I
blank corr). The blank corrected intensities were corrected further by changing the dead time (DT), in seconds, using Equation (1).
The 208Pb/207Pb and 208Pb/206Pb ratios were determined using the blank and dead time corrected intensities (IDT corr) plotted against the signal intensity for 208Pb. The optimal deadtime value for each isotope ratio is when the slope squared is minimised. This value can be readily found using the Microsoft Excel Solver function. The average optimised dead time for the 208Pb/207Pb and 208Pb/206Pb ratios was calculated for the measurement session and set in the instrumental software prior to sample analysis; this ranged from 36–42 × 10−9 s across all measurement sessions.
It was determined that the optimal sample Pb concentration for isotope ratio analysis was 7 µg L−1. Based on the total Pb concentrations, aliquots of the stock sample digests were prepared at this concentration in 5% HNO3 and dosed with Tl to 5 µg L−1 with a 1000 µg L−1 Tl standard stock solution. Where Pb concentrations in the stock sample solutions were insufficient to achieve a Pb concentration of 7 µg L−1, they were analysed without further dilution. Along with the samples, a standard solution of 7 µg L−1 SRM981 Pb spiked with 5 µg L−1 Tl was prepared in 5% HNO3.
The raw signal intensities for each isotope for each run were exported into an Excel file for off-line data processing. They were blank corrected and the following ratios were determined:
205Tl/
203Tl,
208Pb/
207Pb,
208Pb/
206Pb,
204PbHg/
208Pb,
200Hg/
208Pb,
207Pb/
206Pb,
204PbHg/
207Pb,
200Hg/
207Pb,
204PbHg/
206Pb, and
200Hg/
206Pb. Thallium was used for mass bias correction as it is very close in mass to Pb isotopes without having any isobaric interference. Mass bias per unit mass was determined by comparing the measured
205Tl/
203Tl ratio (
205Tl/
203Tl
meas) to the known reference value (
205Tl/
203Tl
ref) of 2.3875. The mass discrimination factor (K) was estimated using the linear law, as shown in Equation (2).
where
205Tl
mass and
203Tl
mass correspond to the atomic masses of
205Tl and
203Tl, respectively. The calculated mass bias correction factor for each run was used to calculate mass bias corrected isotope ratios (IR
corr) for each of the measured isotope ratios (IR
meas) using Equation (3).
where Δm is the difference in the atomic mass between the two isotopes in the ratio. The mass bias corrected
200Hg/
20xPb ratios (where ‘
x’ represents 6, 7, or 8) were then used, with the known
204Hg/
200Hg reference value (
204Hg/
200Hg
ref) of 0.2974, to correct for the isobaric interference of
204Hg on the
204PbHg/
20xPb mass bias corrected ratios (
204PbHg/
20xPb
corr) to derive the mass bias corrected
20xPb/
204Pb ratios (
20xPb/
204Pb
corr).
The SRM981 Pb standard was analysed at least once every 10 samples. For each standard analysis, the deviation in the mass bias corrected ratio from the true reference ratio was determined. Linear interpolation of the deviation in the SRM981 Pb standard was used to apply this correction to samples analysed over the measurement session to derive the final, true isotope ratios. The analytical uncertainty was derived by calculating twice the standard error of the mean of the six scans for each of the isotope ratios.
The Pb isotope analysis performed in BGS laboratories used a comparable approach as outlined above with a few small differences. Analysis was performed using an Agilent 8900 ICP-MS instrument equipped with an SPS4 auto-sampler and an ISIS flow injection system. Samples were diluted by an individual factor calculated from Pb concentrations and instrument sensitivity to give a
208Pb
+ count rate of c. 1 × 10
6 cps to optimise counting statistics, within the pulse counting range of the detector c. 1.6 × 10
6 cps. The isotopes
200Hg
+,
204Pb
+,
206Pb
+,
207Pb
+, and
208Pb
+ were acquired with dwell times to give 1000 sweeps per integration and 10 integrations over 5 min. Thallium was not used to correct mass bias as the BGS has previously found that this showed no improvement over only standard-10 × sample-standard bracketing. Quality control was provided by repeatedly analysing an in-house UK ore Pb (Glendenning) throughout the runs for which the BGS has more than five years of isotope ratio data, including from high-precision multi-collector ICP-MS analysis. A summary of the quality control analysis is provided in
Table S2.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}