
Determination of Hydroxy Polycyclic Aromatic Hydrocarbons in Human Urine Using Automated Microextraction by Packed Sorbent and Gas Chromatography–Mass Spectrometry

 , , , and
, , , and
Abstract
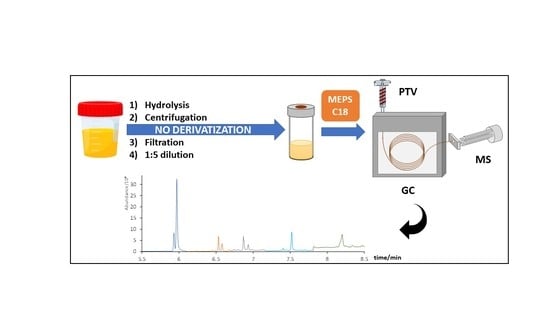
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Standards
2.2. Synthetic Urine and Urine Samples
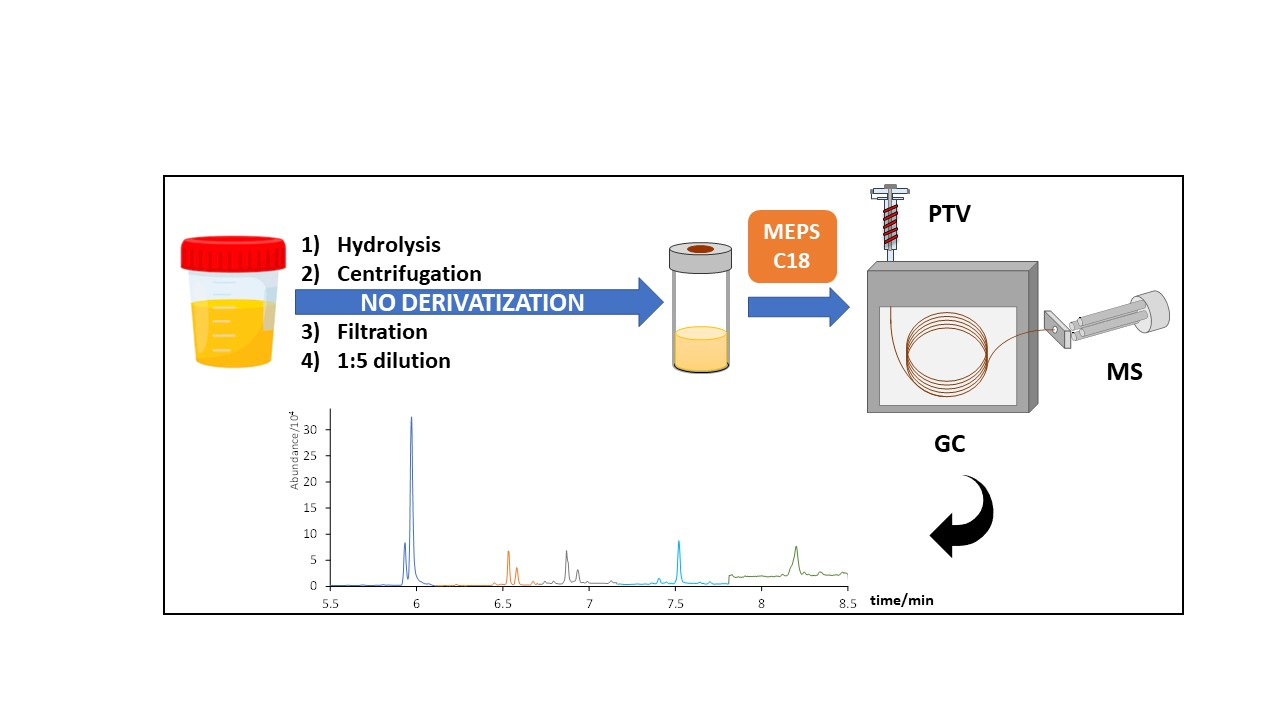
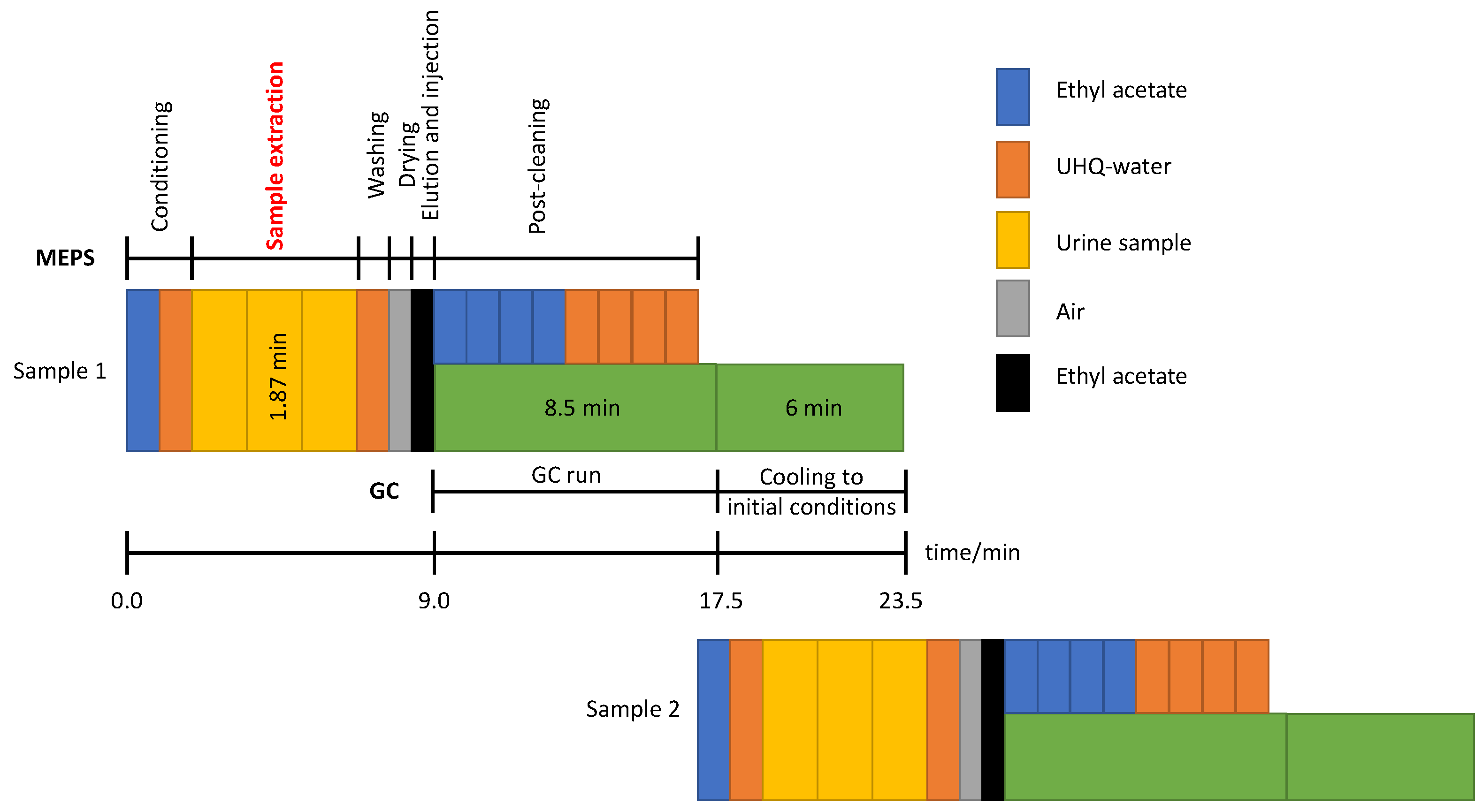
2.3. Hydrolysis, MEPS Extraction Procedure, and Instrumental Conditions
3. Results and Discussion
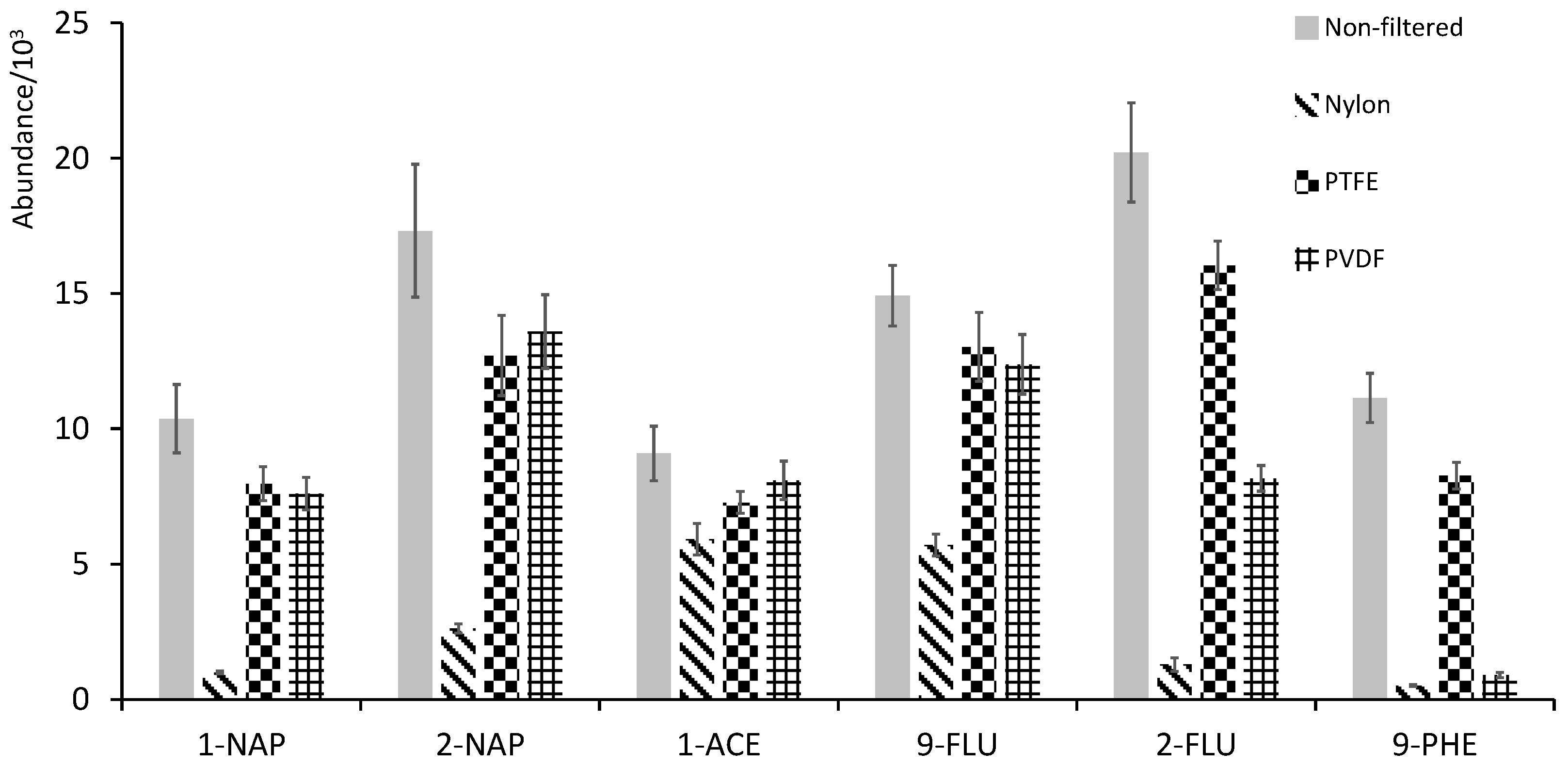
3.1. Optimization of Sample Preparation—Matrix Effect
3.2. Optimization of the MEPS Procedure
3.3. PTV-GC-MS Conditions
3.4. Method Validation
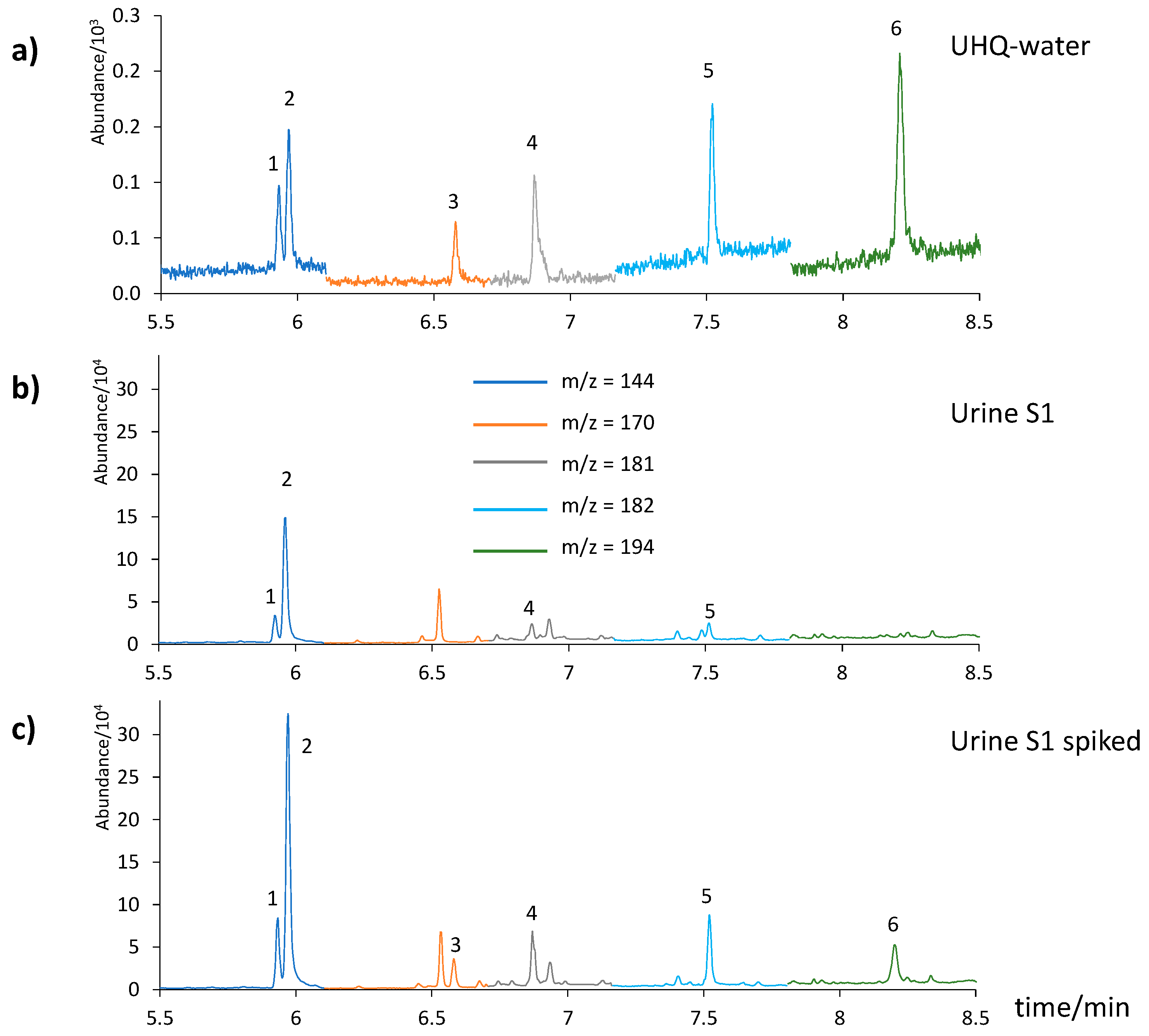
3.5. Analysis of Urine Samples
3.6. Comparison with Other Works
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, K.H.; Jahan, S.A.; Kabir, E.; Brown, R.J. A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Abbas, I.; Badran, G.; Verdin, A.; Ledoux, F.; Roumié, M.; Courcot, D.; Garçon, G. Polycyclic aromatic hydrocarbon derivatives in airborne particulate matter: Sources, analysis and toxicity. Environ. Chem. Lett. 2018, 16, 439–475. [Google Scholar] [CrossRef]
- Moazzen, M.; Ahmadkhaniha, R.; Gorji, M.E.; Yunesian, M.; Rastkari, N. Magnetic solid-phase extraction based on magnetic multi-walled carbon nanotubes for the determination of polycyclic aromatic hydrocarbons in grilled meat samples. Talanta 2013, 115, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Seidel, A. Biomonitoring of polycyclic aromatic hydrocarbons in human urine. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 778, 31–47. [Google Scholar] [CrossRef]
- Romanoff, L.C.; Li, Z.; Young, K.J.; Blakely, N.C.; Patterson, D.G.; Sandau, C.D. Automated solid-phase extraction method for measuring urinary polycyclic aromatic hydrocarbon metabolites in human biomonitoring using isotope-dilution gas chromatography high-resolution mass spectrometry. J. Chromatogr. B 2006, 835, 47–54. [Google Scholar] [CrossRef]
- Gao, Y.; Li, L.; Zhang, X.; Wang, X.; Ji, W.; Zhao, J.; Ozaki, Y. CTAB-triggered Ag aggregates for reproducible SERS analysis of urinary polycyclic aromatic hydrocarbon metabolites. Chem. Commun. 2019, 55, 2146–2149. [Google Scholar] [CrossRef] [PubMed]
- Marie-Desvergne, C.; Maitre, A.; Bouchard, M.; Ravanat, J.L.; Viau, C. Evaluation of DNA adducts, DNA and RNA oxidative lesions, and 3-hydroxybenzo(a)pyrene as biomarkers of DNA damage in lung following intravenous injection of the parent compound in rats. Chem. Res. Toxicol. 2010, 23, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Grova, N.; Fays, F.; Hardy, E.M.; Appenzeller, B.M.R. New insights into urine-based assessment of polycyclic aromatic hydrocarbon-exposure from a rat model: Identification of relevant metabolites and influence of elimination kinetics. Environ. Pollut. 2017, 228, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Lai, Y.; Liu, C.W.; Ru, H. Towards Mass Spectrometry-Based Chemical Exposome: Current Approaches, Challenges, and Future Directions. Toxics 2019, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Luo, Y.; Bi, J.; Li, H.; Lin, J.-M. Quantification of selected monohydroxy metabolites of polycyclic aromatic hydrocarbons in human urine. Sci. China Chem. 2015, 58, 1579–1584. [Google Scholar] [CrossRef]
- Diaz de Leon-Martinez, L.; Flores-Ramirez, R.; Rodriguez-Aguilar, M.; Berumen-Rodriguez, A.; Perez-Vazquez, F.J.; Diaz-Barriga, F. Analysis of urinary metabolites of polycyclic aromatic hydrocarbons in precarious workers of highly exposed occupational scenarios in Mexico. Environ. Sci. Pollut. Res. Int. 2021, 28, 23087–23098. [Google Scholar] [CrossRef] [PubMed]
- Woudneh, M.B.; Benskin, J.P.; Grace, R.; Hamilton, M.C.; Magee, B.H.; Hoeger, G.C.; Forsberg, N.D.; Cosgrove, J.R. Quantitative determination of hydroxy polycylic aromatic hydrocarbons as a biomarker of exposure to carcinogenic polycyclic aromatic hydrocarbons. J. Chromatogr. A 2016, 1454, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhu, S.; Zheng, Z.; Sun, J.; Zhao, X.E.; Liu, H. 9-Plex ultra high performance liquid chromatography tandem mass spectrometry determination of free hydroxyl polycyclic aromatic hydrocarbons in human plasma and urine. J. Chromatogr. A 2020, 1623, 461182. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Huang, S.; Kuang, Y.; Chen, Z.; Guo, J.; Cui, S.; Zheng, J.; Ouyang, G. Facile fabrication of composited solid phase microextraction thin membranes for sensitive detections of trace hydroxylated polycyclic aromatic hydrocarbons in human urine. Anal. Chim. Acta 2021, 1158, 338422. [Google Scholar] [CrossRef]
- Luo, K.; Gao, Q.; Hu, J. Derivatization method for sensitive determination of 3-hydroxybenzo[a]pyrene in human urine by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. A 2015, 1379, 51–55. [Google Scholar] [CrossRef]
- Li, Z.; Romanoff, L.C.; Trinidad, D.A.; Hussain, N.; Jones, R.S.; Porter, E.N.; Patterson, D.G., Jr.; Sjodin, A. Measurement of urinary monohydroxy polycyclic aromatic hydrocarbons using automated liquid-liquid extraction and gas chromatography/isotope dilution high-resolution mass spectrometry. Anal. Chem. 2006, 78, 5744–5751. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.M.; del Nogal Sánchez, M.; Pavón, J.L.P.; Cordero, B.M. Determination of polycyclic aromatic hydrocarbons in human biological samples: A critical review. TrAC Trends Anal. Chem. 2019, 113, 194–209. [Google Scholar] [CrossRef]
- Martín Santos, P.; Jiménez Carracedo, C.; del Nogal Sánchez, M.; Pérez Pavón, J.L.; Moreno Cordero, B. A sensitive and automatic method based on microextraction by packed sorbents for the determination of polycyclic aromatic hydrocarbons in saliva samples. Microchem. J. 2020, 152, 104274. [Google Scholar] [CrossRef]
- Martín Santos, P.; del Nogal Sánchez, M.; Pérez Pavón, J.L.; Moreno Cordero, B. Quantitative and qualitative analysis of polycyclic aromatic hydrocarbons in urine samples using a non-separative method based on mass spectrometry. Talanta 2018, 181, 373–379. [Google Scholar] [CrossRef]
- Yang, B.C.; Fang, S.F.; Wan, X.J.; Luo, Y.; Zhou, J.Y.; Li, Y.; Li, Y.J.; Wang, F.; Huang, O.P. Quantification of monohydroxylated polycyclic aromatic hydrocarbons in human urine samples using solid-phase microextraction coupled with glass-capillary nanoelectrospray ionization mass spectrometry. Anal. Chim. Acta 2017, 973, 68–74. [Google Scholar] [CrossRef]
- van de Merbel, N.C. Quantitative determination of endogenous compounds in biological samples using chromatographic techniques. TrAC Trends Anal. Chem. 2008, 27, 924–933. [Google Scholar] [CrossRef]
- Gonzalez Paredes, R.M.; Garcia Pinto, C.; Perez Pavon, J.L.; Moreno Cordero, B. Derivatization coupled to headspace programmed-temperature vaporizer gas chromatography with mass spectrometry for the determination of amino acids: Application to urine samples. J. Sep. Sci. 2016, 39, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Cuadros-Rodriguez, L.; Bagur-Gonzalez, M.G.; Sanchez-Vinas, M.; Gonzalez-Casado, A.; Gomez-Saez, A.M. Principles of analytical calibration/quantification for the separation sciences. J. Chromatogr. A 2007, 1158, 33–46. [Google Scholar] [CrossRef]
- Krupcik, J.; Majek, P.; Gorovenko, R.; Blasko, J.; Kubinec, R.; Sandra, P. Considerations on the determination of the limit of detection and the limit of quantification in one-dimensional and comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2015, 1396, 117–130. [Google Scholar] [CrossRef]
- Wilhelm, M.; Hardt, J.; Schulz, C.; Angerer, J.; Human Biomonitoring Commission of the German Federal Environment Agency. New reference value and the background exposure for the PAH metabolites 1-hydroxypyrene and 1- and 2-naphthol in urine of the general population in Germany: Basis for validation of human biomonitoring data in environmental medicine. Int. J. Hyg. Environ. Health 2008, 211, 447–453. [Google Scholar] [CrossRef]
- Shang, T.; Zhao, L.-J.; Li, P.; Zeng, X.-Y.; Yu, Z.-Q. Determination of Ten Kinds of Monohydroxylated Polycyclic Aromatic Hydrocarbons in Human Urine by Supported Liquid Extraction Followed by Liquid Chromatography-Tandem Mass Spectrometry. Chin. J. Anal. Chem. 2019, 47, 876–882. [Google Scholar] [CrossRef]
- Fan, R.; Wang, D.; Ramage, R.; She, J. Fast and simultaneous determination of urinary 8-hydroxy-2’-deoxyguanosine and ten monohydroxylated polycyclic aromatic hydrocarbons by liquid chromatography/tandem mass spectrometry. Chem. Res. Toxicol. 2012, 25, 491–499. [Google Scholar] [CrossRef]
- Nguyen, K.; Pitiranggon, M.; Wu, H.C.; John, E.M.; Santella, R.M.; Terry, M.B.; Yan, B. Improvement on recovery and reproducibility for quantifying urinary mono-hydroxylated polycyclic aromatic hydrocarbons (OH-PAHs). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2022, 1192, 123113. [Google Scholar] [CrossRef]
- Peña, J.; Fernández Laespada, M.E.; García Pinto, C.; Pérez Pavón, J.L. Multiple headspace sampling coupled to a programmed temperature vaporizer—Gas chromatograph-mass spectrometer for the determination of polycyclic aromatic hydrocarbons in water and saliva. Microchem. J. 2021, 161, 105822. [Google Scholar] [CrossRef]
- Martín Santos, P.; Campo, L.; Olgiati, L.; Polledri, E.; del Nogal Sánchez, M.; Fustinoni, S. Development of a method to profile 2- to 4-ring polycyclic aromatic hydrocarbons in saliva samples from smokers and non-smokers by headspace-solid-phase microextraction-gas chromatography-triple quadrupole tandem mass spectrometry. J. Chromatogr. B 2020, 1152, 122273. [Google Scholar] [CrossRef]
- Hua, L.; Guo, S.; Wang, Y.; Sun, H.; Zhao, H. Simultaneous determination of multiple isomeric hydroxylated polycyclic aromatic hydrocarbons in urine by using ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2021, 1184, 122983. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
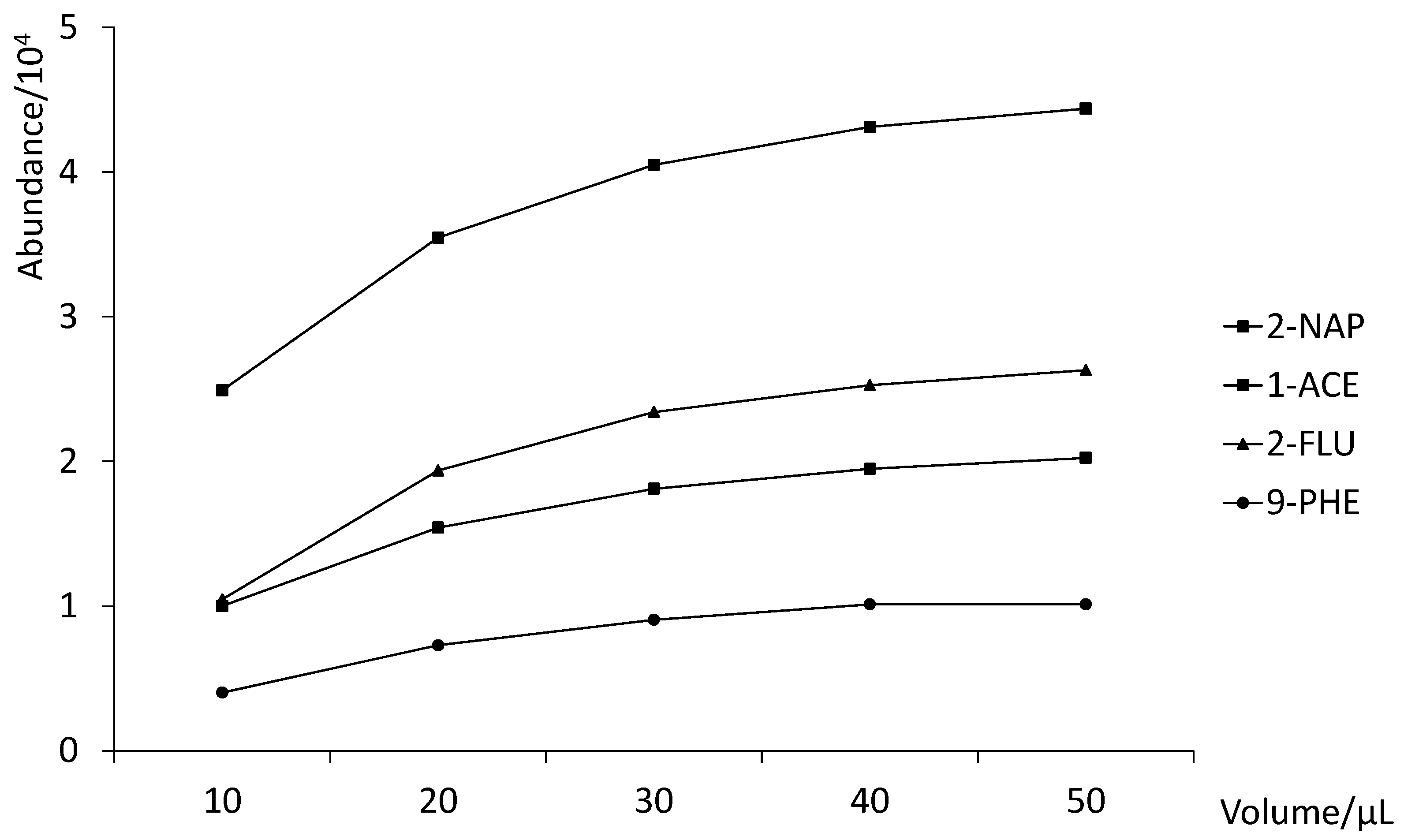{kind=link}
{kind=link}
| Analyte | UHQ-Water | Synthetic Urine | S1 | S2 | S6 |
|---|---|---|---|---|---|
| 1-NAP | 42.0 ± 0.8 | 35 ± 1 | 58 ± 2 | 50 ± 2 | 27 ± 8 |
| 2-NAP | 75 ± 2 | 66 ± 3 | 111 ± 6 | 106 ± 3 | 143 ± 9 |
| 1-ACE | 48 ± 2 | 42 ± 2 | 67 ± 3 | 73 ± 2 | 57 ± 6 |
| 9-FLU | 57 ± 2 | 48 ± 2 | 94 ± 3 | 104 ± 4 | 124 ± 5 |
| 2-FLU | 72 ± 2 | 63 ± 3 | 94 ± 7 | 98 ± 5 | 102 ± 2 |
| 9-PHE | 44 ± 2 | 27 ± 1 | 16 ± 2 | 14 ± 5 | 19 ± 3 |
| Compound | tR/min | Quantifier and Qualifier Ions | SIM Group |
|---|---|---|---|
| 1-NAP | 5.89 | 144, 115, 116 | 1 |
| 2-NAP | 5.93 | 144, 115, 116 | |
| 1-ACE | 6.56 | 170, 169, 152 | 2 |
| 9-FLU | 6.85 | 181, 182, 152 | 3 |
| 2-FLU | 7.48 | 182, 181, 152 | |
| 9-PHE | 8.16 | 194, 165, 166 | 4 |
| Compound | Calibration Range (ng L−1) | Linear Equation | R2 | LOD (ng L−1) | LOQ (ng L−1) | Intra-Day Repeatability (RSD, %) | Inter-Day Repeatability (RSD, %) | ||
|---|---|---|---|---|---|---|---|---|---|
| Low Level a | High Level b | Low Level a | High Level b | ||||||
| 1-NAP | LOQ-800 | y = (42 ± 1) x + (2 ± 3) 102 | 0.9985 | 14.9 | 49.7 | 9.7 | 4.9 | 23.4 | 8.3 |
| 2-NAP | LOQ-500 | y = (75 ± 2) x + (0 ± 6) 102 | 0.9956 | 11.6 | 38.7 | 7.1 | 4.3 | 13.3 | 4.9 |
| 1-ACE | LOQ-1200 | y = (48 ± 2) x − (0 ± 1) 103 | 0.9930 | 14.6 | 48.6 | 5.7 | 2.7 | 13.0 | 3.3 |
| 9-FLU | LOQ-1200 | y = (57 ± 2) x + (0 ± 1) 103 | 0.9957 | 10.1 | 33.6 | 5.9 | 2.1 | 11.9 | 6.0 |
| 2-FLU | LOQ-1200 | y = (72 ± 2) x − (0 ± 1) 103 | 0.9954 | 10.8 | 36.0 | 9.0 | 4.5 | 12.0 | 11.5 |
| 9-PHE | LOQ-18000 | y = (44 ± 2) x − (0 ± 1) 104 | 0.9932 | 39.6 | 132.2 | 9.2 | 5.7 | 10.8 | 14.3 |
| Compound | Calibration Range (µg L−1) | R2 | LOD * (µg L−1) | LOQ * (µg L−1) | Intra-Day Repeatability (RSD, %) | Inter-Day Repeatability (RSD, %) | ||
|---|---|---|---|---|---|---|---|---|
| Low Level a | High Level b | Low Level a | High Level b | |||||
| 1-NAP | LOQ-62 | 0.9977 | 0.6–4.4 | 2.2–14.7 | 7.0 | 8.5 | 12.7 | 7.6 |
| 2-NAP | LOQ-375 | 0.9989 | 0.7–2.6 | 2.3–8.5 | 6.3 | 4.8 | 11.1 | 9.4 |
| 1-ACE | LOQ-62 | 0.9947 | 0.7–7.3 | 2.2–24.0 | 6.0 | 5.1 | 10.6 | 9.6 |
| 9-FLU | LOQ-75 | 0.9955 | 0.7–5.4 | 2.2–18.3 | 5.8 | 3.7 | 11.8 | 10.3 |
| 2-FLU | LOQ-60 | 0.9990 | 0.5–2.1 | 1.5–7.0 | 5.7 | 6.3 | 19.0 | 14.9 |
| 9-PHE | LOQ-125 | 0.9939 | 6.1–19.4 | 20.6–65.6 | 7.9 | 6.5 | 14.7 | 8.9 |
| Compound | UHQ-Water | S4 | S6 | |||
|---|---|---|---|---|---|---|
| Low Level a | High Level b | Low Level a | High Level b | Low Level a | High Level b | |
| 1-NAP | 97 | 89 | 79 | 78 | 68 | 64 |
| 2-NAP | 78 | 74 | 52 | 60 | 49 | 52 |
| 1-ACE | 52 | 59 | 43 | 52 | 36 | 41 |
| 9-FLU | 66 | 64 | 57 | 63 | 51 | 53 |
| 2-FLU | 68 | 81 | 57 | 75 | 51 | 62 |
| 9-PHE | 48 | 70 | 15 | 17 | 11 | 15 |
| Compound | Added Concentration (µg L−1) | Found Concentration (µg L−1) | Accuracy (%) |
|---|---|---|---|
| 1-NAP | 3.2 | 3.1 ± 0.2 | 97 |
| 2-NAP | 9.9 | 9 ± 1 | 91 |
| 1-ACE | 3.2 | 2.8 ± 0.2 | 88 |
| 9-FLU | 2 | 1.8 ± 0.1 | 90 |
| 2-FLU | 2.1 | 2.1 ± 0.1 | 100 |
| 9-PHE | 10.0 | 11 ± 3 | 110 |
| Subject | 1-NAP | 2-NAP | 9-FLU | 2-FLU |
|---|---|---|---|---|
| S1 | 17.7 ± 0.9 | 56 ± 5 | 3.8 ± 0.7 | 4.2 ± 0.5 |
| S2 | 6 ± 2 | 37 ± 6 | ND | 4.3 ± 0.4 |
| S3 | 13 ± 3 | 52 ± 9 | 7 ± 2 | 3 ± 2 |
| S4 | 7 ± 1 | 9 ± 2 | ND | <LOQ |
| S5 | <LOQ | 14 ± 3 | 3.2 ± 0.5 | ND |
| S6 | ND | 20 ± 3 | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-García, S.; Matilla-González, H.; Peña, J.; Nogal Sánchez, M.d.; Casas-Ferreira, A.M.; Pérez Pavón, J.L. Determination of Hydroxy Polycyclic Aromatic Hydrocarbons in Human Urine Using Automated Microextraction by Packed Sorbent and Gas Chromatography–Mass Spectrometry. Int. J. Environ. Res. Public Health 2022, 19, 13089. https://doi.org/10.3390/ijerph192013089
García-García S, Matilla-González H, Peña J, Nogal Sánchez Md, Casas-Ferreira AM, Pérez Pavón JL. Determination of Hydroxy Polycyclic Aromatic Hydrocarbons in Human Urine Using Automated Microextraction by Packed Sorbent and Gas Chromatography–Mass Spectrometry. International Journal of Environmental Research and Public Health. 2022; 19(20):13089. https://doi.org/10.3390/ijerph192013089
Chicago/Turabian StyleGarcía-García, Samuel, Héctor Matilla-González, Javier Peña, Miguel del Nogal Sánchez, Ana María Casas-Ferreira, and José Luis Pérez Pavón. 2022. "Determination of Hydroxy Polycyclic Aromatic Hydrocarbons in Human Urine Using Automated Microextraction by Packed Sorbent and Gas Chromatography–Mass Spectrometry" International Journal of Environmental Research and Public Health 19, no. 20: 13089. https://doi.org/10.3390/ijerph192013089