
Compositional Data Analysis of 16S rRNA Gene Sequencing Results from Hospital Airborne Microbiome Samples

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection Methodology for Bioaerosol Samples
2.2. Description of the Sampling Locations
2.3. Methodology for DNA Extraction and 16S rRNA Gene Metabarcoding Approach
2.4. Statistical Techniques for Compositional Data Analysis
3. Results and Discussion
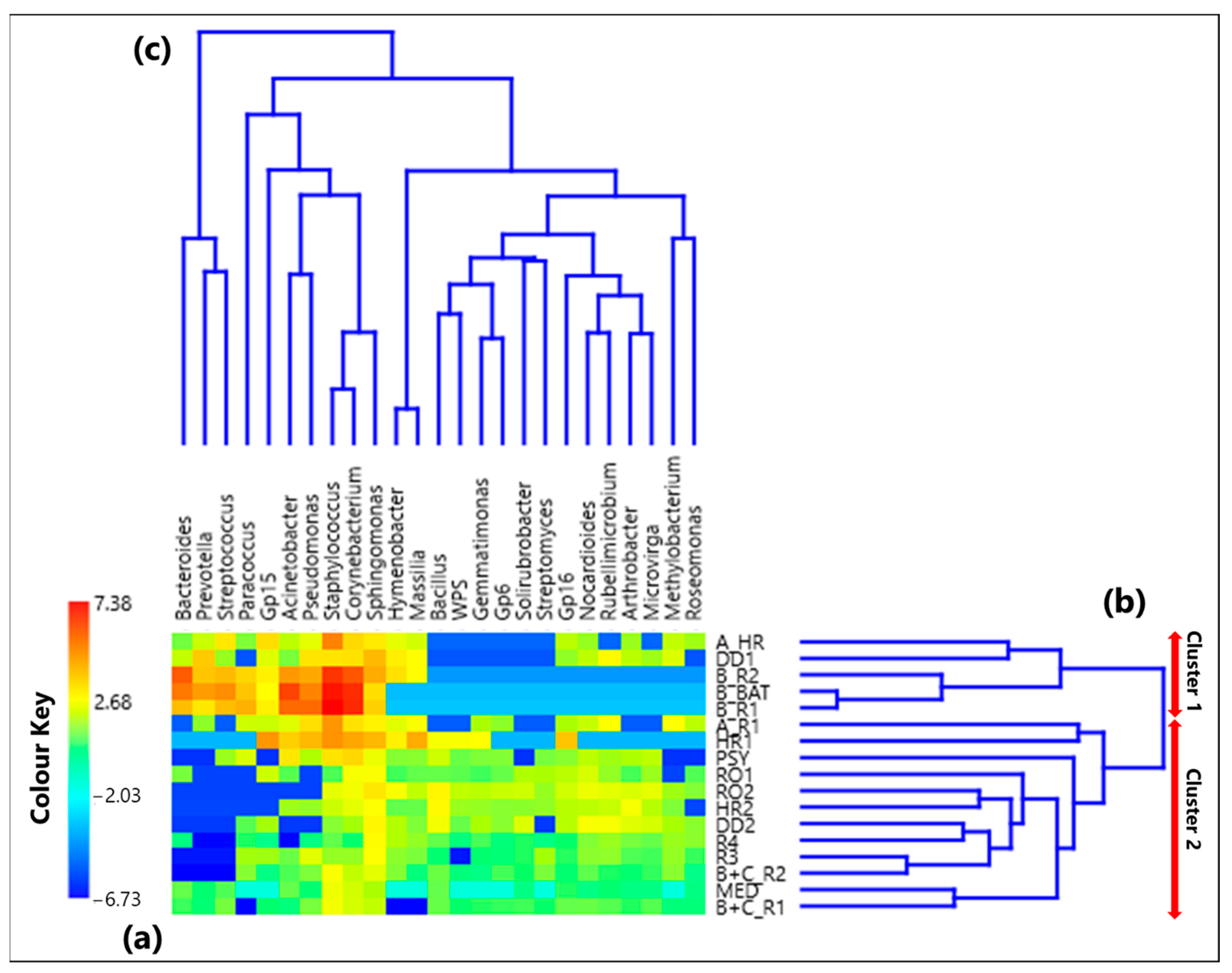
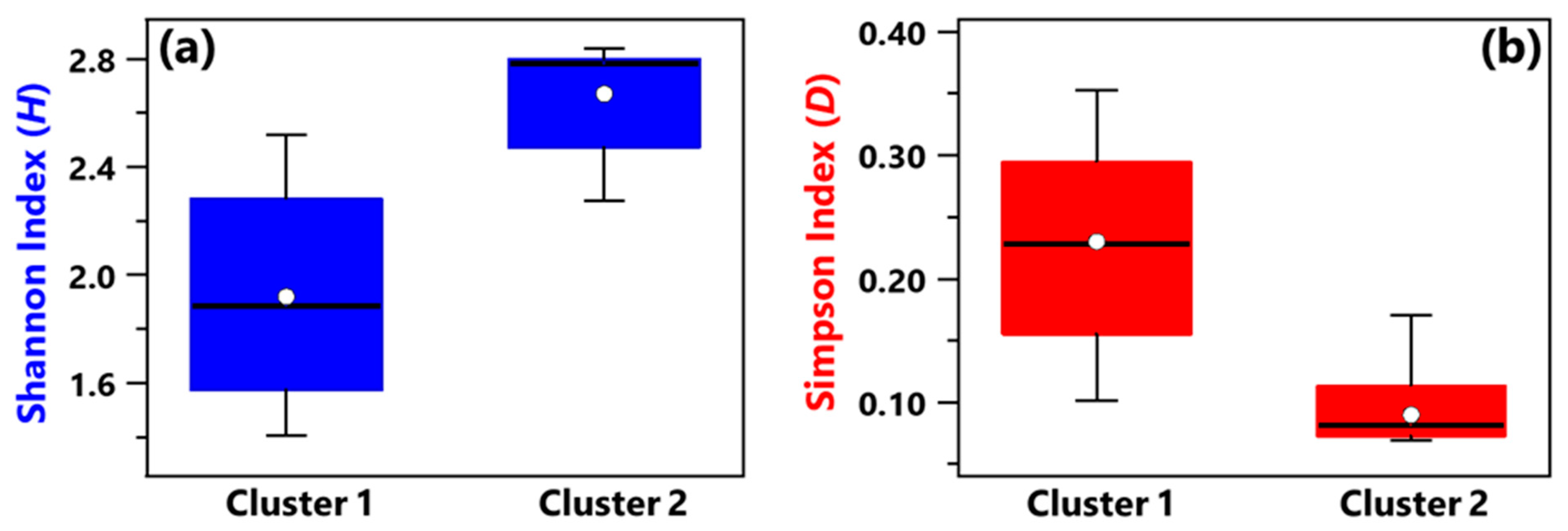
3.1. Centered Log-Ratio Heatmap of Selected Bacterial Genera and Within-Sample Alpha-Diversity
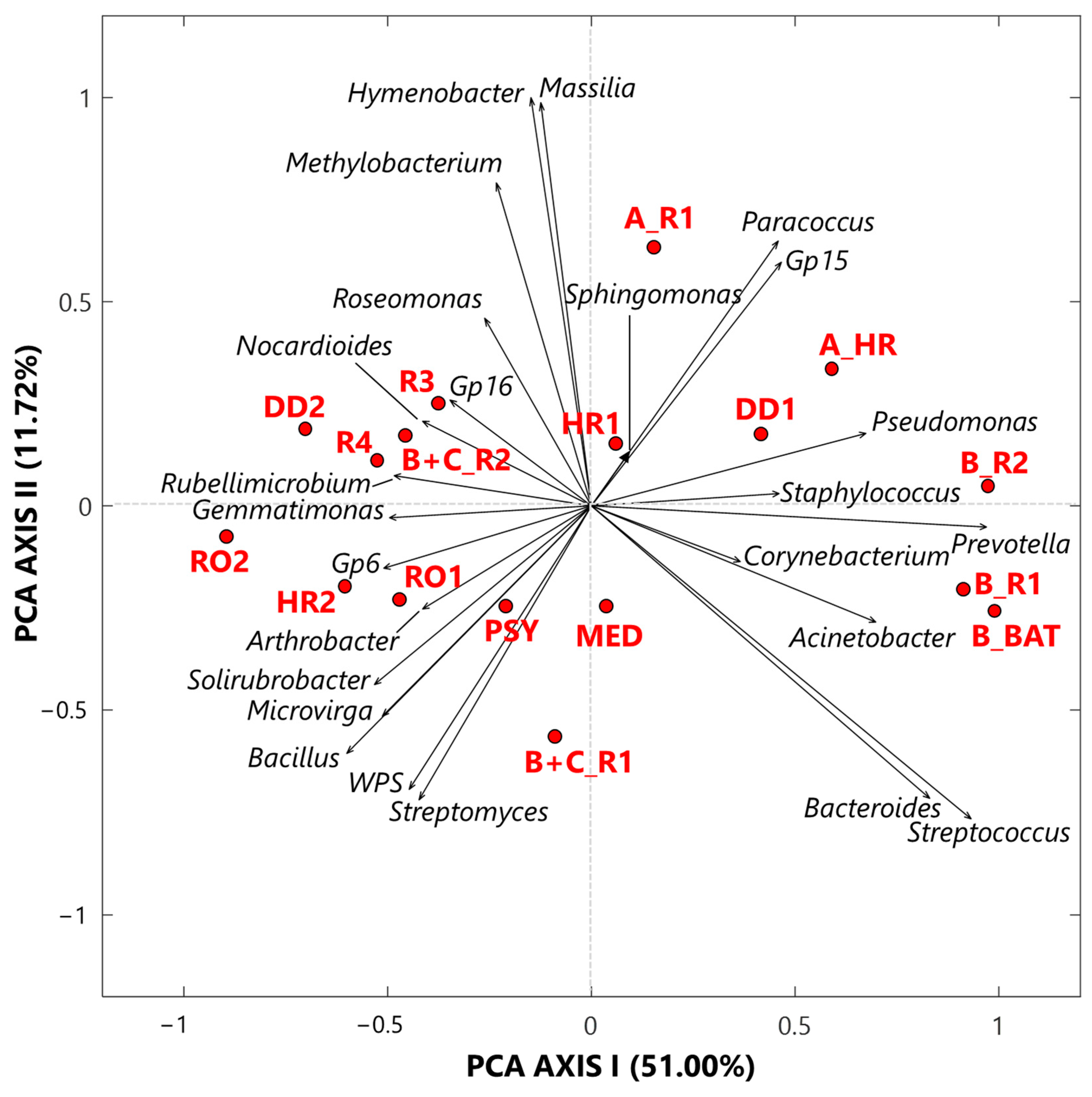
3.1.1. Singular Value Decomposition PCA by Score and Loading Plots at the Genus-Level
3.1.2. Proportionality between Genera by the ρ Metrics
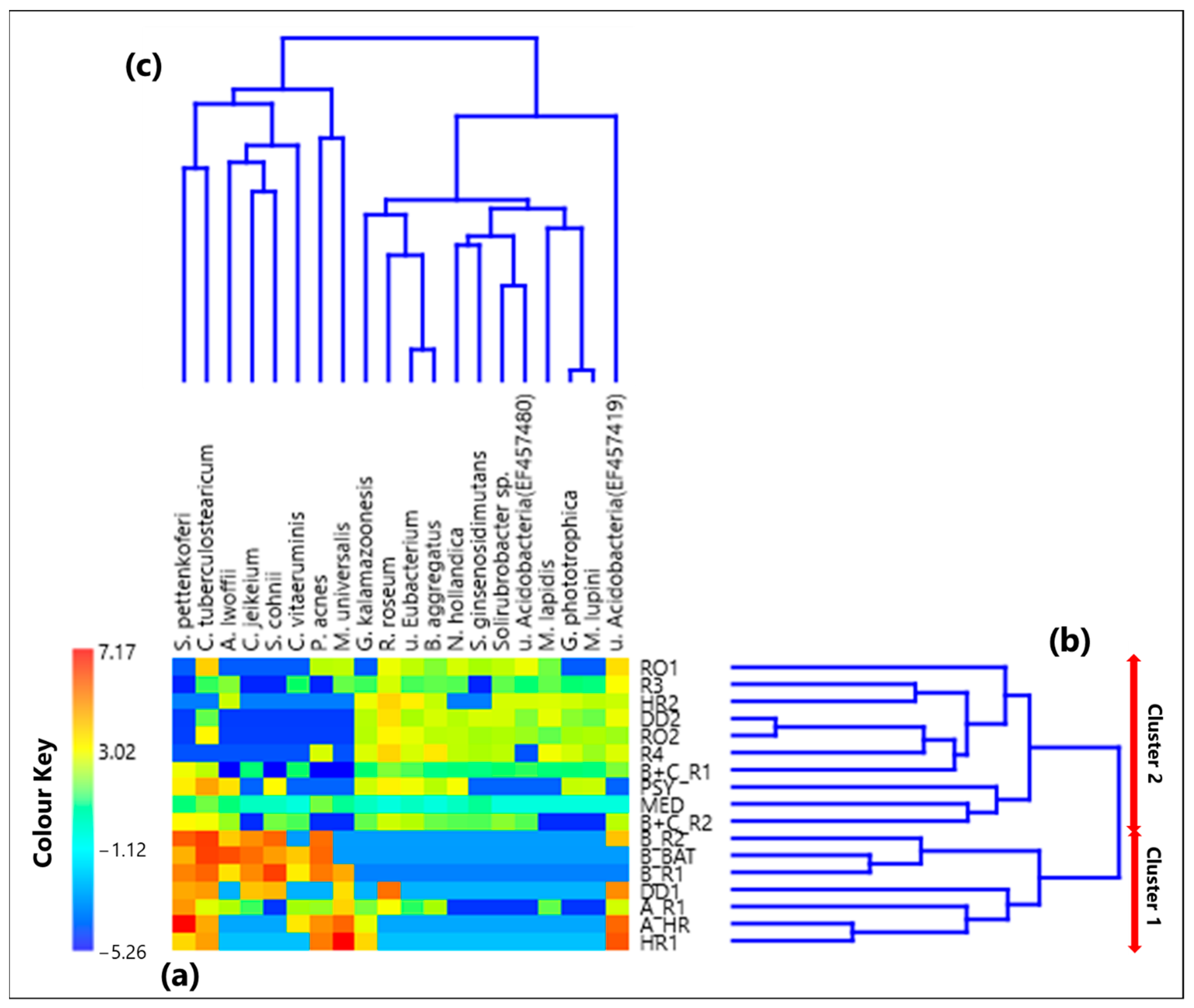
3.2. Centered Log-Ratio Heatmap of Selected Bacterial Species and Within-Sample Alpha-Diversity
3.2.1. Singular Value Decomposition PCA by Score and Loading Plots at the Species Level
3.2.2. Proportionality between Species by the ρ Metrics
4. Conclusions
- Eight and six samples were collected at the ID ward, in rooms with and without COVID-19 patients, respectively. Moreover, a sample (PSY) collected at the psychiatry department and two outdoor samples (RO1 and RO2) collected on the roof of the ID department were also analyzed to compare their bacterial profiles with the corresponding ones of indoor samples from the ID ward.
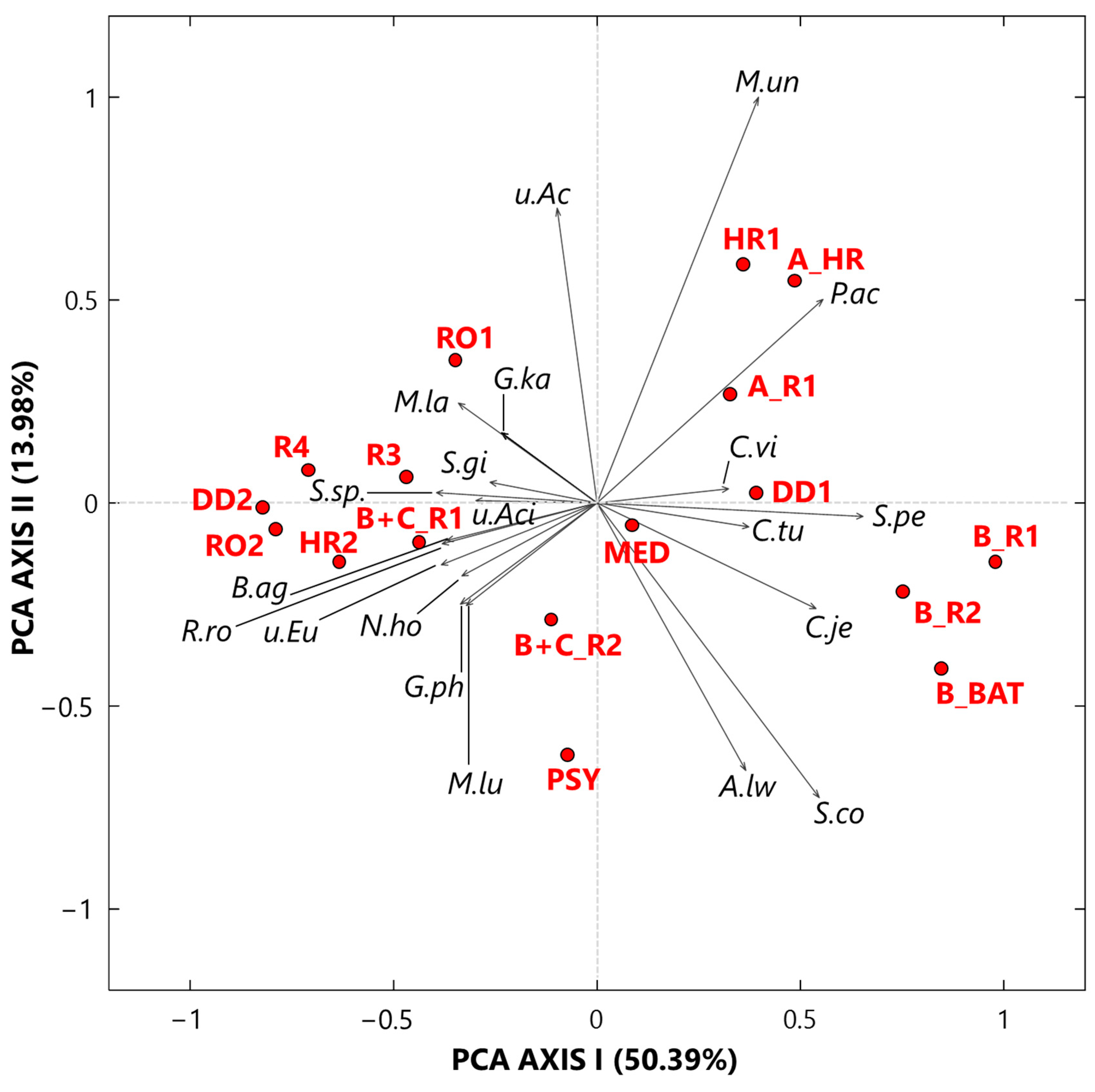
- Twenty-five genera, selected from the ones reaching the largest read number in each sample and common to at least 50% of the 17 collected samples, were analyzed by the compositional approach. More specifically, the SVD-PCA applied to CLR dataset has been used to investigate the relationship among collected samples and selected bacterial genera.
- The SVD-PCA score plot has shown that all samples could be divided in two groups: Cluster 1, mainly consisting of samples collected in rooms occupied by COVID-19 patients, and Cluster 2, which included samples mostly collected in rooms without any COVID-19 patients, as well as outdoor samples.
- The SVD-PCA loading plot has highlighted the different genus structure associated with the samples of Cluster 1 and 2, respectively. Sphingomonas, Paracoccus, Gp15, Pseudomonas, Staphylococcus, Prevotella, Corynebacterium, and Acinetobacter genera were mainly associated with Cluster 1 samples, and they can be responsible for different types of nosocomial infections.
- In contrast, Gp16, Nocardioides, Rubellimicrobium, Arthrobacter, and Solirubrobacter were among the non-pathogenic genera isolated from soil and associated with Cluster 2 samples.
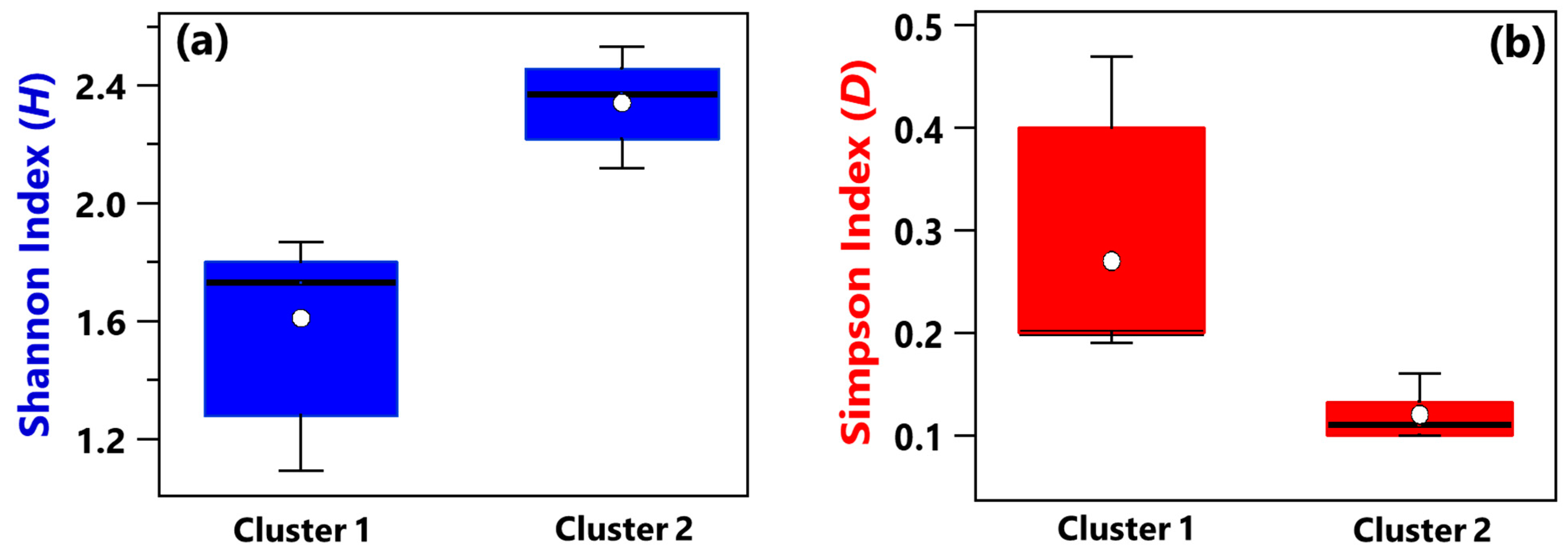
- Shannon and Simpson indices calculated at the genus level have shown that, on average, Cluster 1 samples were characterized by smaller diversity and richness/evenness than Cluster 2 samples.
- The ρ metrics showed few significant positive values between genera associated with Cluster 1 samples. More specifically, positive significant ρ values were found between Corynebacterium and Staphylococcus (0.92), Acinetobacter and Pseudomonas (0.77), Bacteroides and Prevotella (0.78), Bacteroides and Streptococcus (0.76), and Prevotella and Streptococcus (0.83). Moreover, it has been found that Corynebacterium and Staphylococcus were characterized by significantly negative ρ proportionality with some non-pathogenic genera associated with Cluster 2 samples.
- Significant positive ρ metrics values have also been found among some non-pathogenic bacteria associated with the Cluster 2 samples, as the ones between Hymenobacter and Massilia (0.98), and Bacillus and Gemmatimonas (0.78), as well as Microvirga (0.69), Gp6 (0.66), Solirubrobacter (0.71), WPS (0.85), and Streptomyces (0.66).
- Twenty bacterial species were also selected and analyzed by the SVD-PCA applied to the CLR-transformed species dataset. Then, the score and loading plots allowed dividing all samples into two clusters characterized by different bacterial species.
- Cluster 1 included all the samples collected in rooms with COVID-19 patients A and B, while Cluster 2 was mostly consisted of samples collected in rooms without COVID-19 patients. Propionibacterium acnes, Corynebacterium vitaeruminis, Staphylococcus pettenkoferi, Corynebacterium tuberculostearicum, and Corynebacterium jeikeium were the main species associated with Cluster 1 samples. Except for Corynebacterium vitaeruminis, which has been proved to be safe and non-pathogenic, all the other detected species have frequently been identified in hospitals as agents of nosocomial infections.
- Non-pathogenic species were mainly associated with Cluster 2 samples, such as Rubellimicrobium roseum, which was reported as one of the most ubiquitous soil and organic material-dwelling bacteria in outdoor particulate matter.
- Shannon and Simpson index mean values associated with Cluster 1 samples also featured a smaller diversity and richness/evenness than Cluster 2 samples.
- The ρ metrics also revealed strong proportionality between bacterial species of Cluster 1 samples, while negative relationships were found with non-pathogenic species detected in Cluster 2.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the Analysis of High-Throughput Sequencing Datasets: Characterizing RNA-Seq, 16S RRNA Gene Sequencing and Selective Growth Experiments by Compositional Data Analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Hayes, M.G.; MacDonald, J.; Desai, D.K.; Allward, N.; Jones, C.M.A.; Wright, R.J.; Dhanani, A.S.; Comeau, A.M.; et al. Microbiome Differential Abundance Methods Produce Different Results across 38 Datasets. Nat. Commun. 2022, 13, 342. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J.; Chen, D.-G. Compositional Analysis of Microbiome Data. In Statistical Analysis of Microbiome Data with R; Springer: Singapore, 2018; pp. 331–393. [Google Scholar] [CrossRef]
- Kleine Bardenhorst, S.; Berger, T.; Klawonn, F.; Vital, M.; Karch, A.; Rübsamen, N. Data Analysis Strategies for Microbiome Studies in Human Populations-a Systematic Review of Current Practice. mSystems 2021, 6, 1. [Google Scholar] [CrossRef]
- Gloor, G.B.; Reid, G. Compositional Analysis: A Valid Approach to Analyze Microbiome High-Throughput Sequencing Data. Can. J. Microbiol. 2016, 62, 692–703. [Google Scholar] [CrossRef]
- Aitchison, J. The Statistical Analysis of Compositional Data; Chapman and Hall: London, UK, 1986. [Google Scholar]
- Aitchison, J. Principal Component Analysis of Compositional Data. Biometrika 1983, 70, 57–65. [Google Scholar] [CrossRef]
- Aitchison, J. Reducing the Dimensionality of Compositional Data Sets. Math. Geol. 1984, 16, 617–635. [Google Scholar] [CrossRef]
- Van Den Boogaart, K.G.; Tolosana-Delgado, R. Analyzing Compositional Data with R; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Pawlowsky-Glahn, V.; Egozcue, J.J.; Tolosana-Delgado, R. Modeling and Analysis of Compositional Data: Pawlowsky-Glahn/Modelling and Analysis of Compositional Data, 1st ed.; John Wiley & Sons: Nashville, TN, USA, 2015. [Google Scholar]
- Xia, Y.; Sun, J. Hypothesis Testing and Statistical Analysis of Microbiome. Genes Dis. 2017, 4, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Macklaim, J.M.; Vu, M.; Fernandes, A.D. Compositional Uncertainty Should Not Be Ignored in High-Throughput Sequencing Data Analysis. Austrian J. Stat. 2016, 45, 73–87. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and Microbial Differential Abundance Strategies Depend upon Data Characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef]
- Greenacre, M.; Martínez-Álvaro, M.; Blasco, A. Compositional Data Analysis of Microbiome and Any-Omics Datasets: A Validation of the Additive Logratio Transformation. Front. Microbiol. 2021, 12, 727398. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J.; Barceló-Vidal, C.; Martín-Fernández, J.A.; Pawlowsky-Glahn, V. Logratio Analysis and Compositional Distance. Math. Geol. 2000, 32, 271–275. [Google Scholar] [CrossRef]
- Robinson, J.M.; Cando-Dumancela, C.; Antwis, R.E.; Cameron, R.; Liddicoat, C.; Poudel, R.; Weinstein, P.; Breed, M.F. Exposure to Airborne Bacteria Depends upon Vertical Stratification and Vegetation Complexity. Sci. Rep. 2021, 11, 9516. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J.; Greenacre, M. Biplots of Compositional Data. J. R. Stat. Soc. Ser. C Appl. Stat. 2002, 51, 375–392. [Google Scholar] [CrossRef]
- Bian, G.; Gloor, G.B.; Gong, A.; Jia, C.; Zhang, W.; Hu, J.; Zhang, H.; Zhang, Y.; Zhou, Z.; Zhang, J.; et al. The Gut Microbiota of Healthy Aged Chinese Is Similar to That of the Healthy Young. mSphere 2017, 2, 5. [Google Scholar] [CrossRef]
- Wang, Y.; Randolph, T.W.; Shojaie, A.; Ma, J. The generalized matrix decomposition biplot and its application to microbiome data. mSystems 2019, 4, e00504-19. [Google Scholar] [CrossRef]
- Grześkowiak, Ł.; Dadi, T.H.; Zentek, J.; Vahjen, W. Developing Gut Microbiota Exerts Colonisation Resistance to Clostridium (Syn. Clostridioides) Difficile in Piglets. Microorganisms 2019, 7, 218. [Google Scholar] [CrossRef]
- Satten, G.A.; Tyx, R.E.; Rivera, A.J.; Stanfill, S. Restoring the Duality between Principal Components of a Distance Matrix and Linear Combinations of Predictors, with Application to Studies of the Microbiome. PLoS ONE 2017, 12, e0168131. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef]
- Lovell, D.; Pawlowsky-Glahn, V.; Egozcue, J.J.; Marguerat, S.; Bähler, J. Proportionality: A Valid Alternative to Correlation for Relative Data. PLoS Comput. Biol. 2015, 11, e1004075. [Google Scholar] [CrossRef] [PubMed]
- Erb, I.; Notredame, C. How Should We Measure Proportionality on Relative Gene Expression Data? Theory Biosci. 2016, 135, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Erb, I. Partial Correlations in Compositional Data Analysis. Appl. Comput. Geosci. 2020, 6, 100026. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Squair, J.W.; Foster, L.J. Evaluating Measures of Association for Single-Cell Transcriptomics. Nat. Methods 2019, 16, 381–386. [Google Scholar] [CrossRef]
- Quinn, T.P.; Richardson, M.F.; Lovell, D.; Crowley, T.M. Propr: An R-Package for Identifying Proportionally Abundant Features Using Compositional Data Analysis. Sci. Rep. 2017, 7, 16252. [Google Scholar] [CrossRef]
- Quinn, T.P.; Erb, I.; Richardson, M.F.; Crowley, T.M. Understanding Sequencing Data as Compositions: An Outlook and Review. Bioinformatics 2018, 34, 2870–2878. [Google Scholar] [CrossRef]
- Egozcue, J.J.; Pawlowsky-Glahn, V.; Gloor, G.B. Linear Association in Compositional Data Analysis. Austrian J. Stat. 2018, 47, 3–31. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, J.; Shi, W.; Du, N.; Xu, X.; Zhang, Y.; Ji, P.; Zhang, F.; Jia, Z.; Wang, Y.; et al. Dysbiosis of Maternal and Neonatal Microbiota Associated with Gestational Diabetes Mellitus. Gut 2018, 67, 1614–1625. [Google Scholar] [CrossRef]
- Matchado, M.S.; Lauber, M.; Reitmeier, S.; Kacprowski, T.; Baumbach, J.; Haller, D.; List, M. Network Analysis Methods for Studying Microbial Communities: A Mini Review. Comput. Struct. Biotechnol. J. 2021, 19, 2687–2698. [Google Scholar] [CrossRef]
- Nabwera, H.M.; Espinoza, J.L.; Worwui, A.; Betts, M.; Okoi, C.; Sesay, A.K.; Bancroft, R.; Agbla, S.C.; Jarju, S.; Bradbury, R.S.; et al. Interactions between Fecal Gut Microbiome, Enteric Pathogens, and Energy Regulating Hormones among Acutely Malnourished Rural Gambian Children. EBioMedicine 2021, 73, 103644. [Google Scholar] [CrossRef]
- Bakir-Gungor, B.; Hacılar, H.; Jabeer, A.; Nalbantoglu, O.U.; Aran, O.; Yousef, M. Inflammatory Bowel Disease Biomarkers of Human Gut Microbiota Selected via Different Feature Selection Methods. PeerJ 2022, 10, e13205. [Google Scholar] [CrossRef] [PubMed]
- Romay, F.J.; Liu, B.Y.H.; Chae, S.-J. Experimental Study of Electrostatic Capture Mechanisms in Commercial Electret Filters. Aerosol Sci. Technol. 1998, 28, 224–234. [Google Scholar] [CrossRef]
- Shu, H.; Xiangchao, C.; Peng, L.; Hui, G. Study on Electret Technology of Air Filtration Material. IOP Conf. Ser. Earth Environ. Sci. 2017, 100, 012110. [Google Scholar] [CrossRef]
- Barrett, L.W.; Rousseau, A.D. Aerosol Loading Performance of Electret Filter Media. Am. Ind. Hyg. Assoc. J. 1998, 59, 532–539. [Google Scholar] [CrossRef]
- King, P.; Pham, L.K.; Waltz, S.; Sphar, D.; Yamamoto, R.T.; Conrad, D.; Taplitz, R.; Torriani, F.; Forsyth, R.A. Longitudinal Metagenomic Analysis of Hospital Air Identifies Clinically Relevant Microbes. PLoS ONE 2016, 11, e0160124. [Google Scholar] [CrossRef]
- Bøifot, K.O.; Gohli, J.; Skogan, G.; Dybwad, M. Performance Evaluation of High-Volume Electret Filter Air Samplers in Aerosol Microbiome Research. Environ. Microbiome 2020, 15, 14. [Google Scholar] [CrossRef]
- Jaing, C.; Thissen, J.; Morrison, M.; Dillon, M.B.; Waters, S.M.; Graham, G.T.; Be, N.A.; Nicoll, P.; Verma, S.; Caro, T.; et al. Sierra Nevada sweep: Metagenomic measurements of bioaerosols vertically distributed across the troposphere. Sci. Rep. 2020, 10, 1. [Google Scholar] [CrossRef]
- Ginn, O.; Rocha-Melogno, L.; Bivins, A.; Lowry, S.; Cardelino, M.; Nichols, D.; Tripathi, S.N.; Soria, F.; Andrade, M.; Bergin, M.; et al. Detection and Quantification of Enteric Pathogens in Aerosols near Open Wastewater Canals in Cities with Poor Sanitation. Environ. Sci. Technol. 2021, 55, 14758–14771. [Google Scholar] [CrossRef]
- Ginn, O.; Berendes, D.; Wood, A.; Bivins, A.; Rocha-Melogno, L.; Deshusses, M.A.; Tripathi, S.N.; Bergin, M.H.; Brown, J. Open Waste Canals as Potential Sources of Antimicrobial Resistance Genes in Aerosols in Urban Kanpur, India. Am. J. Trop. Med. Hyg. 2021, 104, 1761–1767. [Google Scholar] [CrossRef]
- Pepin, B.; Williams, T.; Polson, D.; Gauger, P.; Dee, S. Survival of swine pathogens in compost formed from preprocessed carcasses. Transbound. Emerg. Dis. 2020, 68, 2239–2249. [Google Scholar] [CrossRef]
- McCumber, A.W.; Kim, Y.J.; Isikhuemhen, O.S.; Tighe, R.M.; Gunsch, C.K. The Environment Shapes Swine Lung Bacterial Communities. Sci. Total Environ. 2021, 758, 143623. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wu, X.; Zhang, Y.; Xia, J.; Li, M.; Feng, Y.; Yu, X.; Duan, J.; Weng, X.; Chen, Y.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Contamination in Air and Environment in Temporary COVID-19 ICU Wards. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Borges, J.T.; Nakada, L.Y.K.; Maniero, M.G.; Guimarães, J.R. SARS-CoV-2: A Systematic Review of Indoor Air Sampling for Virus Detection. Environ. Sci. Pollut. Res. Int. 2021, 28, 40460–40473. [Google Scholar] [CrossRef]
- Romano, S.; Di Salvo, M.; Rispoli, G.; Alifano, P.; Perrone, M.R.; Talà, A. Airborne Bacteria in the Central Mediterranean: Structure and Role of Meteorology and Air Mass Transport. Sci. Total Environ. 2019, 697, 134020. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Alishum, A. DADA2 formatted 16S rRNA gene sequences for both bacteria & archaea. Zenodo 2019. [Google Scholar] [CrossRef]
- Lubbe, S.; Filzmoser, P.; Templ, M. Comparison of Zero Replacement Strategies for Compositional Data with Large Numbers of Zeros. Chemometr. Intell. Lab. Syst. 2021, 210, 104248. [Google Scholar] [CrossRef]
- Martín-Fernández, J.A.; Barceló-Vidal, C.; Pawlowsky-Glahn, V. Dealing with zeros and missing values in compositional data sets using nonparametric imputation. Math. Geol. 2003, 35, 253–278. [Google Scholar] [CrossRef]
- Martín-Fernández, J.; Barceló-Vidal, C.; Pawlowsky-Glahn, V.; Buccianti, A.; Nardi, G.; Potenza, R. Measures of difference for compositional data and hierarchical clustering methods. Proc. IAMG 1998, 98, 526–531. [Google Scholar]
- Escobar-Zepeda, A.; Vera-Ponce de León, A.; Sanchez-Flores, A. The Road to Metagenomics: From Microbiology to DNA Sequencing Technologies and Bioinformatics. Front. Genet. 2015, 6, 348. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-R.; Shin, J.; Guevarra, R.; Lee, J.H.; Kim, D.W.; Seol, K.-H.; Lee, J.-H.; Kim, H.B.; Isaacson, R. Deciphering Diversity Indices for a Better Understanding of Microbial Communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.J. (Ed.) University of British Columbia; Species diversity measures. In Ecological Methodology; Vancouver, BC, Canada, 2014; pp. 532–593. [Google Scholar]
- Ribeiro, L.F.; Lopes, E.M.; Kishi, L.T.; Ribeiro, L.F.C.; Menegueti, M.G.; Gaspar, G.G.; Silva-Rocha, R.; Guazzaroni, M.-E. Microbial Community Profiling in Intensive Care Units Expose Limitations in Current Sanitary Standards. Front. Public Health 2019, 7, 240. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Troise, O.; Donaldson, H.; Mughal, N.; Moore, L.S.P. Bacterial and Fungal Coinfection among Hospitalized Patients with COVID-19: A Retrospective Cohort Study in a UK Secondary-Care Setting. Clin. Microbiol. Infect. 2020, 26, 1395–1399. [Google Scholar] [CrossRef]
- Lax, S.; Sangwan, N.; Smith, D.; Larsen, P.; Handley, K.M.; Richardson, M.; Guyton, K.; Krezalek, M.; Shogan, B.D.; Defazio, J.; et al. Bacterial Colonization and Succession in a Newly Opened Hospital. Sci. Transl. Med. 2017, 9, eaah6500. [Google Scholar] [CrossRef]
- Zhang, W.; Mo, G.; Yang, J.; Hu, X.; Huang, H.; Zhu, J.; Zhang, P.; Xia, H.; Xie, L. Community Structure of Environmental Microorganisms Associated with COVID-19 Affected Patients. Aerobiologia 2021, 37, 575–583. [Google Scholar] [CrossRef]
- Sirivongrangson, P.; Kulvichit, W.; Payungporn, S.; Pisitkun, T.; Chindamporn, A.; Peerapornratana, S.; Pisitkun, P.; Chitcharoen, S.; Sawaswong, V.; Worasilchai, N.; et al. Endotoxemia and Circulating Bacteriome in Severe COVID-19 Patients. Intensive Care Med. Exp. 2020, 8, 72. [Google Scholar] [CrossRef]
- Chezganova, E.; Efimova, O.; Sakharova, V.; Efimova, A.; Sozinov, S.; Kutikhin, A.; Ismagilov, Z.; Brusina, E. Ventilation-Associated Particulate Matter Is a Potential Reservoir of Multidrug-Resistant Organisms in Health Facilities. Life 2021, 11, 639. [Google Scholar] [CrossRef]
- Hewitt, K.M.; Mannino, F.L.; Gonzalez, A.; Chase, J.H.; Caporaso, J.G.; Knight, R.; Kelley, S.T. Bacterial Diversity in Two Neonatal Intensive Care Units (NICUs). PLoS ONE 2013, 8, e54703. [Google Scholar] [CrossRef]
- Brooks, B.; Firek, B.A.; Miller, C.S.; Sharon, I.; Thomas, B.C.; Baker, R.; Morowitz, M.J.; Banfield, J.F. Microbes in the Neonatal Intensive Care Unit Resemble Those Found in the Gut of Premature Infants. Microbiome 2014, 2, 1. [Google Scholar] [CrossRef]
- Hu, H.; Johani, K.; Gosbell, I.B.; Jacombs, A.S.W.; Almatroudi, A.; Whiteley, G.S.; Deva, A.K.; Jensen, S.; Vickery, K. Intensive Care Unit Environmental Surfaces Are Contaminated by Multidrug-Resistant Bacteria in Biofilms: Combined Results of Conventional Culture, Pyrosequencing, Scanning Electron Microscopy, and Confocal Laser Microscopy. J. Hosp. Infect. 2015, 91, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Schwebke, I.; Kampf, G. How Long Do Nosocomial Pathogens Persist on Inanimate Surfaces? A Systematic Review. BMC Infect. Dis. 2006, 6, 130. [Google Scholar] [CrossRef] [PubMed]
- Majed, R.; Faille, C.; Kallassy, M.; Gohar, M. Bacillus Cereus Biofilms-Same, Only Different. Front. Microbiol. 2016, 7, 1054. [Google Scholar] [CrossRef] [PubMed]
- Magill, S.S.; O’Leary, E.; Janelle, S.J.; Thompson, D.L.; Dumyati, G.; Nadle, J.; Wilson, L.E.; Kainer, M.A.; Lynfield, R.; Greissman, S.; et al. Changes in Prevalence of Health Care-Associated Infections in U.s. Hospitals. N. Engl. J. Med. 2018, 379, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, Y.; Shen, H.; Lv, Y.; Zheng, W.; Wang, J. Higher Prevalence of Multi-Antimicrobial Resistant Bacteroides Spp. Strains Isolated at a Tertiary Teaching Hospital in China. Infect. Drug Resist. 2020, 13, 1537–1546. [Google Scholar] [CrossRef]
- König, E.; Ziegler, H.P.; Tribus, J.; Grisold, A.J.; Feierl, G.; Leitner, E. Surveillance of Antimicrobial Susceptibility of Anaerobe Clinical Isolates in Southeast Austria: Bacteroides Fragilis Group Is on the Fast Track to Resistance. Antibiotics 2021, 10, 479. [Google Scholar] [CrossRef]
- Bastiaens, G.J.H.; Cremers, A.J.H.; Coolen, J.P.M.; Nillesen, M.T.; Boeree, M.J.; Hopman, J.; Wertheim, H.F.L. Nosocomial Outbreak of Multi-Resistant Streptococcus Pneumoniae Serotype 15A in a Centre for Chronic Pulmonary Diseases. Antimicrob. Resist. Infect. Control 2018, 7, 158. [Google Scholar] [CrossRef]
- Weyant, R.S.; Whitney, A.M. Roseomonas. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1–9. [Google Scholar]
- Guo, X.; Xie, C.; Wang, L.; Li, Q.; Wang, Y. Biodegradation of Persistent Environmental Pollutants by Arthrobacter Sp. Environ. Sci. Pollut. Res. Int. 2019, 26, 8429–8443. [Google Scholar] [CrossRef]
- Whitman, W.B. Solirubrobacter. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1–5. [Google Scholar]
- Pastuszka, J.S.; Marchwinska-Wyrwal, E.; Wlazlo, A. Bacterial Aerosol in Silesian Hospitals: Preliminary Results. Pol. J. Environ. Stud. 2005, 14, 883–890. [Google Scholar]
- Okten, S.; Asan, A. Airborne Fungi and Bacteria in Indoor and Outdoor Environment of the Pediatric Unit of Edirne Government Hospital. Environ. Monit. Assess. 2012, 184, 1739–1751. [Google Scholar] [CrossRef]
- Ling, S.; Hui, L. Evaluation of the Complexity of Indoor Air in Hospital Wards Based on PM2.5, Real-Time PCR, Adenosine Triphosphate Bioluminescence Assay, Microbial Culture and Mass Spectrometry. BMC Infect. Dis. 2019, 19, 646. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Kim, T.; Ryoo, S. Detection of Airborne Bacteria from Patient Spaces in Tuberculosis Hospital. Int. J. Mycobacteriol. 2020, 9, 293–295. [Google Scholar] [CrossRef]
- Stearns, J.C.; Davidson, C.J.; McKeon, S.; Whelan, F.J.; Fontes, M.E.; Schryvers, A.B.; Bowdish, D.M.E.; Kellner, J.D.; Surette, M.G. Culture and Molecular-Based Profiles Show Shifts in Bacterial Communities of the Upper Respiratory Tract That Occur with Age. ISME J. 2015, 9, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Schenck, L.P.; Surette, M.G.; Bowdish, D.M.E. Composition and Immunological Significance of the Upper Respiratory Tract Microbiota. FEBS Lett. 2016, 590, 3705–3720. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, B.C.; Binnerup, S.J.; Gillings, M. Microcolony Cultivation on a Soil Substrate Membrane System Selects for Previously Uncultured Soil Bacteria. Appl. Environ. Microbiol. 2005, 71, 8714–8720. [Google Scholar] [CrossRef]
- Nagy, M.L.; Pérez, A.; Garcia-Pichel, F. The Prokaryotic Diversity of Biological Soil Crusts in the Sonoran Desert (Organ Pipe Cactus National Monument, AZ). FEMS Microbiol. Ecol. 2005, 54, 233–245. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of Root Colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar] [CrossRef]
- Ten, L.N.; Han, Y.E.; Park, K.I.; Kang, I.-K.; Han, J.-S.; Jung, H.-Y. Hymenobacter jeollabukensis Sp. Nov., Isolated from Soil. J. Microbiol. 2018, 56, 500–506. [Google Scholar] [CrossRef]
- Samaké, A.; Bonin, A.; Jaffrezo, J.-L.; Taberlet, P.; Weber, S.; Uzu, G.; Jacob, V.; Conil, S.; Martins, J.M.F. High Levels of Primary Biogenic Organic Aerosols Are Driven by Only a Few Plant-Associated Microbial Taxa. Atmos. Chem. Phys. 2020, 20, 5609–5628. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The Skin Microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Perry, A.; Lambert, P. Propionibacterium Acnes: Infection beyond the Skin. Expert Rev. Anti. Infect. Ther. 2011, 9, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Bargel, H.; Raßmann, N.; Scheibel, T. Microbial Repellence Properties of Engineered Spider Silk Coatings Prevent Biofilm Formation of Opportunistic Bacterial Strains. MRS Commun. 2021, 11, 356–362. [Google Scholar] [CrossRef]
- Jones, M.; Kishore, M.K.; Redfern, D. Propionibacterium Acnes Infection of the Elbow. J. Shoulder Elb. Surg. 2011, 20, e22–e25. [Google Scholar] [CrossRef] [PubMed]
- Zeller, V.; Ghorbani, A.; Strady, C.; Leonard, P.; Mamoudy, P.; Desplaces, N. Propionibacterium Acnes: An Agent of Prosthetic Joint Infection and Colonization. J. Infect. 2007, 55, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.E.; Hennicke, C.; Byers, K.; Welch, W.C. Postoperative Discitis Due to Propionibacterium Acnes: A Case Report and Review of the Literature. Surg. Neurol. 2005, 63, 538–541; discussion 541. [Google Scholar] [CrossRef]
- Colombo, M.; Castilho, N.P.A.; Todorov, S.D.; Nero, L.A. Beneficial and Safety Properties of a Corynebacterium Vitaeruminis Strain Isolated from the Cow Rumen. Probiotics Antimicrob. Proteins 2017, 9, 157–162. [Google Scholar] [CrossRef]
- Dobinson, H.C.; Anderson, T.P.; Chambers, S.T.; Doogue, M.P.; Seaward, L.; Werno, A.M. Antimicrobial Treatment Options for Granulomatous Mastitis Caused by Corynebacterium Species. J. Clin. Microbiol. 2015, 53, 2895–2899. [Google Scholar] [CrossRef]
- Trülzsch, K.; Rinder, H.; Trcek, J.; Bader, L.; Wilhelm, U.; Heesemann, J. “Staphylococcus pettenkoferi,” a Novel Staphylococcal Species Isolated from Clinical Specimens. Diagn. Microbiol. Infect. Dis. 2002, 43, 175–182. [Google Scholar] [CrossRef]
- Hashi, A.A.; Delport, J.A.; Elsayed, S.; Silverman, M.S. Staphylococcus Pettenkoferi Bacteremia: A Case Report and Review of the Literature. Can. J. Infect. Dis. Med. Microbiol. 2015, 26, 319–322. [Google Scholar] [CrossRef]
- Strong, C.; Cosiano, M.; Cabezas, M.; Barwatt, J.W.; Tillekeratne, L.G. Staphylococcus Pettenkoferi Bacteremia in an American Intensive Care Unit. Case Rep. Infect. Dis. 2021, 2021, 5235691. [Google Scholar] [CrossRef]
- Eke, U.A.; Fairfax, M.R.; Mitchell, R.; Taylor, M.; Salimnia, H. Staphylococcus Pettenkoferi-Positive Blood Cultures in Hospitalized Patients in a Multi-Site Tertiary Center. Diagn. Microbiol. Infect. Dis. 2021, 99, 115284. [Google Scholar] [CrossRef] [PubMed]
- Asgin, N.; Otlu, B. Antimicrobial Resistance and Molecular Epidemiology of Corynebacterium Striatum Isolated in a Tertiary Hospital in Turkey. Pathogens 2020, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Hinic, V.; Lang, C.; Weisser, M.; Straub, C.; Frei, R.; Goldenberger, D. Corynebacterium Tuberculostearicum: A Potentially Misidentified and Multiresistant Corynebacterium Species Isolated from Clinical Specimens. J. Clin. Microbiol. 2012, 50, 2561–2567. [Google Scholar] [CrossRef] [PubMed]
- Rathinavelu, S.; Zavros, Y.; Merchant, J.L. Acinetobacter Lwoffii Infection and Gastritis. Microbes Infect. 2003, 5, 651–657. [Google Scholar] [CrossRef]
- Szewczyk, E.M.; Rózalska, M.; Cieślikowski, T.; Nowak, T. Plasmids of Staphylococcus Cohnii Isolated from the Intensive-Care Unit. Folia Microbiol. 2004, 49, 123–131. [Google Scholar] [CrossRef]
- Kováts, N.; Horváth, E.; Hubai, K.; Hoffer, A.; Jancsek-Turóczi, B.; Fekete, C. Exotic Airborne Bacteria Identified in Urban Resuspended Dust by next Generation Sequencing. E3S Web Conf. 2019, 99, 04009. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Date (dd/mm/yy) | At the Genus Level | At the Species Level | ||
|---|---|---|---|---|---|
| Shannon Index (H) | Simpson Index (D) | Shannon Index (H) | Simpson Index (D) | ||
| A_HR | 30/04/20 | 2.05 | 0.24 | 1.28 | 0.40 |
| A_R1 | 01/05/20 | 2.29 | 0.17 | 1.87 | 0.26 |
| B_R1 | 05/05/20 | 1.41 | 0.35 | 1.79 | 0.20 |
| B_R2 | 07/05/20 | 1.89 | 0.21 | 1.72 | 0.20 |
| B_BAT | 06/05/20 | 1.74 | 0.23 | 1.73 | 0.20 |
| B+C_R1 | 17/05/20 | 2.83 | 0.08 | 2.53 | 0.10 |
| B+C_R2 | 21/05/20 | 2.79 | 0.08 | 2.45 | 0.10 |
| HR1 | 01/05/20 | 2.27 | 0.12 | 1.09 | 0.47 |
| HR2 | 15/05/20 | 2.78 | 0.07 | 2.27 | 0.12 |
| R3 | 02/05/20 | 2.84 | 0.07 | 2.48 | 0.11 |
| R4 | 04/06/20 | 2.80 | 0.09 | 2.34 | 0.11 |
| MED | 03/05/20 | 2.43 | 0.12 | 2.15 | 0.14 |
| RO1 | 08/05/20 | 2.79 | 0.08 | 2.24 | 0.13 |
| RO2 | 16/07/20 | 2.78 | 0.07 | 2.44 | 0.10 |
| PSY | 11/07/20 | 2.60 | 0.10 | 2.12 | 0.16 |
| * DD1 | 07/05/20 | 2.52 | 0.10 | 1.80 | 0.19 |
| * DD2 | 07/05/20 | 2.78 | 0.07 | 2.40 | 0.11 |
| Bacterial Genera | Positive Correlations | Negative Correlations |
|---|---|---|
| Corynebacterium | Staphylococcus (0.92) | Nocardioides (−0.78), Arthrobacter (−0.66), Rubellimicrobium (−0.66) |
| Staphylococcus | Nocardioides (−0.78), Arthrobacter (−0.74), Rubellimicrobium (−0.75), Microvirga (−0.79), Gp6 (−0.66), Solirubrobacter (−0.67) | |
| Acinetobacter | Pseudomonas (0.77) | |
| Pseudomonas | Solirubrobacter (−0.70) | |
| Hymenobacter | Massilia (0.98) | |
| Nocardioides | Arthrobacter (0.75), Rubellimicrobium (0.79) | |
| Arthrobacter | Microvirga (0.82) | |
| Rubellimicrobium | Microvirga (0.75), Gp6 (0.73) | |
| Bacillus | Gemmatimonas (0.78), Microvirga (0.69), Gp6 (0.66), Solirubrobacter (0.71), WPS (0.85), Streptomyces (0.66) | Prevotella (−0.78) |
| Gemmatimonas | Gp6 (0.83), WPS (0.68) | Bacteroides (−0.66), Prevotella (−0.68), Streptococcus (−0.67) |
| Bacteroides | Prevotella (0.78), Streptococcus (0.76) | |
| Prevotella | Streptococcus (0.83) |
| Bacterial Species | Positive Correlations | Negative Correlations |
|---|---|---|
| Corynebacterium tuberculostearicum | Uncultured eubacterium (−0.66), Blastococcus aggregatus (−0.69), Modestobacter lapidis (−0.72), Solirubrobacter sp. (−0.66) | |
| Rubellimicrobium roseum | Uncultured eubacterium (0.75), Blastococcus aggregatus (0.76) | Propionibacterium acnes (−0.71) |
| Staphylococcus pettenkoferi | Corynebacterium jeikeium (0.66) | Modestobacter lapidis (−0.71), Solirubrobacter sp. (−0.76) |
| Uncultured eubacterium | Blastococcus aggregatus (0.98), Nitrolancea hollandica (0.67), Solirubrobacter sp. (0.68) | Corynebacterium jeikeium (−0.69) |
| Blastococcus aggregatus | Modestobacter lapidis (0.68) | Corynebacterium jeikeium (−0.67) |
| Nitrolancea hollandica | Solirubrobacter ginsenosidimutans (0.66) | |
| Modestobacter lapidis | Staphylococcus cohnii (−0.73) | |
| Solirubrobacter sp. | Solirubrobacter ginsenosidimutans (0.69), uncultured Acidobacteria(EF457480) (0.83) | |
| Corynebacterium jeikeium | Staphylococcus cohnii (0.68) | |
| Gemmatimonas phototrophica | Microvirga lupini (0.99) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrone, M.R.; Romano, S.; De Maria, G.; Tundo, P.; Bruno, A.R.; Tagliaferro, L.; Maffia, M.; Fragola, M. Compositional Data Analysis of 16S rRNA Gene Sequencing Results from Hospital Airborne Microbiome Samples. Int. J. Environ. Res. Public Health 2022, 19, 10107. https://doi.org/10.3390/ijerph191610107
Perrone MR, Romano S, De Maria G, Tundo P, Bruno AR, Tagliaferro L, Maffia M, Fragola M. Compositional Data Analysis of 16S rRNA Gene Sequencing Results from Hospital Airborne Microbiome Samples. International Journal of Environmental Research and Public Health. 2022; 19(16):10107. https://doi.org/10.3390/ijerph191610107
Chicago/Turabian StylePerrone, Maria Rita, Salvatore Romano, Giuseppe De Maria, Paolo Tundo, Anna Rita Bruno, Luigi Tagliaferro, Michele Maffia, and Mattia Fragola. 2022. "Compositional Data Analysis of 16S rRNA Gene Sequencing Results from Hospital Airborne Microbiome Samples" International Journal of Environmental Research and Public Health 19, no. 16: 10107. https://doi.org/10.3390/ijerph191610107