Diesel Particulate Matter 2.5 Induces Epithelial-to-Mesenchymal Transition and Upregulation of SARS-CoV-2 Receptor during Human Pluripotent Stem Cell-Derived Alveolar Organoid Development

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Stepwise Differentiation of hPSCs into AECs
2.3. Generation of Multicellular AOs from hPSCs
2.4. PM Preparation and Treatment
2.5. Cell Viability Detected Using Neutral Red Assay
2.6. Hematoxylin and Eosin and Immunofluorescence Staining
2.7. Western Blot Analysis
2.8. RNA Extraction and Quantitative Real-Time qPCR
2.9. Statistical Analysis
3. Results
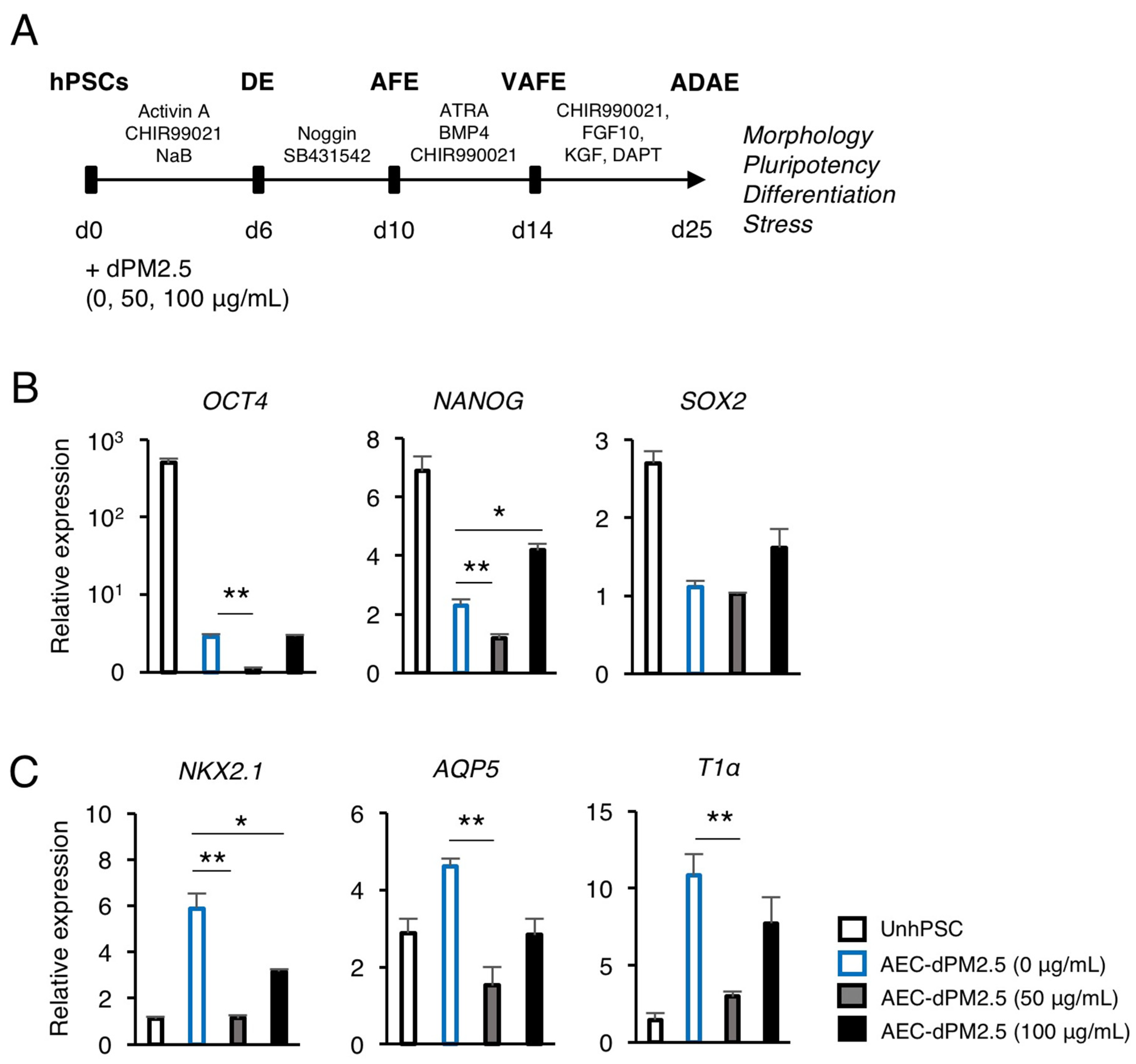
3.1. dPM2.5 Disturbed hPSC Differentiation towards AECs
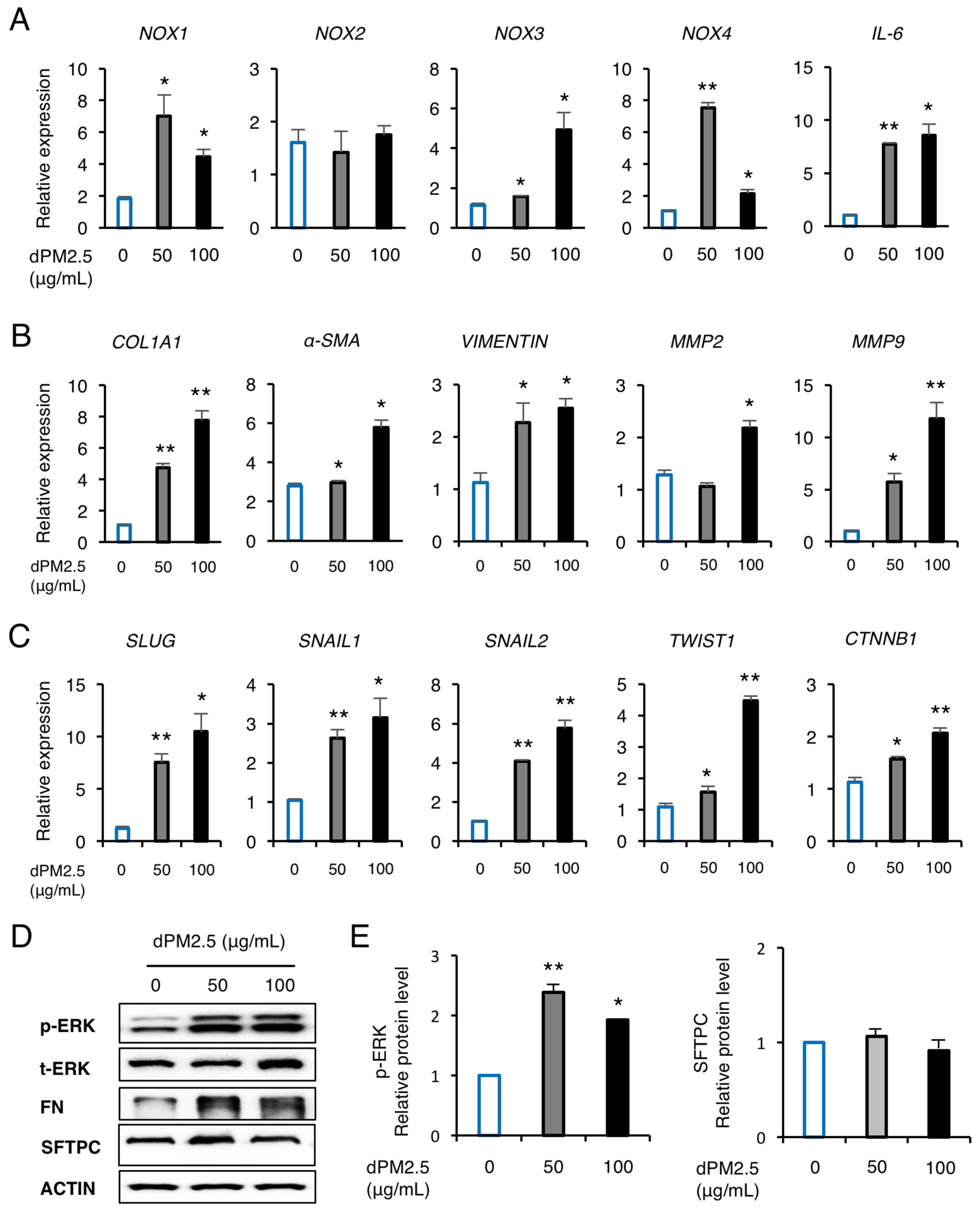
3.2. dPM2.5 Promoted EMT during hPSC-Derived AEC Differentiation via Activation of ERK Signaling
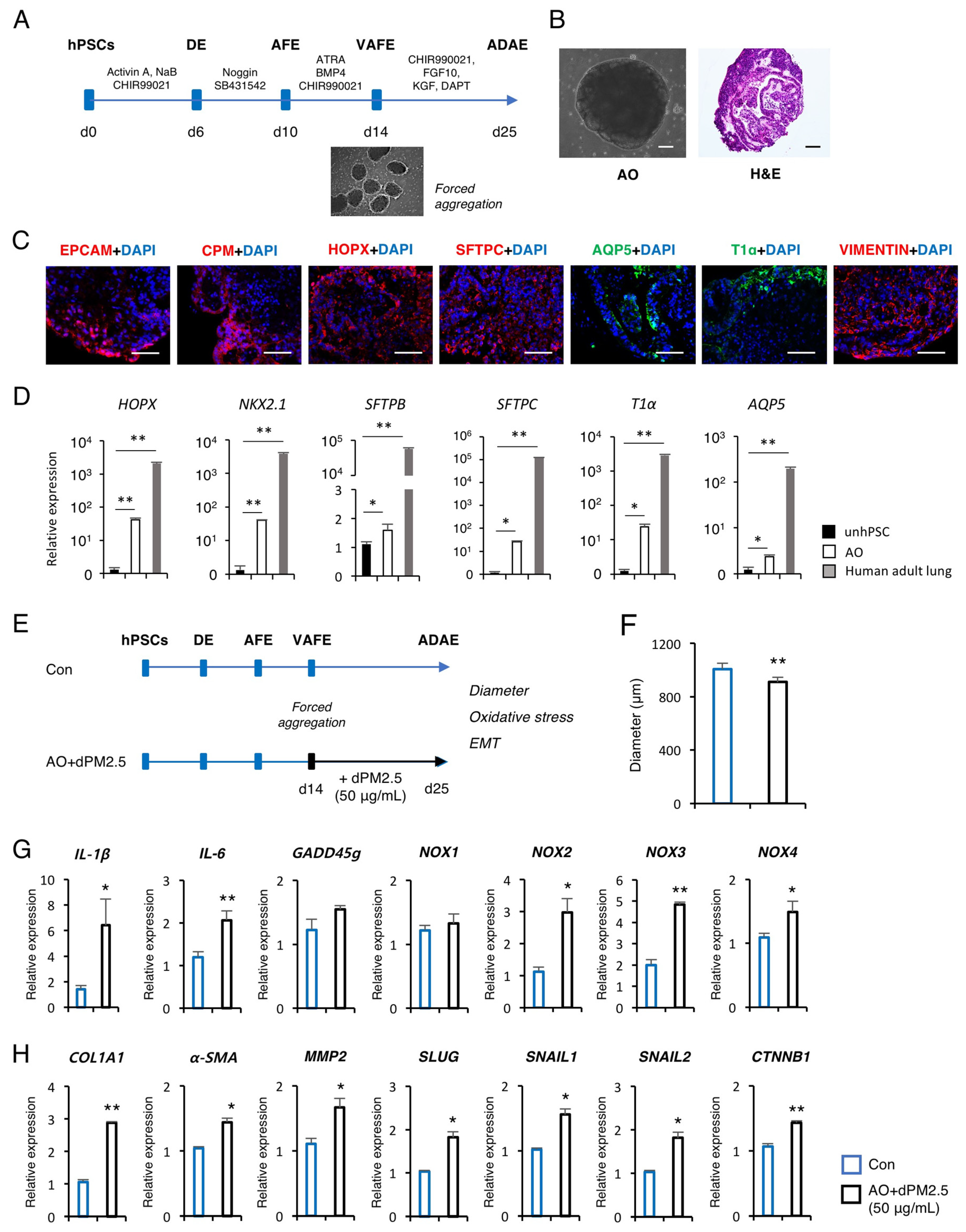
3.3. dPM2.5 Induced EMT during 3D Alveolar Development
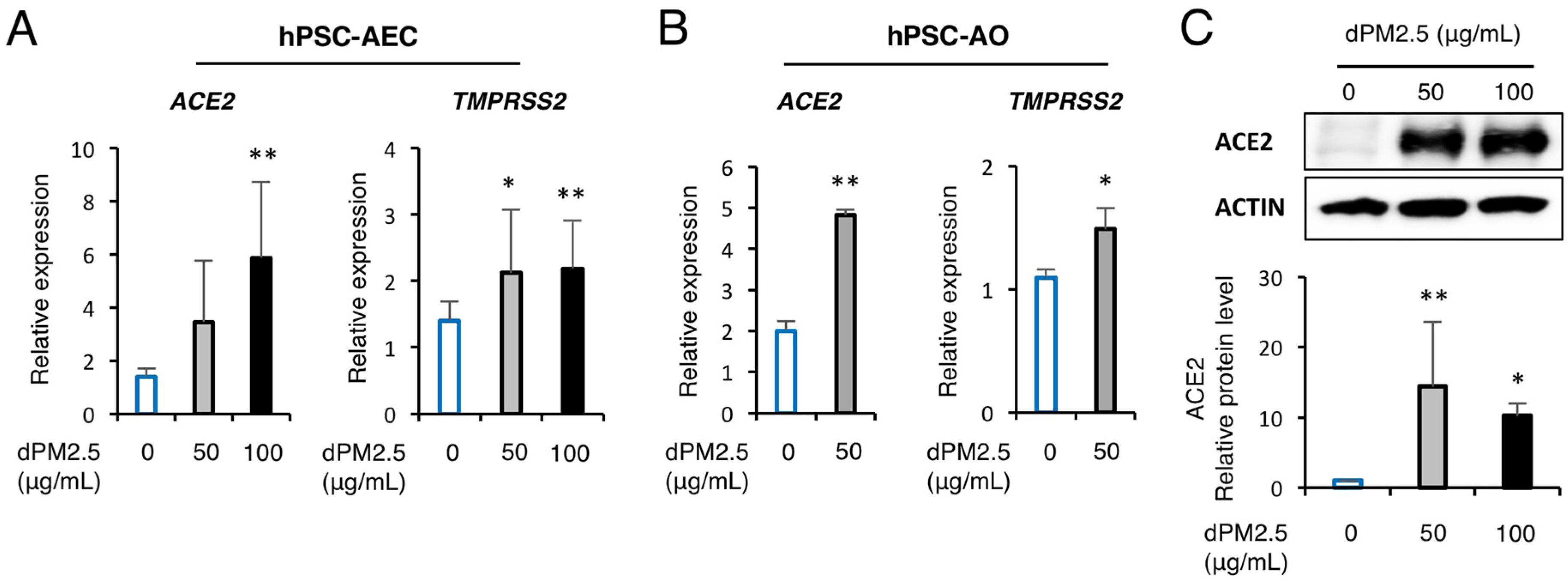
3.4. dPM2.5 Enhanced ACE2 and TMPRSS2 Expression in hPSC-Derived AECs and AOs
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kyung, S.Y.; Jeong, S.H. Particulate-Matter Related Respiratory Diseases. Tuberc. Respir. Dis. 2020, 83, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.A.; Demers, P.A.; Karr, C.J.; Koehoorn, M.; Lencar, C.; Tamburic, L.; Brauer, M. Effect of early life exposure to air pollution on development of childhood asthma. Environ. Health Perspect. 2010, 118, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Q.; Lu, C.; Norback, D.; Bornehag, C.G.; Zhang, Y.; Liu, W.; Yuan, H.; Sundell, J. Early life exposure to ambient air pollution and childhood asthma in China. Environ. Res. 2015, 143, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Chiu, Y.H.; Coull, B.A.; Kloog, I.; Schwartz, J.; Lee, A.; Wright, R.O.; Wright, R.J. Prenatal Particulate Air Pollution and Asthma Onset in Urban Children. Identifying Sensitive Windows and Sex Differences. Am. J. Respir. Crit. Care Med. 2015, 192, 1052–1059. [Google Scholar]
- Lavigne, E.; Donelle, J.; Hatzopoulou, M.; Van Ryswyk, K.; van Donkelaar, A.; Martin, R.V.; Chen, H.; Stieb, D.M.; Gasparrini, A.; Crighton, E.; et al. Spatiotemporal Variations in Ambient Ultrafine Particles and the Incidence of Childhood Asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Korten, I.; Ramsey, K.; Latzin, P. Air pollution during pregnancy and lung development in the child. Paediatr. Respir. Rev. 2017, 21, 38–46. [Google Scholar] [CrossRef]
- Jedrychowski, W.A.; Perera, F.P.; Maugeri, U.; Mroz, E.; Klimaszewska-Rembiasz, M.; Flak, E.; Edwards, S.; Spengler, J.D. Effect of prenatal exposure to fine particulate matter on ventilatory lung function of preschool children of non-smoking mothers. Paediatr. Périnat. Epidemiol. 2010, 24, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, L.; Carugno, M.; Bollati, V. Particulate matter exposure shapes DNA methylation through the lifespan. Clin. Epigenetics 2019, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Huang, S.; Du, L.; Sun, W.; Yu, Z.; Zhou, Y.; Chen, J.; Li, X.; Yu, B.; Chen, D. Expression of HMGB1 in maternal exposure to fine particulate air pollution induces lung injury in rat offspring assessed with micro-CT. Chem. Biol. Interact. 2018, 280, 64–69. [Google Scholar] [CrossRef]
- Cai, C.; Huang, J.; Lin, Y.; Miao, W.; Chen, P.; Chen, X.; Wang, J.; Chen, M. Particulate matter 2.5 induced arrhythmogenesis mediated by TRPC3 in human induced pluripotent stem cell-derived cardiomyocytes. Arch. Toxicol. 2019, 93, 1009–1020. [Google Scholar] [CrossRef]
- Cheng, Z.; Liang, X.; Liang, S.; Yin, N.; Faiola, F. A human embryonic stem cell-based in vitro model revealed that ultrafine carbon particles may cause skin inflammation and psoriasis. J. Environ. Sci. 2020, 87, 194–204. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wang, Y.M.; Zhao, Z.M.; Zhou, W.; Yuan, X.Y.; Jia, L.; Zhao, J.; Peng, S.Q. Oxidative damage related to PM2.5 exposure in human embryonic stem cell-derived fibroblasts. Zhonghua Yu Fang Yi Xue Za Zhi 2016, 50, 705–709. [Google Scholar] [PubMed]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibel, S.L.; Winquist, A.; Tseu, I.; Wang, J.; Luo, D.; Shojaie, S.; Nathan, N.; Snyder, E.; Post, M. Reversal of Surfactant Protein B Deficiency in Patient Specific Human Induced Pluripotent Stem Cell Derived Lung Organoids by Gene Therapy. Sci. Rep. 2019, 9, 13450. [Google Scholar] [CrossRef] [Green Version]
- Strikoudis, A.; Cieślak, A.; Loffredo, L.; Chen, Y.W.; Patel, N.; Saqi, A.; Lederer, D.J.; Snoeck, H.W. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep. 2019, 27, 3709–3723 e5. [Google Scholar] [CrossRef] [Green Version]
- Korogi, Y.; Gotoh, S.; Ikeo, S.; Yamamoto, Y.; Sone, N.; Tamai, K.; Konishi, S.; Nagasaki, T.; Matsumoto, H.; Ito, I.; et al. In Vitro Disease Modeling of Hermansky-Pudlak Syndrome Type 2 Using Human Induced Pluripotent Stem Cell-Derived Alveolar Organoids. Stem Cell Rep. 2019, 12, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Gotoh, S.; Korogi, Y.; Seki, M.; Konishi, S.; Ikeo, S.; Sone, N.; Nagasaki, T.; Matsumoto, H.; Muro, S.; et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat. Methods 2017, 14, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Na, S.; An, B.; Yang, S.R.; Kim, W.J.; Ha, K.S.; Han, E.T.; Park, W.S.; Lee, C.M.; Lee, J.Y.; et al. Paracrine influence of human perivascular cells on the proliferation of adenocarcinoma alveolar epithelial cells. Korean J. Physiol. Pharmacol. 2017, 21, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.J.; Kim, H.J.; Rhie, B.H.; Lee, M.R.; Choi, M.J.; Hong, S.H.; Kim, K.S. Maintenance of hPSCs under Xeno-Free and Chemically Defined Culture Conditions. Int. J. Stem Cells 2019, 12, 484–496. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.H.; Rampalli, S.; Lee, J.B.; McNicol, J.; Collins, T.; Draper, J.S.; Bhatia, M. Cell fate potential of human pluripotent stem cells is encoded by histone modifications. Cell Stem Cell 2011, 9, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Rasaei, R.; Kim, E.; Kim, J.Y.; Na, S.; Kim, J.H.; Heo, J.; Shin, D.M.; Choi, S.S.; Hong, S.H. Regulation of JAM2 Expression in the Lungs of Streptozotocin-Induced Diabetic Mice and Human Pluripotent Stem Cell-Derived Alveolar Organoids. Biomedicines 2020, 8, 346. [Google Scholar] [CrossRef] [PubMed]
- Borenfreund, E.; Puerner, J.A. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 2010, 61, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiemstra, P.S.; Grootaers, G.; van der Does, A.M.; Krul, C.A.M.; Kooter, I.M. Human lung epithelial cell cultures for analysis of inhaled toxicants: Lessons learned and future directions. Toxicol. Vitr. 2018, 47, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, S.; Ito, I.; Nagasaki, T.; Yamamoto, Y.; Konishi, S.; Korogi, Y.; Matsumoto, H.; Muro, S.; Hirai, T.; Funato, M.; et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Rep. 2014, 3, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Heo, H.R.; Kim, J.; Kim, W.J.; Yang, S.R.; Han, S.S.; Lee, S.J.; Hong, Y.; Hong, S.H. Human pluripotent stem cell-derived alveolar epithelial cells are alternatives for in vitro pulmotoxicity assessment. Sci. Rep. 2019, 9, 505. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, Y.; Hou, T.; Liao, J.; Zhang, C.; Sun, C.; Wang, G. PM2.5 induces EMT and promotes CSC properties by activating Notch pathway in vivo and vitro. Ecotoxicol. Environ. Saf. 2019, 178, 159–167. [Google Scholar] [CrossRef]
- Tang, W.; Du, L.; Sun, W.; Yu, Z.; He, F.; Chen, J.; Li, X.; Li, X.; Yu, L.; Chen, D. Maternal exposure to fine particulate air pollution induces epithelial-to-mesenchymal transition resulting in postnatal pulmonary dysfunction mediated by transforming growth factor-beta/Smad3 signaling. Toxicol. Lett. 2017, 267, 11–20. [Google Scholar] [CrossRef]
- Thevenot, P.T.; Saravia, J.; Jin, N.; Giaimo, J.D.; Chustz, R.E.; Mahne, S.; Kelley, M.A.; Hebert, V.Y.; Dellinger, B.; Dugas, T.R.; et al. Radical-containing ultrafine particulate matter initiates epithelial-to-mesenchymal transitions in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2013, 48, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Park, C.; Hong, S.H.; Kim, G.Y.; Song, K.S.; Hyun, J.W.; et al. Diesel particulate matter2.5 promotes epithelial-mesenchymal transition of human retinal pigment epithelial cells via generation of reactive oxygen species. Environ. Pollut. 2020, 262, 114301. [Google Scholar] [CrossRef]
- Fattorini, D.; Regoli, F. Role of the chronic air pollution levels in the Covid-19 outbreak risk in Italy. Environ. Pollut. 2020, 264, 114732. [Google Scholar] [CrossRef] [PubMed]
- Conticini, E.; Frediani, B.; Caro, D. Can atmospheric pollution be considered a co-factor in extremely high level of SARS-CoV-2 lethality in Northern Italy? Environ. Pollut. 2020, 261, 114465. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Nethery, R.C.; Sabath, M.B.; Braun, D.; Dominici, F. Air pollution and COVID-19 mortality in the United States: Strengths and limitations of an ecological regression analysis. Sci. Adv. 2020, 6, eabd4049. [Google Scholar] [CrossRef] [PubMed]
- Leparskiĭ, E.A. The WHO European Regional Bureau: The main trends in its activities. Pediatriia 1988, 8, 66–70. [Google Scholar]
- Frontera, A.; Cianfanelli, L.; Vlachos, K.; Landoni, G.; Cremona, G. Severe air pollution links to higher mortality in COVID-19 patients: The “double-hit” hypothesis. J. Infect. 2020, 81, 255–259. [Google Scholar] [CrossRef]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev. Cell 2020, 53, 514–529 e3. [Google Scholar] [CrossRef]
- Chakladar, J.; Shende, N.; Li, W.T.; Rajasekaran, M.; Chang, E.Y.; Ongkeko, W.M. Smoking-Mediated Upregulation of the Androgen Pathway Leads to Increased SARS-CoV-2 Susceptibility. Int. J. Mol. Sci. 2020, 21, 3627. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, X.; Li, R.; Zheng, M.; Yang, S.; Dai, L.; Wu, A.; Hu, C.; Huang, Y.; Xie, M.; et al. Overexpression of the SARS-CoV-2 receptor ACE2 is induced by cigarette smoke in bronchial and alveolar epithelia. J. Pathol. 2020. [Google Scholar] [CrossRef]
- Bui, L.T.; Winters, N.I.; Chung, M.I.; Joseph, C.; Gutierrez, A.J.; Habermann, A.C.; Adams, T.S.; Schupp, J.C.; Poli, S.; Peter, L.M.; et al. Single-cell RNA-sequencing reveals dysregulation of molecular programs associated with SARS-CoV-2 severity and outcomes in patients with chronic lung disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Comunian, S.; Dongo, D.; Milani, C.; Palestini, P. Air Pollution and Covid-19: The Role of Particulate Matter in the Spread and Increase of Covid-19’s Morbidity and Mortality. Int. J. Environ. Res. Public Health 2020, 17, 4487. [Google Scholar] [CrossRef]
- Parajuli, N.; Ramprasath, T.; Patel, V.B.; Wang, W.; Putko, B.; Mori, J.; Oudit, G.Y. Targeting angiotensin-converting enzyme 2 as a new therapeutic target for cardiovascular diseases. Can. J. Physiol. Pharmacol. 2014, 92, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Aztatzi-Aguilar, O.G.; Uribe-Ramírez, M.; Arias-Montaño, J.A.; Barbier, O.; De Vizcaya-Ruiz, A. Acute and subchronic exposure to air particulate matter induces expression of angiotensin and bradykinin-related genes in the lungs and heart: Angiotensin-II type-I receptor as a molecular target of particulate matter exposure. Part. Fibre Toxicol. 2015, 12, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Takano, H. Pulmonary surfactant itself must be a strong defender against SARS-CoV-2. Med. Hypotheses 2020, 144, 110020. [Google Scholar] [CrossRef]
- Yue, H.; Ji, X.; Li, G.; Hu, M.; Sang, N. Maternal Exposure to PM2.5 Affects Fetal Lung Development at Sensitive Windows. Environ. Sci. Technol. 2020, 54, 316–324. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Sequence 5′ to 3′ | Product Size (bp) | |
|---|---|---|---|
| T1α | F | TGC GAA AAA TGT CGG GAA GG | 51 |
| R | GGC GTA ACC CTT CAG CTC TT | ||
| SFTPB | F | GCC ATA CCA CAG GCA ATG CT | 80 |
| R | TGC TGC TCC ACA AAT TGC TT | ||
| SFTPC | F | CCT TCT TAT CGT GGT GGT GGT | 96 |
| R | TCT CCG TGT GTT TCT GGC TCA T | ||
| HOPX | F | GCC TTT CCG AGG AGG AGA C | 97 |
| R | TCT GTG ACG GAT CTG CAC TC | ||
| AQP5 | F | ACT GGG TTT TCT GGG TAG GG | 172 |
| R | ATG GTC TTC TTC CGC TCT TC | ||
| NKX2.1 | F | AGC ACA CGA CTC CGT TCT CA | 75 |
| R | CCT CCA TGC CCA CTT TCT TG | ||
| VIMENTIN | F | CCA GGC AAA GCA GGA GTC | 212 |
| R | CGA AGG TGA CGA GCC ATT | ||
| α-SMA | F | GAC GAA GCA CAG AGC AAA AG | 70 |
| R | AGT TGG TGA TGA TGC CAT GT | ||
| COL1A1 | F | AAG GGT GAG ACA GGC GAA CA | 70 |
| R | GAC CCT GGA GGC CAG AGA AG | ||
| CTNNB1 | F | AAA ATG GCA GTG CGT TTA | 99 |
| R | TTT GAA GGC AGT CTG TCG TA | ||
| TWIST1 | F | AGC AAG ATT CAG ACC CTC AAG | 145 |
| R | ATC CTC CAG ACC GAG AAG G | ||
| SNAIL1 | F | TTT ACC TTC CAG CAG CCC TA | 73 |
| R | GAC AGA GTC CCA GAT GAG CA | ||
| SNAIL2 | F | CTG TGG GGA CAT GAA CTG TG | 115 |
| R | AGG GTC TGG GGA AAC TCG | ||
| IL-1α | F | CCA ACG GGA AGG TTC TGA AG | 70 |
| R | GCC TCC AGG TCA TCA TCA GT | ||
| IL-1β | F | CTG TCC TGC GTG TTG AAA GA | 179 |
| R | TTC TGC TTG AGA GGT GCT GA | ||
| IL-6 | F | AGC CCT GAG AAA GGA GAC AT | 85 |
| R | TGG AAG GTT CAG GTT GTT TT | ||
| TNF-α | F | AAC CTC CTC TCT GCC ATC AA | 185 |
| R | CCA AAG TAG ACC TGC CCA GA | ||
| NOX1 | F | AGG GCT TTC GAA CAA CAA TA | 104 |
| R | CCA GCA CAG CCA CTT CAT AC | ||
| NOX2 | F | AAC TGC TGG AGA GCC AGA TG | 101 |
| R | GCA AAG TGA TTG GCC TGA GA | ||
| NOX3 | F | GCT ATG CAG AAT GGC AGA CA | 101 |
| R | TAC AAG ACC ACA GGG CCT AA | ||
| NOX4 | F | CTT TTG GAA GTC CAT TTG AG | 231 |
| R | GTC TGT TCT CTT GCC AAA AC | ||
| OCT4 | F | TCG AGA ACC GAG TGA GAG G | 125 |
| R | GAA CCA CAC TCG GAC CAC A | ||
| SOX2 | F | GCA CAT GAA GGA GCA CCC GGA TTA | 86 |
| R | GTG GTC CTT CTT GTG CTG C | ||
| NANOG | F | CAA AGG CAA ACA ACC CAC TT | 158 |
| R | TCT GCT GGA GGC TGA GGT AT | ||
| GADD45g | F | CAG ATC CAT TTT ACG CTG ATC CA | 209 |
| R | TCC TCG CAA AAC AGG CTG AG | ||
| SLUG | F | TGT GAC AAG GAA TAT GTG AGC C | 203 |
| R | TGA GCC CTC AGA TTT GAC CTG | ||
| MMP2 | F | AGC GAG TGG ATG CCG CCT TTA A | 138 |
| R | CAT TCC AGG CAT CTG CGA TGA G | ||
| MMP9 | F | CTC GAA CTT TGA CAG CGA CA | 187 |
| R | GCC ATT CAC GTC GTC CTT AT | ||
| ACE2 | F | GGGATCAGAGATCGGAAGAAGAAA | 124 |
| R | AGGAGGTCTGAACATCATCAGTG | ||
| TMPRSS2 | F | AATCGGTGTGTTCGCCTCTAC | 106 |
| R | CGTAGTTCTCGTTCCAGTCGT | ||
| GAPDH | F | GGC ATG GAC TGT GGT CAT GA | 87 |
| R | TGC ACC ACC AAC TGC TTA GC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Kim, J.; Kim, W.J.; Choi, Y.H.; Yang, S.-R.; Hong, S.-H. Diesel Particulate Matter 2.5 Induces Epithelial-to-Mesenchymal Transition and Upregulation of SARS-CoV-2 Receptor during Human Pluripotent Stem Cell-Derived Alveolar Organoid Development. Int. J. Environ. Res. Public Health 2020, 17, 8410. https://doi.org/10.3390/ijerph17228410
Kim J-H, Kim J, Kim WJ, Choi YH, Yang S-R, Hong S-H. Diesel Particulate Matter 2.5 Induces Epithelial-to-Mesenchymal Transition and Upregulation of SARS-CoV-2 Receptor during Human Pluripotent Stem Cell-Derived Alveolar Organoid Development. International Journal of Environmental Research and Public Health. 2020; 17(22):8410. https://doi.org/10.3390/ijerph17228410
Chicago/Turabian StyleKim, Jung-Hyun, Jeeyoung Kim, Woo Jin Kim, Yung Hyun Choi, Se-Ran Yang, and Seok-Ho Hong. 2020. "Diesel Particulate Matter 2.5 Induces Epithelial-to-Mesenchymal Transition and Upregulation of SARS-CoV-2 Receptor during Human Pluripotent Stem Cell-Derived Alveolar Organoid Development" International Journal of Environmental Research and Public Health 17, no. 22: 8410. https://doi.org/10.3390/ijerph17228410