
De Novo Transcriptome Analysis by PacBio SMRT-Seq and Illumina RNA-Seq Provides New Insights into Polyphenol Biosynthesis in Chinese Olive Fruit

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Measurement of Total Phenolic Content (TPC)
2.3. RNA Sample Preparation
2.4. PacBio Library Construction and Single-Molecule Sequencing
2.5. Illumina RNA-Seq Library Construction and Sequencing
2.6. Data Analyses of Single-Molecule Sequencing Data
2.7. Quality Assessment of Transcriptome Assembly
2.8. Differentially Expressed Genes (DEGs) Analysis and Functional Annotation
2.9. Gene Validation and Quantitative Real-Time PCR (RT-qPCR) Analysis
3. Results
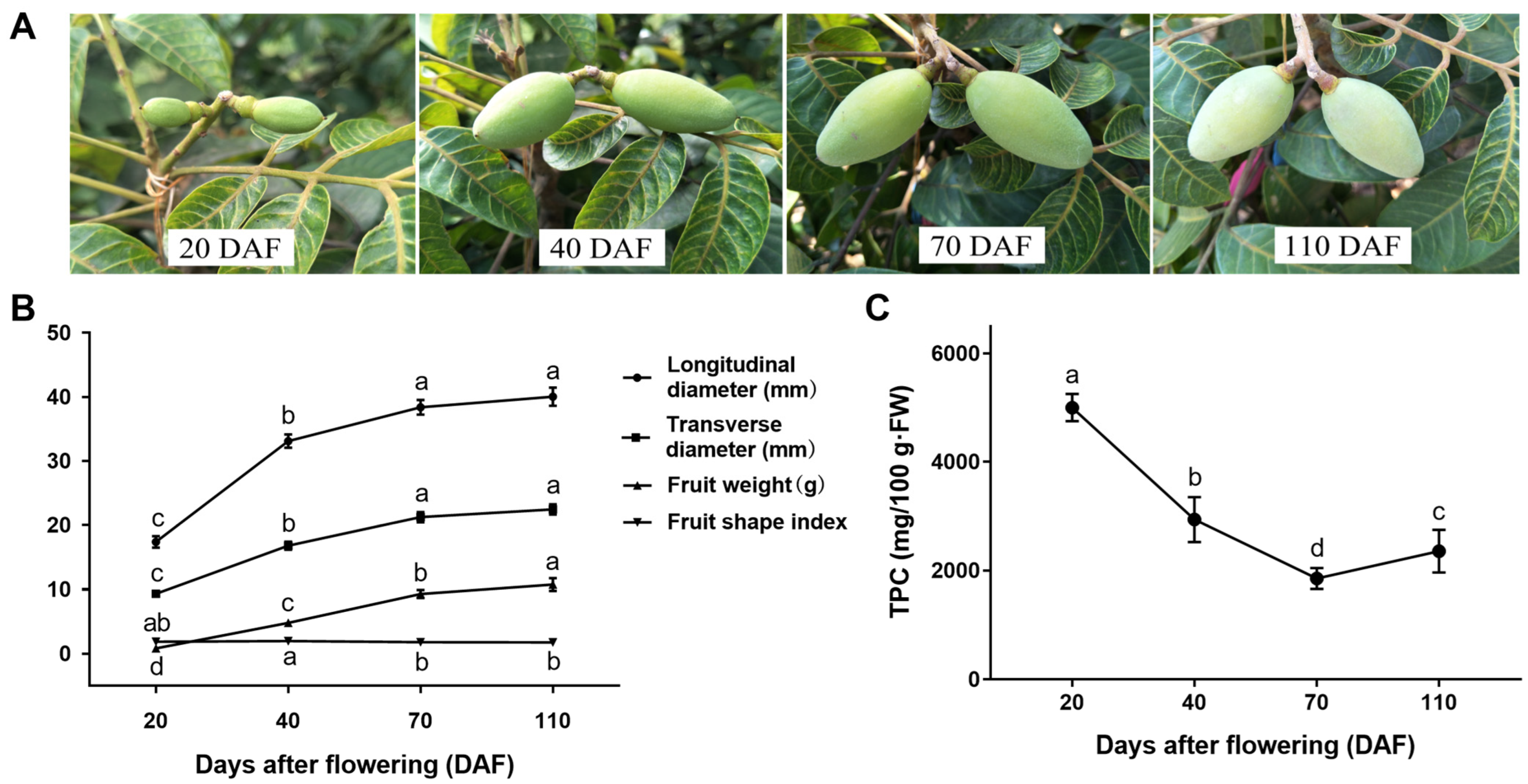
3.1. Growth and Development and TPC of C. Album Fruits
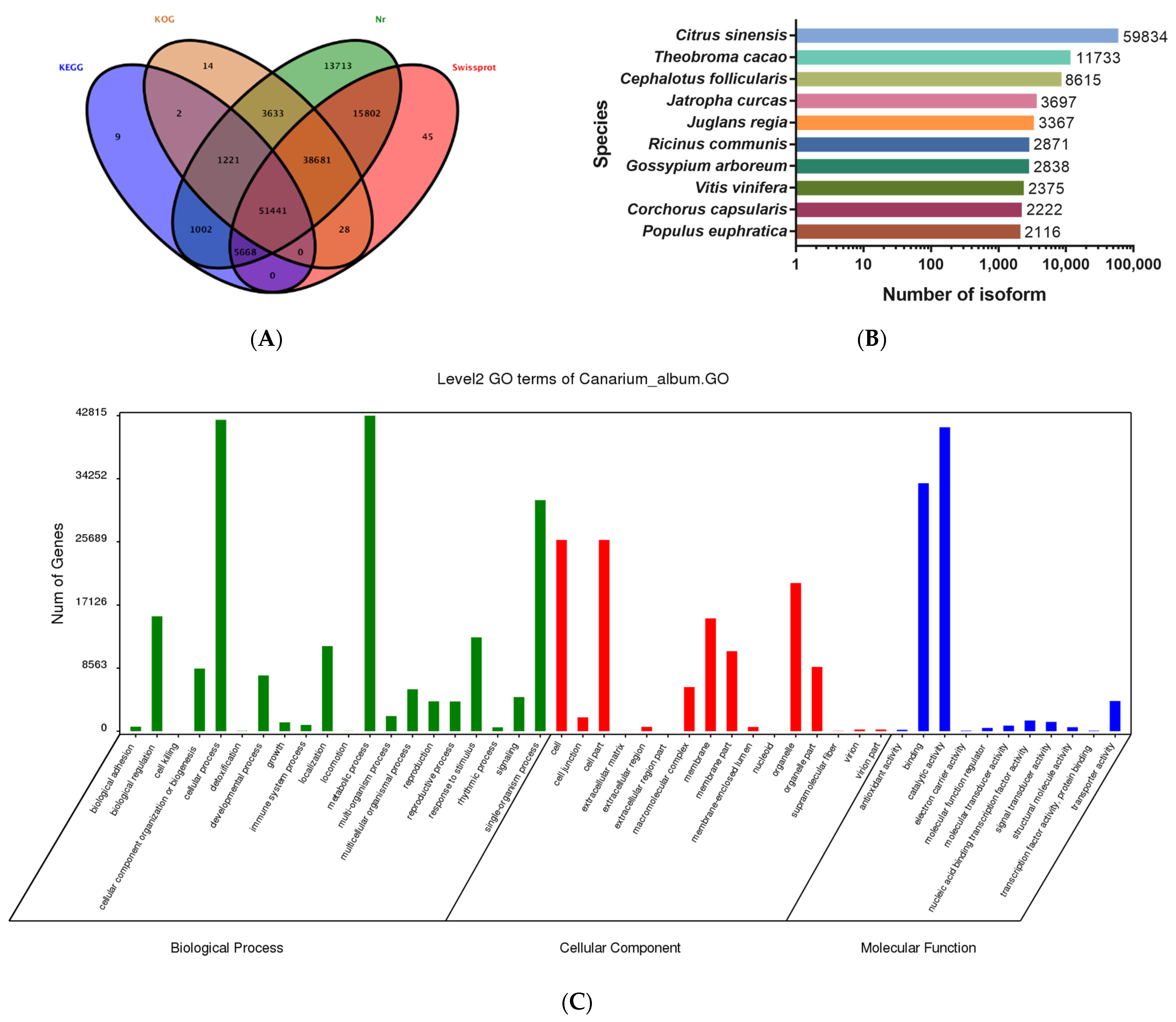
3.2. PacBio SMRT Sequencing Captures Full-Length Transcripts of C. album
3.3. Quality Evaluation of Illumina RNA-Seq
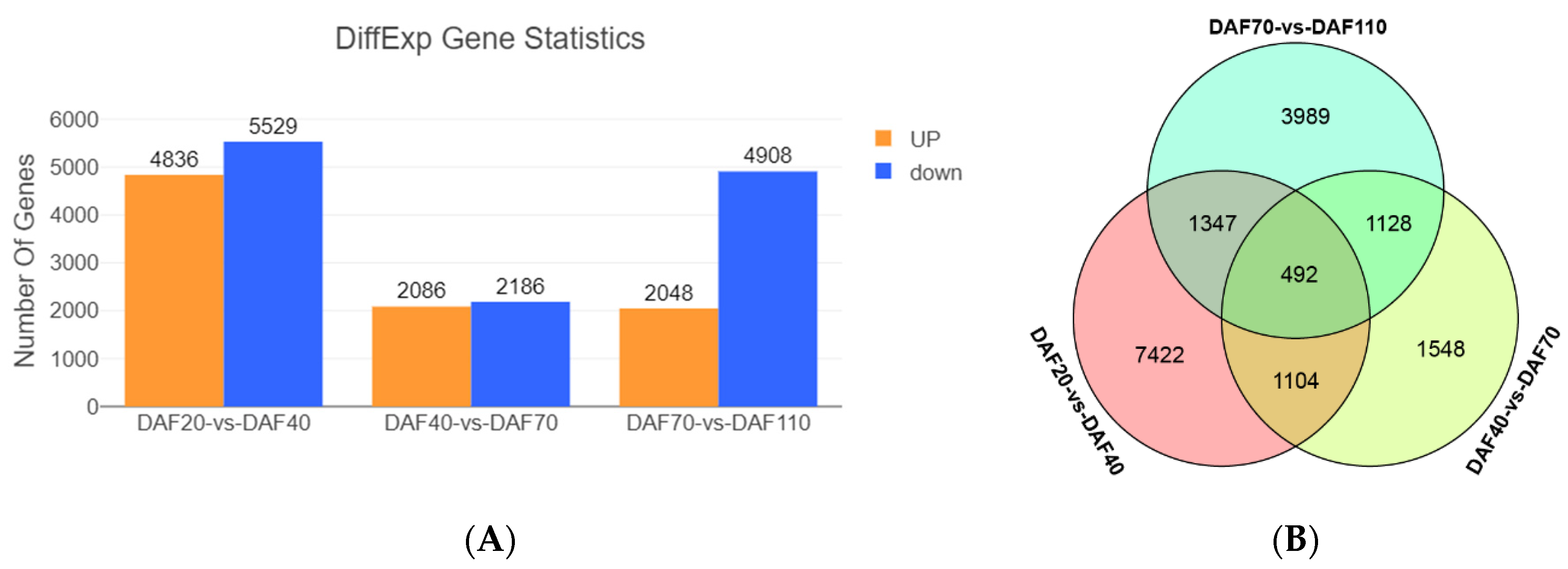
3.4. DEGs Analysis
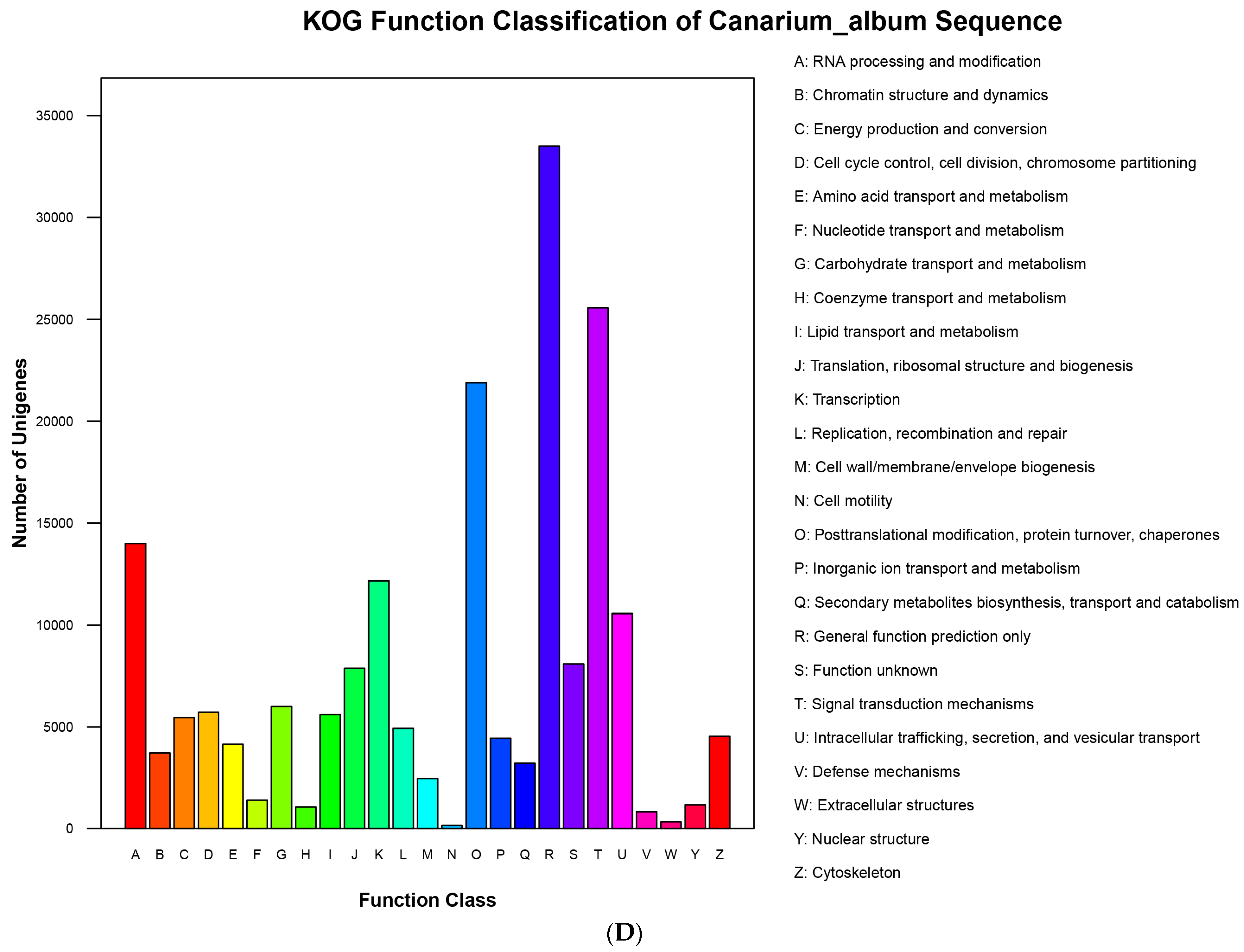
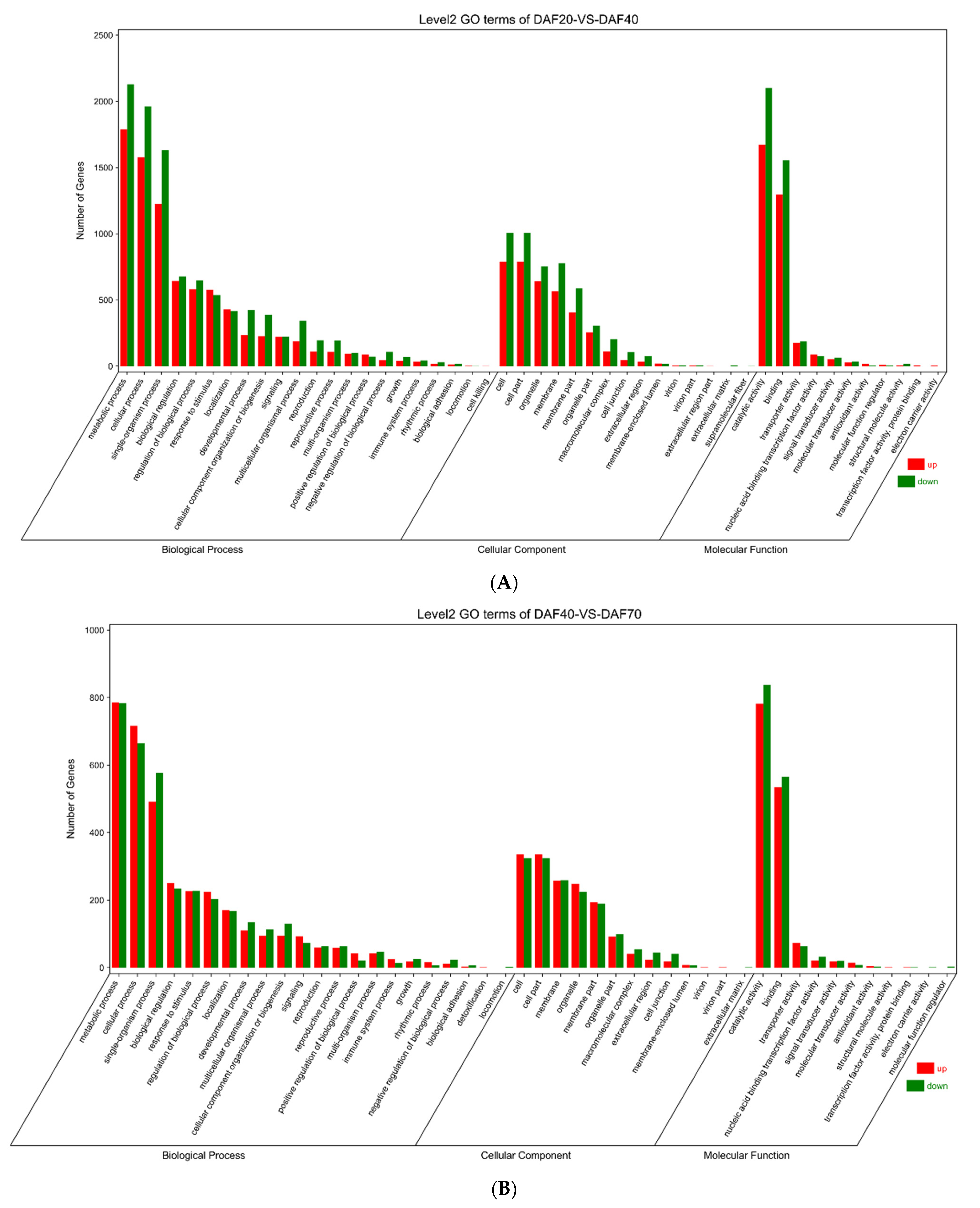
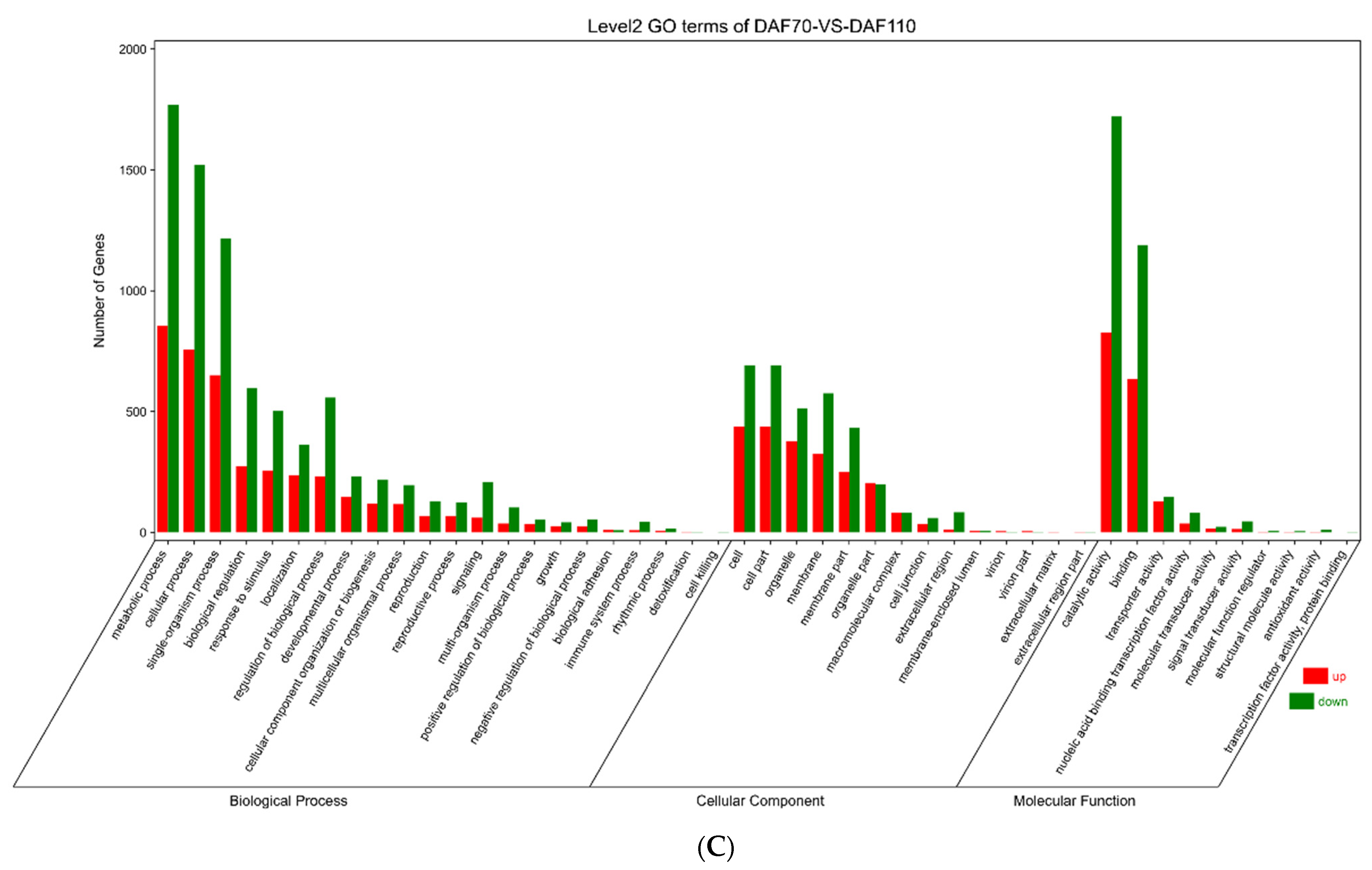
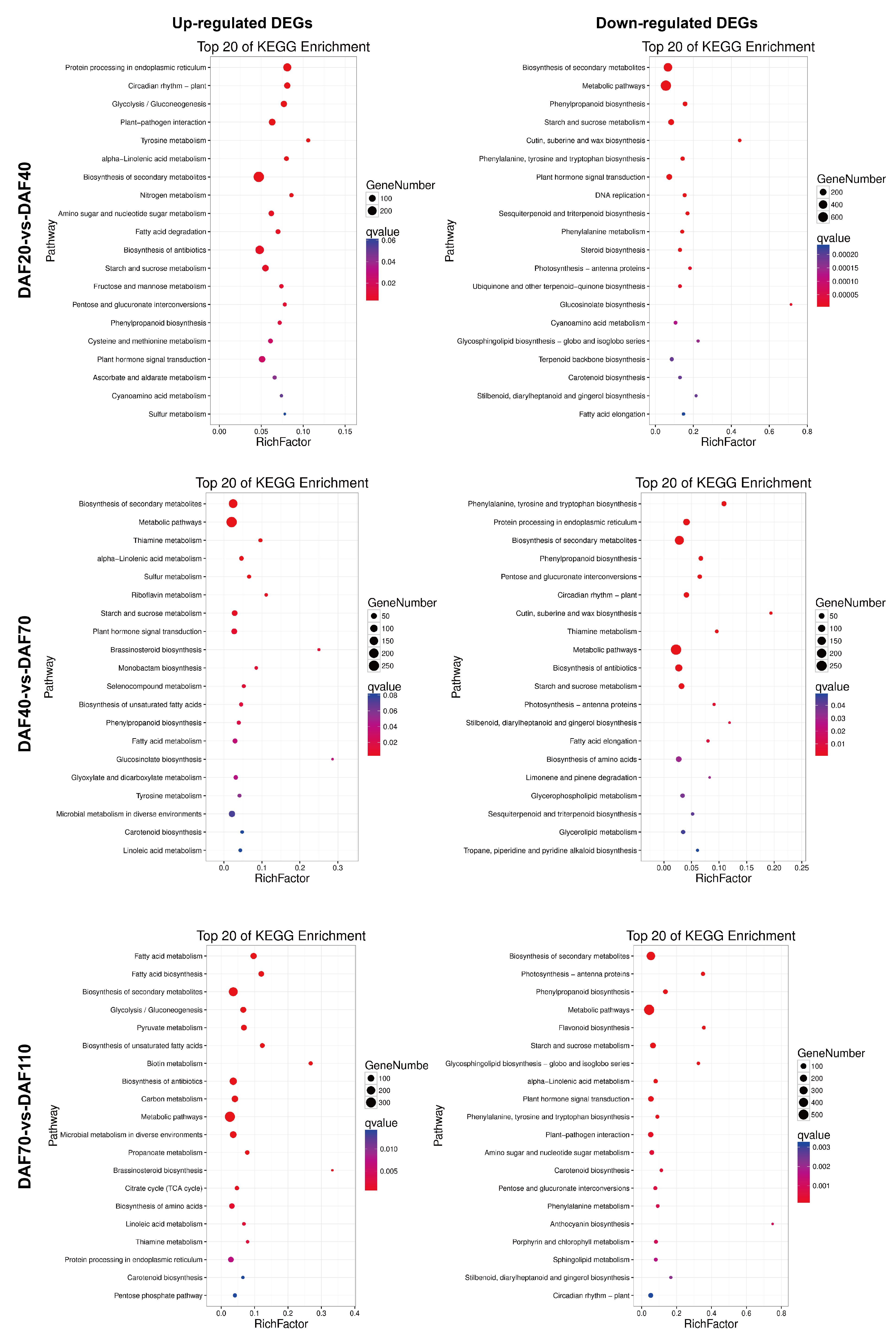
3.5. Functional Annotation of DEGs
3.6. Identification of DEGs in Major Polyphenol Biosynthetic Pathway
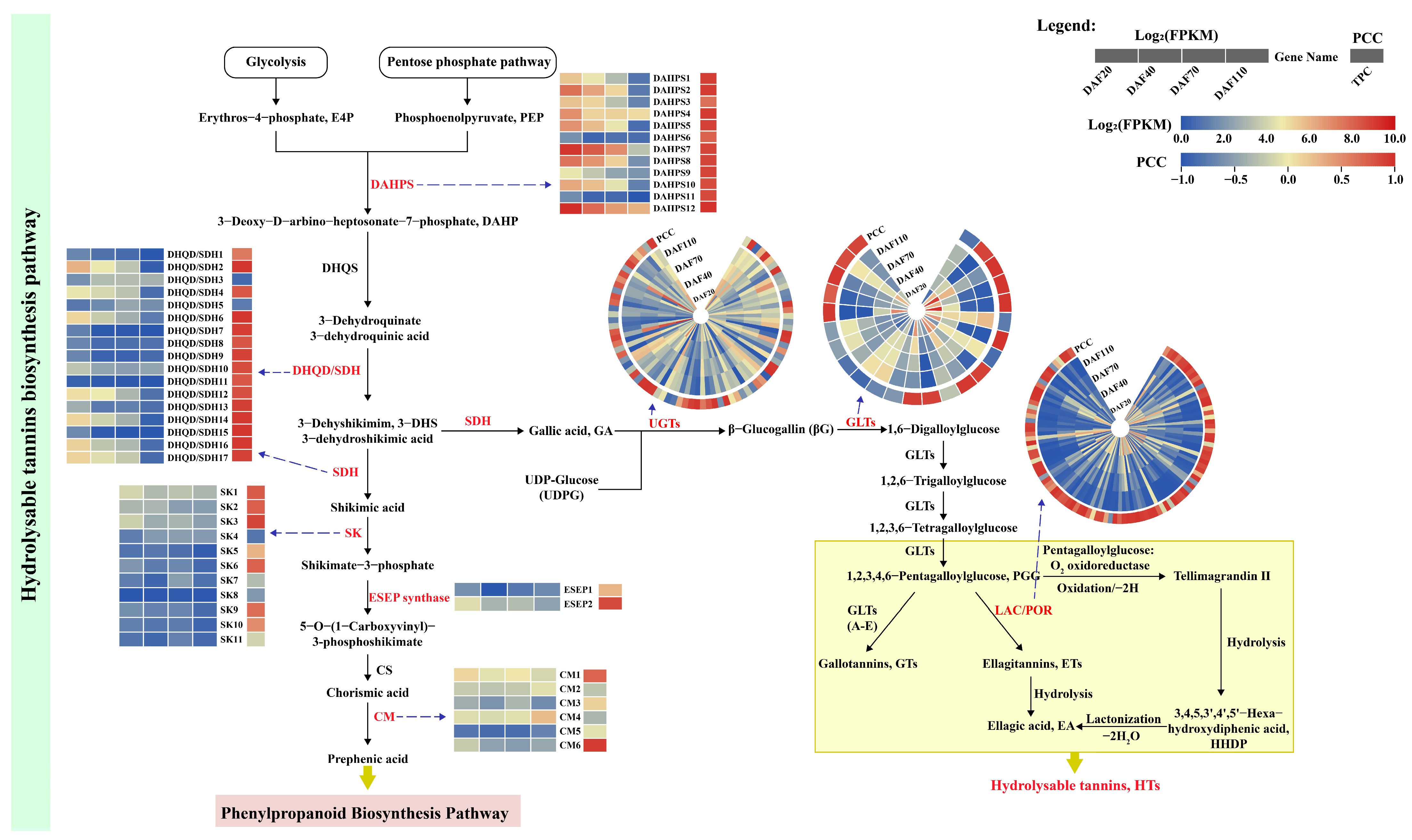
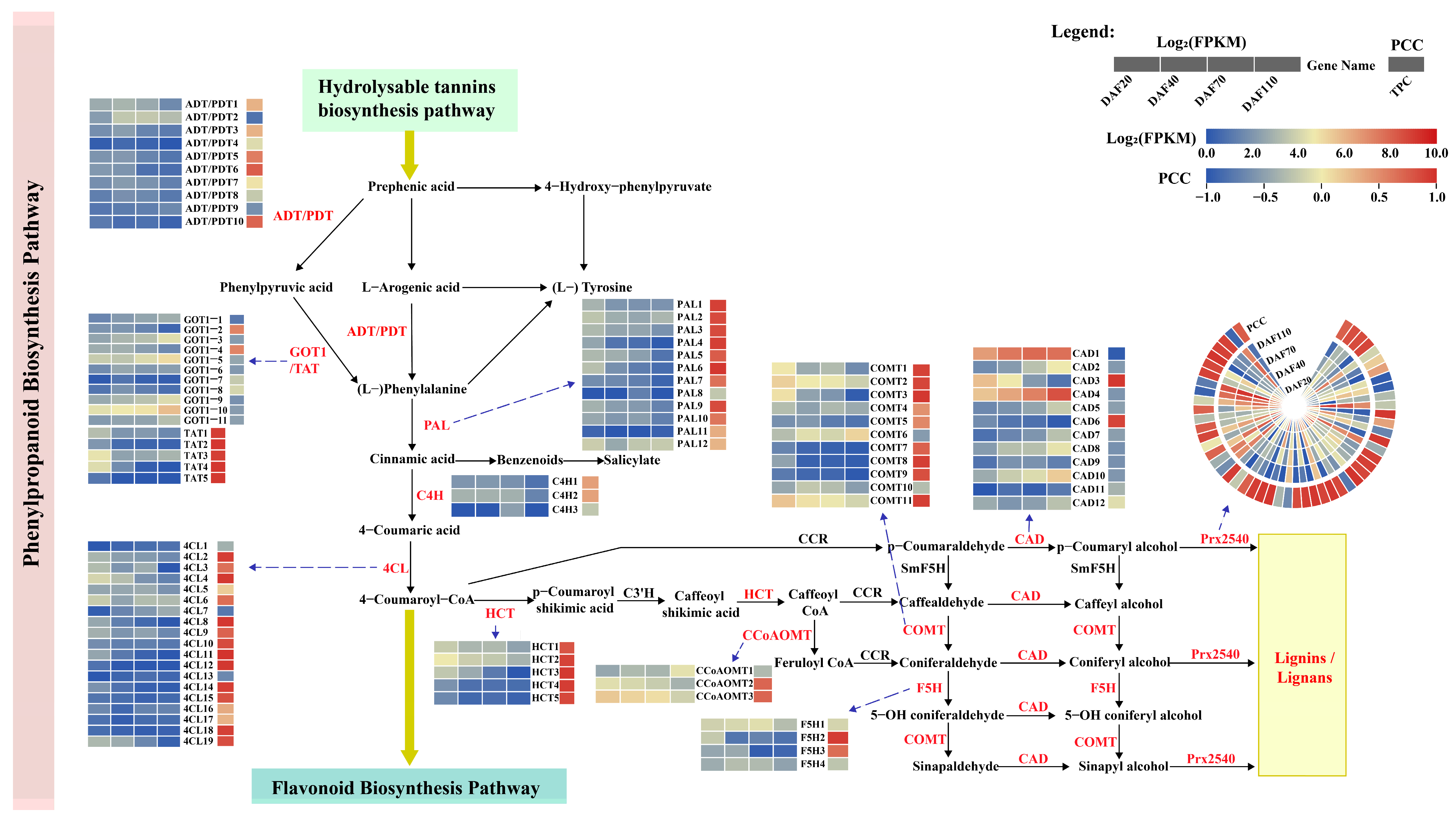
3.6.1. Identification of DEGs Related to HT Biosynthesis Pathway
3.6.2. Identification of DEGs Related to the Phenylpropanoid Biosynthesis Pathway
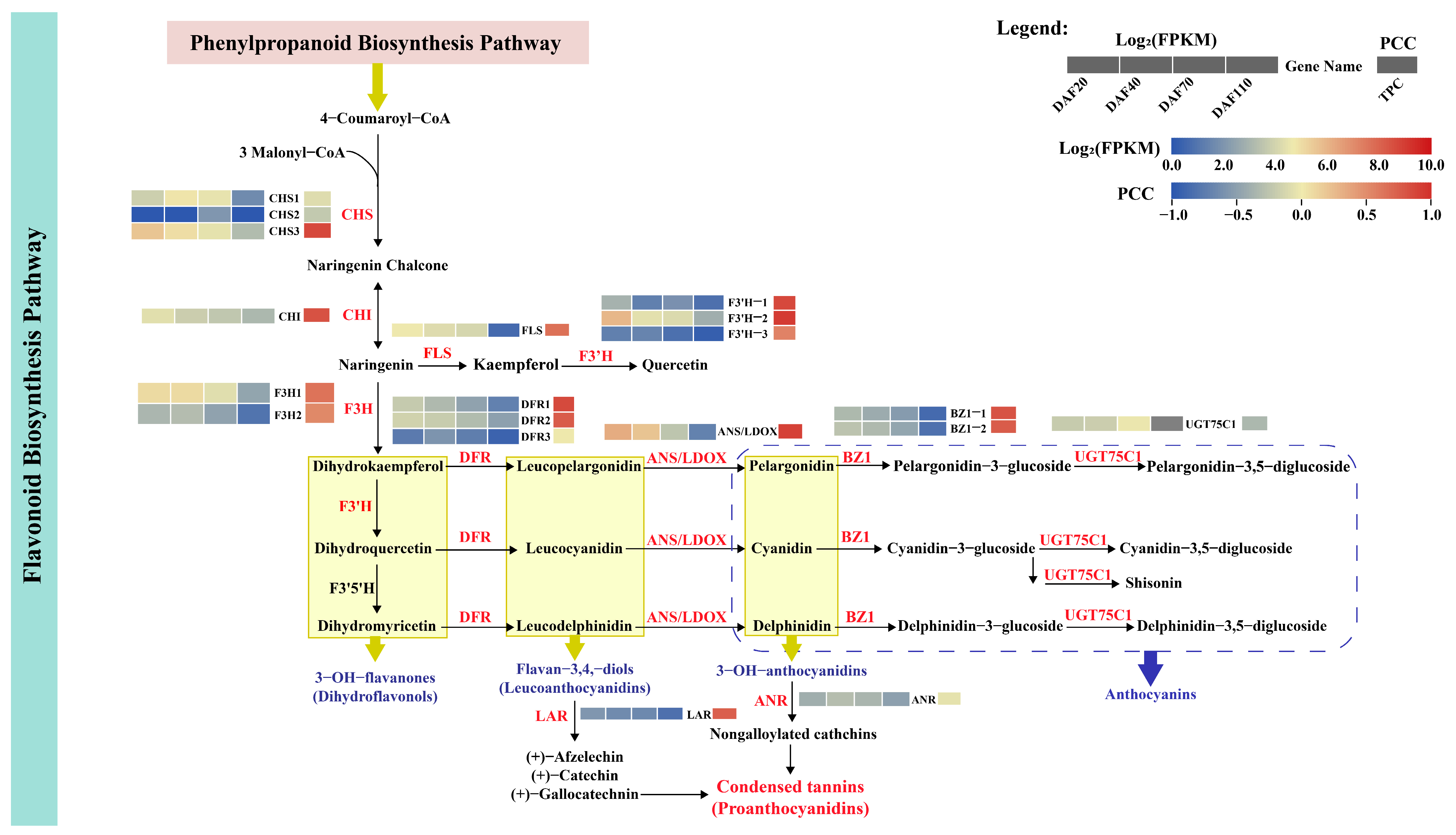
3.6.3. Identification of DEGs Related to the Flavonoid Biosynthesis Pathway
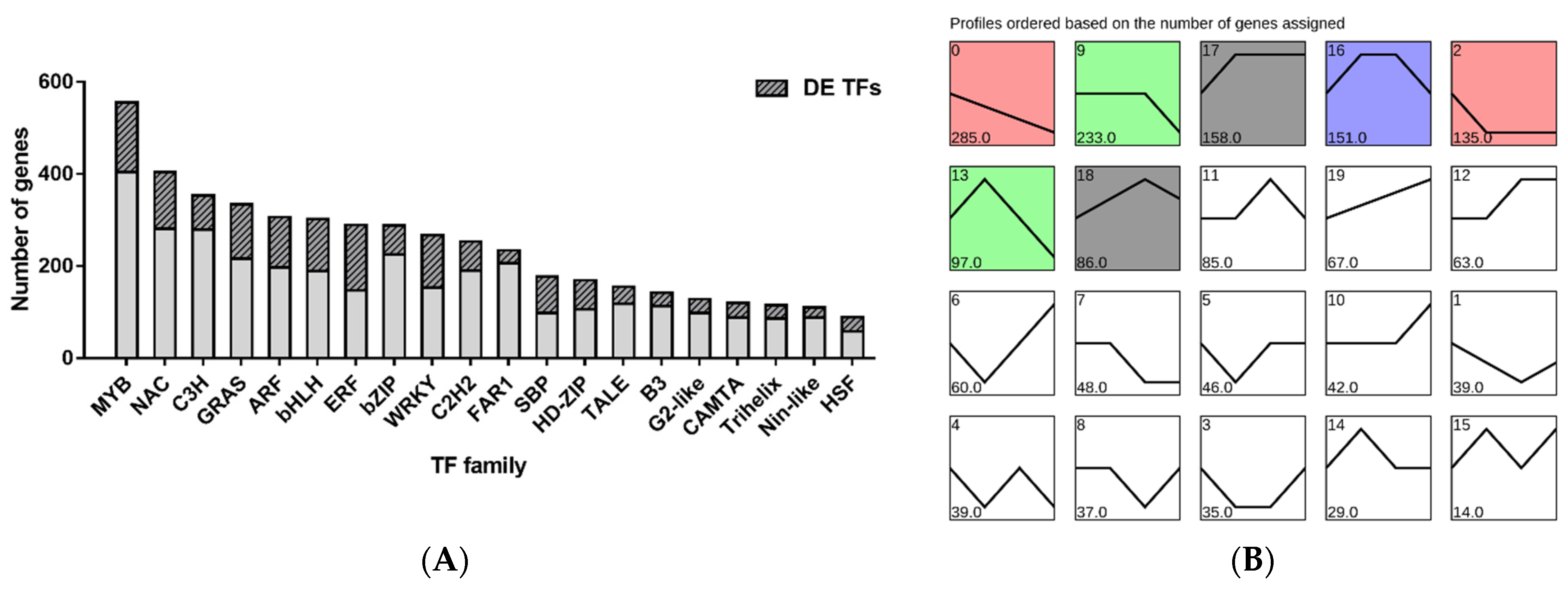
3.7. Identification and Analysis of TFs
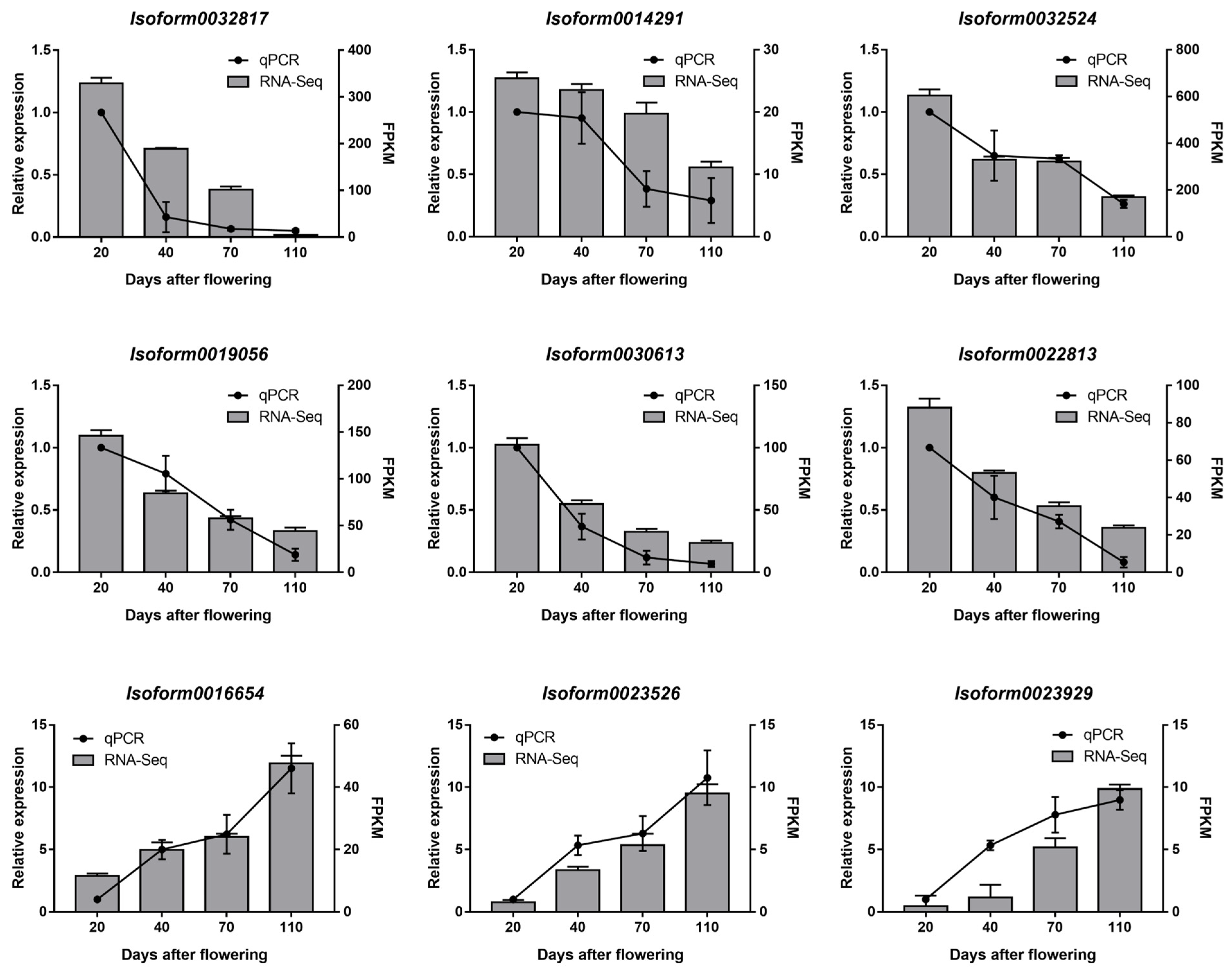
3.8. RT-qPCR for Validation of RNA-Seq
4. Discussion
4.1. Combined Sequencing Approaches Provided Comprehensive Transcriptome Information of C. album
4.2. Differentially Expressed Transcripts Reveal the Metabolism and Regulation of Polyphenols of C. album Fruit
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| 4CL | 4-coumarate CoA ligase |
| ANR | anthocyanidin reductase |
| ANS | anthocyanidin synthase |
| BZ1 | anthocyanidin 3-O-glucosyltransferase |
| C4H | cinnamic acid-4-hydroxylase |
| CAD | cinnamyl alcohol dehydrogenase |
| CCS | circular consensus sequence |
| CHI | chalcone isomerase |
| CHS | Chalcone synthase |
| CM | chorismate mutase |
| CTs | Condensed tannins |
| DAF | Days after flowering |
| DAHP | 3-deoxy-D-arabino-heptulosonate-7-phosphate |
| DAHPS | 3-deoxy-7-phosphoheptulonate synthase |
| DEGs | differentially expressed genes |
| DFR | dihydroflavonol 4-reductase |
| DHQD/DHQ | 3-dehydroquinate dehydratase I |
| E4P | erythrose-4-phosphate |
| EPSP | 3-phosphoshikimate 1-carboxyvinyltransferase |
| F3H | naringenin 3-dioxygenase |
| F3’H | flavonoid 3’-monooxygenase |
| FC | Fold change |
| FDR | False discovery rate |
| FLNC | Full-length non-chimeric |
| FLS | flavonol synthase |
| FPKM | Fragments per kilobase of transcript per million fragments mapped |
| GA | Gallic acid |
| GLTs | galloyltransferases |
| GO | Gene Ontology |
| HCT | O-hydroxycinnamoyltransferase |
| HTs | hydrolysable tannins |
| KEGG | The Kyoto Encyclopedia of Genes and Genomes |
| KOG | Eukaryotic clusters of orthologous groups |
| LACs | laccases |
| LAR | leucoanthocyanidin reductase |
| LDOX | leucocyanidin dioxygenase |
| LRS | Long-read sequencing |
| nFL | Non-full-length |
| NGS | Next-generation sequencing |
| Nr | Non-redundant protein |
| PAL | phenylalanine ammonia-lyase |
| PCA | Principal component analysis |
| PCC | Pearson correlation coefficient |
| PEP | phosphoenolpyruvate |
| PGG | pentagalloylglucose |
| RT-qPCR | Quantitative real-time PCR |
| SDH | shikimate dehydrogenase |
| SK | shikimate kinase |
| SMRT | Single-molecule real-time |
| TFs | Transcription factors |
| TGS | Third-generation sequencing |
| TPC | Total phenolic content |
| UGTs | UDP-glucosyltransferases |
| βG | β-glucogallin |
References
- Raven, P.H.; Zhang, L.; Al-Shehbaz, I.A. Flora of China; Science Press & Missouri Botanical Garden Press: Beijing, China; St. Louis, MO, USA, 2008. [Google Scholar]
- Mei, Z.; Zhang, X.; Liu, X.; Imani, S.; Fu, J. Genetic analysis of Canarium album in different areas of China by improved RAPD and ISSR. Comptes Rendus Biol. 2017, 340, 558–564. [Google Scholar] [CrossRef] [PubMed]
- He, C.N. Canarium album (Lour.) Raeusch. (Qingguo, Chinese Olive). In Dietary Chinese Herbs; Springer: Vienna, Austria, 2015; pp. 307–313. [Google Scholar] [CrossRef]
- Chen, Y.M.; Lin, B.; Lin, Y.F.; Sang, Y.Y.; Lin, M.S.; Fan, Z.Q.; Chen, Y.H.; Wang, H.; Lin, H.T. Involvements of membrane lipid and phenolic metabolism in reducing browning and chilling injury of cold-stored Chinese olive by γ-aminobutyric acid treatment. Postharvest Biol. Technol. 2024, 209, 112664. [Google Scholar] [CrossRef]
- Kuo, C.T.; Liu, T.H.; Hsu, T.H.; Lin, F.Y.; Chen, H.Y. Antioxidant and antiglycation properties of different solvent extracts from Chinese olive (Canarium album L.) fruit. Asian Pac. J. Trop. Med. 2015, 8, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Mogana, R.; Wiart, C. Canarium L.: A Phytochemical and Pharmacological Review. J. Pharm. Res. 2011, 4, 2482–2489. [Google Scholar]
- He, Z.Y.; Xia, W.S. Nutritional composition of the kernels from Canarium album L. Food Chem. 2007, 102, 808–811. [Google Scholar] [CrossRef]
- He, Z.Y.; Xia, W.S.; Chen, J. Isolation and structure elucidation of phenolic compounds in Chinese olive (Canarium album L.) fruit. Eur. Food Res. Technol. 2008, 226, 1191–1196. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, N.; Ding, H.; Yao, R. Polyphenols contents and antioxidant capacity of 68 Chinese herbals suitable for medical or food uses. Food Res. Int. 2008, 41, 363–370. [Google Scholar] [CrossRef]
- Xiang, Z.-B.; Liu, X.-Y.; He, C.-L.; Lin, S.H. Flavonoids from Canarium album. Asian J. Chem. 2014, 26, 4529–4530. [Google Scholar] [CrossRef]
- Giang, P.M.; Konig, W.A.; Son, P.T. Chemical composition of the resin essential oil of Canarium album from Vietnam. Chem. Nat. Compd. 2006, 42, 523–524. [Google Scholar] [CrossRef]
- Tamai, M.; Watanabe, N.; Someya, M.; Kondoh, H.; Omura, S.; Zhang, P.L.; Chang, R.; Chen, W.M. New hepatoprotective triterpenes from Canarium album. Planta Medica 1989, 55, 44–47. [Google Scholar] [CrossRef]
- Zeng, H.; Miao, S.; Zheng, B.; Lin, S.; Jian, Y.; Chen, S.; Zhang, Y. Molecular Structural Characteristics of Polysaccharide Fractions from Canarium album (Lour.) Raeusch and Their Antioxidant Activities. J. Food Sci. 2015, 80, H2585–H2596. [Google Scholar] [CrossRef]
- Wen, L.; Lin, S.; Zhu, Q.; Wu, D.; Jiang, Y.; Zhao, M.; Sun, J.; Luo, D.; Zeng, S.; Yang, B. Analysis of Chinese Olive Cultivars Difference by the Structural Characteristics of Oligosaccharides. Food Anal. Methods 2013, 6, 1529–1536. [Google Scholar] [CrossRef]
- Li, J.; Wang, R.; Wang, Y.; Zeng, J.; Xu, Z.; Xu, J.; He, X. Anti-Inflammatory Benzofuran Neolignans from the Fruits of Canarium album (Chinese Olive). J. Agric. Food Chem. 2022, 70, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.T.; Hsu, K.M.; Chen, H.J.; Su, N.W.; Liao, Y.C.; Hsieh, S.C. Identification of Scoparone from Chinese Olive Fruit as a Modulator of Macrophage Polarization. J. Agric. Food Chem. 2023, 71, 5195–5207. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, S.; Zhong, R.; Wan, F.; Chen, L.; Liu, L.; Yi, B.; Zhang, H. Olive Fruit Extracts Supplement Improve Antioxidant Capacity via Altering Colonic Microbiota Composition in Mice. Front. Nutr. 2021, 8, 645099. [Google Scholar] [CrossRef]
- He, Z.Y.; Xia, W.S.; Liu, Q.H.; Chen, J. Identification of a new phenolic compound from Chinese olive (Canarium album L.) fruit. Eur. Food Res. Technol. 2009, 228, 339–343. [Google Scholar] [CrossRef]
- He, Z.; Xia, W.S. Analysis of phenolic compounds in Chinese olive (Canarium album L.) fruit by RPHPLC-DAD-ESI-MS. Food Chem. 2007, 105, 1307–1311. [Google Scholar] [CrossRef]
- Fu, L.; Xu, B.T.; Xu, X.R.; Gan, R.Y.; Zhang, Y.; Xia, E.Q.; Li, H.B. Antioxidant capacities and total phenolic contents of 62 fruits. Food Chem. 2011, 129, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Tijjani, H.; Zangoma, M.H.; Mohammed, Z.S.; Obidola, S.M.; Egbuna, C.; Abdulai, S.I. Polyphenols: Classifications, Biosynthesis and Bioactivities. In Functional Foods and Nutraceuticals: Bioactive Components, Formulations and Innovations; Egbuna, C., Dable Tupas, G., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 389–414. [Google Scholar]
- Cai, J.; Wang, N.; Zhao, J.; Zhao, Y.; Xu, R.; Fu, F.; Pan, T.; Yu, Y.; Guo, Z.; She, W. Accumulation of Polyphenolics and Differential Expression of Genes Related to Shikimate Pathway during Fruit Development and Maturation of Chinese Olive (Canarium album). Agronomy 2023, 13, 895. [Google Scholar] [CrossRef]
- Tyagi, K.; Shukla, P.; Rohela, G.K.; Shabnam, A.A.; Gautam, R. Plant Phenolics: Their Biosynthesis, Regulation, Evolutionary Significance, and Role in Senescence. In Plant Phenolics in Sustainable Agriculture; Springer: Berlin/Heidelberg, Germany, 2020; pp. 431–449. [Google Scholar]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Marudamuthu, B.; Sharma, T.; Purru, S.; Soam, S.K.; Rao, C.S. Next-generation sequencing technology: A boon to agriculture. Genet. Resour. Crop Evol. 2023, 70, 353–372. [Google Scholar] [CrossRef]
- Tyagi, P.; Singh, D.; Mathur, S.; Singh, A.; Ranjan, R. Upcoming progress of transcriptomics studies on plants: An overview. Front. Plant Sci. 2022, 13, 1030890. [Google Scholar] [CrossRef]
- Gao, L.; Xu, W.; Xin, T.; Song, J. Application of third-generation sequencing to herbal genomics. Front. Plant Sci. 2023, 14, 1124536. [Google Scholar] [CrossRef] [PubMed]
- Hamim, I.; Sekine, K.-T.; Komatsu, K. How do emerging long-read sequencing technologies function in transforming the plant pathology research landscape? Plant Mol. Biol. 2022, 110, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, X.H.; Ren, C.X.; Wei, B.; Wu, Y.Y.; Wu, Q.H.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 548. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving PacBio Long Read Accuracy by Short Read Alignment. PLoS ONE 2012, 7, e46679. [Google Scholar] [CrossRef]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, W.; Chen, Q.X. Comparative study on three different methods for the determination of total phenolics in Chinese olive. Food Sci. 2014, 35, 204–207. (In Chinese) [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Huang, M.J.; Wen, Z.F.; Chi, Y.B.; Peng, Z.F.; Chen, Q.X. Molecular Cloning and Expression Analysis of Flavonoids 3′-hydroxylase (CaF3′H) in Canarium album. Mol. Plant Breed. 2017, 15, 839–847. (In Chinese) [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Leo, D.J. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Lesschaeve, I.; Noble, A.C. Polyphenols: Factors influencing their sensory properties and their effects on food and beverage preferences. Am. J. Clin. Nutr. 2005, 81, 330S–335S. [Google Scholar] [CrossRef]
- He, M.; Tian, H.; Luo, X.; Qi, X.; Chen, X. Molecular progress in research on fruit astringency. Molecules 2015, 20, 1434–1451. [Google Scholar] [CrossRef]
- Xu, Z.C.; Peters, R.J.; Weirather, J.; Luo, H.M.; Liao, B.S.; Zhang, X.; Zhu, Y.J.; Ji, A.J.; Zhang, B.; Hu, S.N.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Tahara, K.; Nishiguchi, M.; Funke, E.; Miyazawa, S.-I.; Miyama, T.; Milkowski, C. Dehydroquinate dehydratase/shikimate dehydrogenases involved in gallate biosynthesis of the aluminum-tolerant tree species Eucalyptus camaldulensis. Planta 2021, 253, 3. [Google Scholar] [CrossRef]
- Chang, Q.; Su, M.; Chen, Q.; Zeng, B.; Li, H.; Wang, W. Physicochemical properties and antioxidant capacity of Chinese olive (Canarium album L.) cultivars. J. Food Sci. 2017, 82, 1369–1377. [Google Scholar] [CrossRef]
- Torres-Leon, C.; Ventura-Sobrevilla, J.; Serna-Cock, L.; Ascacio-Valdes, J.A.; Contreras-Esquivel, J.; Aguilar, C.N. Pentagalloylglucose (PGG): A valuable phenolic compound with functional properties. J. Funct. Foods 2017, 37, 176–189. [Google Scholar] [CrossRef]
- Qin, G.H.; Xu, C.Y.; Ming, R.; Tang, H.B.; Guyot, R.; Kramer, E.M.; Hu, Y.D.; Yi, X.K.; Qi, Y.J.; Xu, X.Y.; et al. The pomegranate (Punica granatum L.) genome and the genomics of punicalagin biosynthesis. Plant J. 2017, 91, 1108–1128. [Google Scholar] [CrossRef]
- Yuan, Z.H.; Fang, Y.M.; Zhang, T.K.; Fei, Z.J.; Han, F.M.; Liu, C.Y.; Liu, M.; Xiao, W.; Zhang, W.J.; Wu, S.; et al. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol. J. 2018, 16, 1363–1374. [Google Scholar] [CrossRef]
- Grundhofer, P.; Niemetz, R.; Schilling, G.; Gross, G.G. Biosynthesis and subcellular distribution of hydrolyzable tannins. Phytochemistry 2001, 57, 915–927. [Google Scholar] [CrossRef]
- Niemetz, R.; Gross, G.G. Oxidation of pentagalloylglucose to the ellagitannin tellimagrandin II, by a phenol oxidase from Tellima grandiflora leaves. Phytochemistry 2003, 62, 301–306. [Google Scholar] [CrossRef]
- Cheng, X.; Muhammad, A.; Li, G.; Zhang, J.; Cheng, J.; Qiu, J.; Jiang, T.; Jin, Q.; Cai, Y.; Lin, Y. Family-1 UDP glycosyltransferases in pear (Pyrus bretschneideri): Molecular identification, phylogenomic characterization and expression profiling during stone cell formation. Mol. Biol. Rep. 2019, 46, 2153–2175. [Google Scholar] [CrossRef]
- Cui, Y.; Ma, J.; Liu, G.; Wang, N.; Pei, W.; Wu, M.; Li, X.; Zhang, J.; Yu, J. Genome-Wide Identification, Sequence Variation, and Expression of the Glycerol-3-Phosphate Acyltransferase (GPAT) Gene Family in Gossypium. Front. Genet. 2019, 10, 116. [Google Scholar] [CrossRef]
- Xu, X.; Zhou, Y.; Wang, B.; Ding, L.; Wang, Y.; Luo, L.; Zhang, Y.; Kong, W. Genome-wide identification and characterization of laccase gene family in Citrus sinensis. Gene 2019, 689, 114–123. [Google Scholar] [CrossRef]
- Vanholme, R.; De Meester, B.; Ralph, J.; Boerjan, W. Lignin biosynthesis and its integration into metabolism. Curr. Opin. Biotechnol. 2019, 56, 230–239. [Google Scholar] [CrossRef]
- Ni, J.; Zhao, Y.; Tao, R.; Yin, L.; Gao, L.; Strid, A.; Qian, M.; Li, J.; Li, Y.; Shen, J.; et al. Ethylene mediates the branching of the jasmonate-induced flavonoid biosynthesis pathway by suppressing anthocyanin biosynthesis in red Chinese pear fruits. Plant Biotechnol. J. 2020, 18, 1223–1240. [Google Scholar] [CrossRef]
- Zuk, M.; Dzialo, M.; Richter, D.; Dyminska, L.; Matula, J.; Kotecki, A.; Hanuza, J.; Szopa, J. Chalcone Synthase (CHS) Gene Suppression in Flax Leads to Changes in Wall Synthesis and Sensing Genes, Cell Wall Chemistry and Stem Morphology Parameters. Front. Plant Sci. 2016, 7, e0132628. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, J.; Li, S.-S.; Zhang, H.-J.; Feng, C.-Y.; Yin, D.-D.; Wu, R.-Y.; Wang, L.-S. Transcriptome sequencing and metabolite analysis for revealing the blue flower formation in waterlily. BMC Genom. 2016, 17, 897. [Google Scholar] [CrossRef]
- Mitsuda, N.; Ohme-Takagi, M. Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiol. 2009, 50, 1232–1248. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, Q.; Wu, Y.; Chai, X.; Liu, W.; Wang, Y.; Yang, Q.; Wang, Z.; Liu, Z. Full-length transcript sequencing and comparative transcriptomic analysis to evaluate the contribution of osmotic and ionic stress components towards salinity tolerance in the roots of cultivated alfalfa (Medicago sativa L.). BMC Plant Biol. 2019, 19, 32. [Google Scholar] [CrossRef]
- Deng, Y.; Zheng, H.; Yan, Z.C.; Liao, D.Y.; Li, C.L.; Zhou, J.Y.; Liao, H. Full-Length Transcriptome Survey and Expression Analysis of Cassia obtusifolia to Discover Putative Genes Related to Aurantio-Obtusin Biosynthesis, Seed Formation and Development, and Stress Response. Int. J. Mol. Sci. 2018, 19, 2476. [Google Scholar] [CrossRef]
- Sun, M.Y.; Li, J.Y.; Li, D.; Huang, F.J.; Wang, D.; Li, H.; Xing, Q.; Zhu, H.B.; Shi, L. Full-Length Transcriptome Sequencing and Modular Organization Analysis of the Naringin/Neoeriocitrin-Related Gene Expression Pattern in Drynaria roosii. Plant Cell Physiol. 2018, 59, 1398–1414. [Google Scholar] [CrossRef]
- Lai, R.L.; Shen, C.G.; Feng, X.; Gao, M.X.; Zhang, Y.Y.; Wei, X.X.; Chen, Y.T.; Cheng, C.Z.; Wu, R.J. Integrated Metabolomic and Transcriptomic Analysis Reveals Differential Flavonoid Accumulation and Its Underlying Mechanism in Fruits of Distinct Canarium album Cultivars. Foods 2022, 11, 2527. [Google Scholar] [CrossRef]
- Lai, R.L.; Guan, Q.X.; Shen, C.G.; Feng, X.; Zhang, Y.Y.; Chen, Y.T.; Cheng, C.Z.; Wu, R.J. Integrated SRNA-Seq and RNA-Seq Analysis Reveals the Regulatory Roles of miRNAs in the Low-Temperature Responses of Canarium album. Horticulturae 2022, 8, 667. [Google Scholar] [CrossRef]
- Lai, R.L.; Feng, X.; Chen, J.; Zhang, Y.Y.; Wei, X.X.; Chen, Y.T.; Cheng, C.Z.; Wu, R.J. De novo transcriptome assembly and comparative transcriptomic analysis provide molecular insights into low temperature stress response of Canarium album. Sci. Rep. 2021, 11, 10561. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.L.; Feng, X.; Chen, J.; Chen, Y.T.; Wu, R.J. The complete chloroplast genome characterization and phylogenetic analysis of Canarium album. Mitochondrial DNA B Resour. 2019, 4, 2948–2949. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Yang, C.; Chen, J.; Guo, Y.; Li, Y.; Niu, S.; Cao, K.; Chen, Z. Comprehensive identification of the full-length transcripts and alternative splicing related to the secondary metabolism pathways in the tea plant (Camellia sinensis). Sci. Rep. 2019, 9, 2709. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Wang, J.; Cai, J.R.; Zhao, J.Y.; Guo, Z.X.; Pan, T.F.; Yu, Y.; She, W.Q. Enzyme Activities in the Lignin Metabolism of Chinese Olive (Canarium album) with Different Flesh Characteristics. Horticulturae 2022, 8, 408. [Google Scholar] [CrossRef]
- Yang, C.Q.; Fang, X.; Wu, X.M.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Transcriptional regulation of plant secondary metabolism. J. Integr. Plant Biol. 2012, 54, 703–712. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, B.; Qin, L.; Rahman, K.; Zhang, L.; Han, T. Transcription Factor: A Powerful Tool to Regulate Biosynthesis of Active Ingredients in Salvia miltiorrhiza. Front. Plant Sci. 2021, 12, 622011. [Google Scholar] [CrossRef]
- Cao, Y.; Li, K.; Li, Y.; Zhao, X.; Wang, L. MYB Transcription Factors as Regulators of Secondary Metabolism in Plants. Biology 2020, 9, 61. [Google Scholar] [CrossRef]
- Zhou, W.; Shi, M.; Deng, C.; Lu, S.; Huang, F.; Wang, Y.; Kai, G. The methyl jasmonate-responsive transcription factor SmMYB1 promotes phenolic acid biosynthesis in Salvia miltiorrhiza. Hortic. Res. 2021, 8, 10. [Google Scholar] [CrossRef]
- Hao, X.; Pu, Z.; Cao, G.; You, D.; Zhou, Y.; Deng, C.; Shi, M.; Nile, S.H.; Wang, Y.; Zhou, W.; et al. Tanshinone and salvianolic acid biosynthesis are regulated by SmMYB98 in Salvia miltiorrhiza hairy roots. J. Adv. Res. 2020, 23, 1–12. [Google Scholar] [CrossRef]
- Arlotta, C.; Puglia, G.D.; Genovese, C.; Toscano, V.; Karlova, R.; Beekwilder, J.; De Vos, R.C.H.; Raccuia, S.A. MYB5-like and bHLH influence flavonoid composition in pomegranate. Plant Sci. 2020, 298, 110563. [Google Scholar] [CrossRef] [PubMed]
- An, J.P.; Zhang, X.W.; Bi, S.Q.; You, C.X.; Wang, X.F.; Hao, Y.J. The ERF transcription factor MdERF38 promotes drought stress-induced anthocyanin biosynthesis in apple. Plant J. 2020, 101, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Sun, M.; Yuan, T.; Wang, Y.; Shi, M.; Lu, S.; Tang, B.; Pan, J.; Wang, Y.; Kai, G. The AP2/ERF transcription factor SmERF1L1 regulates the biosynthesis of tanshinones and phenolic acids in Salvia miltiorrhiza. Food Chem. 2019, 274, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Wang, Y.; Shi, M.; Hao, X.; Zhao, W.; Wang, Y.; Ren, J.; Kai, G. Transcription Factor SmWRKY1 Positively Promotes the Biosynthesis of Tanshinones in Salvia miltiorrhiza. Front. Plant Sci. 2018, 9, 554. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total number | 135,439 |
| Total length (bp) | 364,052,044 |
| Maximum length (bp) | 11,994 |
| Minimum length (bp) | 56 |
| Average length (bp) | 2687.94 |
| N50 length (bp) | 3224 |
| GC content | 40.47% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Zhang, S.; Xie, Q.; Wang, W.; Lin, Z.; Wang, H.; Yuan, Y.; Chen, Q. De Novo Transcriptome Analysis by PacBio SMRT-Seq and Illumina RNA-Seq Provides New Insights into Polyphenol Biosynthesis in Chinese Olive Fruit. Horticulturae 2024, 10, 293. https://doi.org/10.3390/horticulturae10030293
Ye Q, Zhang S, Xie Q, Wang W, Lin Z, Wang H, Yuan Y, Chen Q. De Novo Transcriptome Analysis by PacBio SMRT-Seq and Illumina RNA-Seq Provides New Insights into Polyphenol Biosynthesis in Chinese Olive Fruit. Horticulturae. 2024; 10(3):293. https://doi.org/10.3390/horticulturae10030293
Chicago/Turabian StyleYe, Qinghua, Shiyan Zhang, Qian Xie, Wei Wang, Zhehui Lin, Huiquan Wang, Yafang Yuan, and Qingxi Chen. 2024. "De Novo Transcriptome Analysis by PacBio SMRT-Seq and Illumina RNA-Seq Provides New Insights into Polyphenol Biosynthesis in Chinese Olive Fruit" Horticulturae 10, no. 3: 293. https://doi.org/10.3390/horticulturae10030293