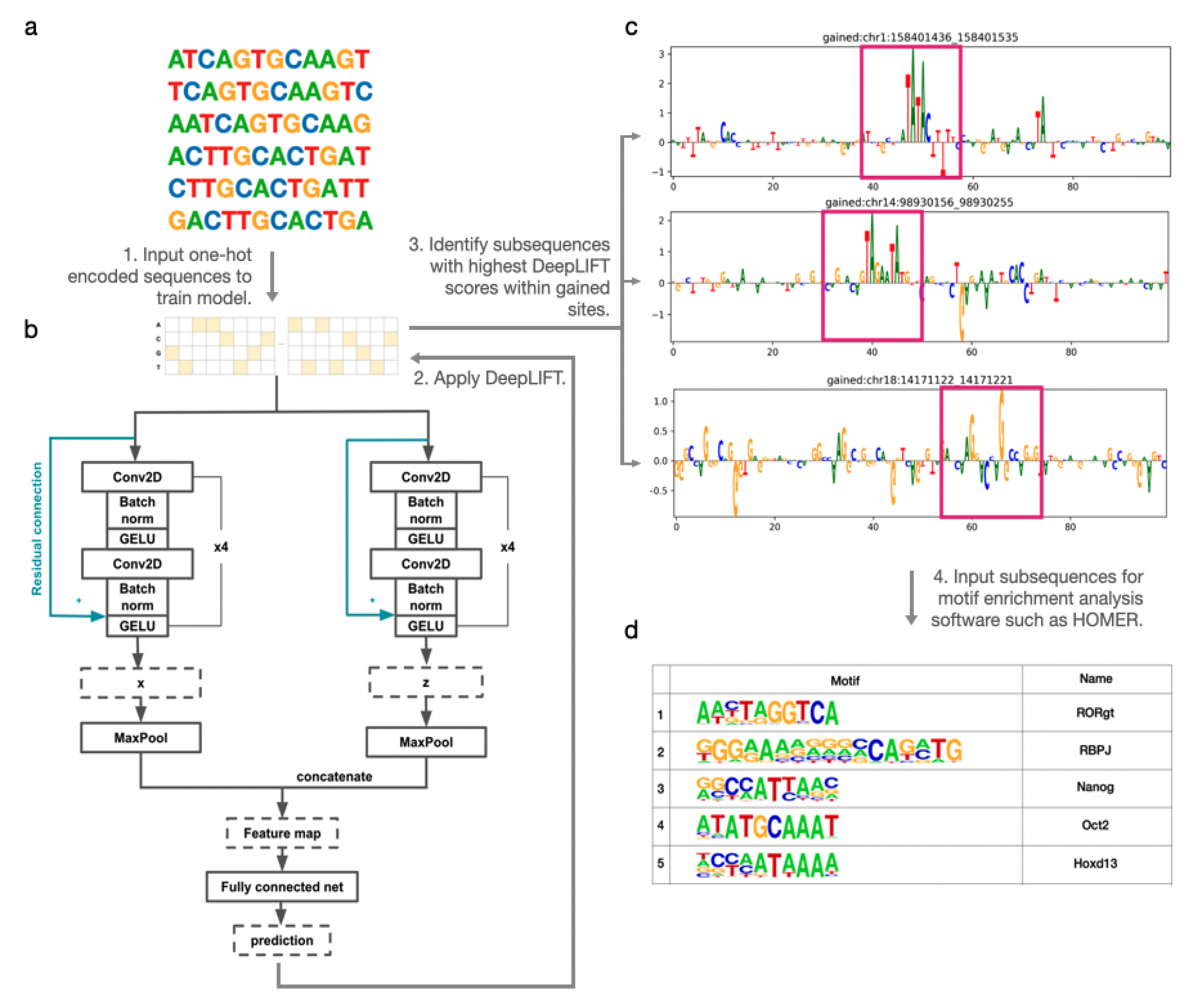
3.1. Performance Evaluation through Simulation
To evaluate the validity of our method and DARDN’s classification ability in detecting crucial features in DNA sequences, we conducted a preliminary test using 25,762 real DNA sequences of a length of 10,000 base pairs (bps) without CTCF binding sites. We replaced any occurrences of the RBPJ consensus sequence (CCTGGGAA) with a random 8 bp combination. Then, we inserted the RBPJ consensus sequence at ten random locations in each of the 33% of the sites ( sites). We trained DARDN on the classification of RBPJ-inserted sequences and achieved 100% accuracy on hold-out data. Subsequently, we used DeepLIFT to assign contribution scores to each base pairs (bps) in the sequence. This approach allowed us evaluation of the performance of our pipeline in accurately identifying inserted RBPJ sequences and assigning relevant scores to each bp.
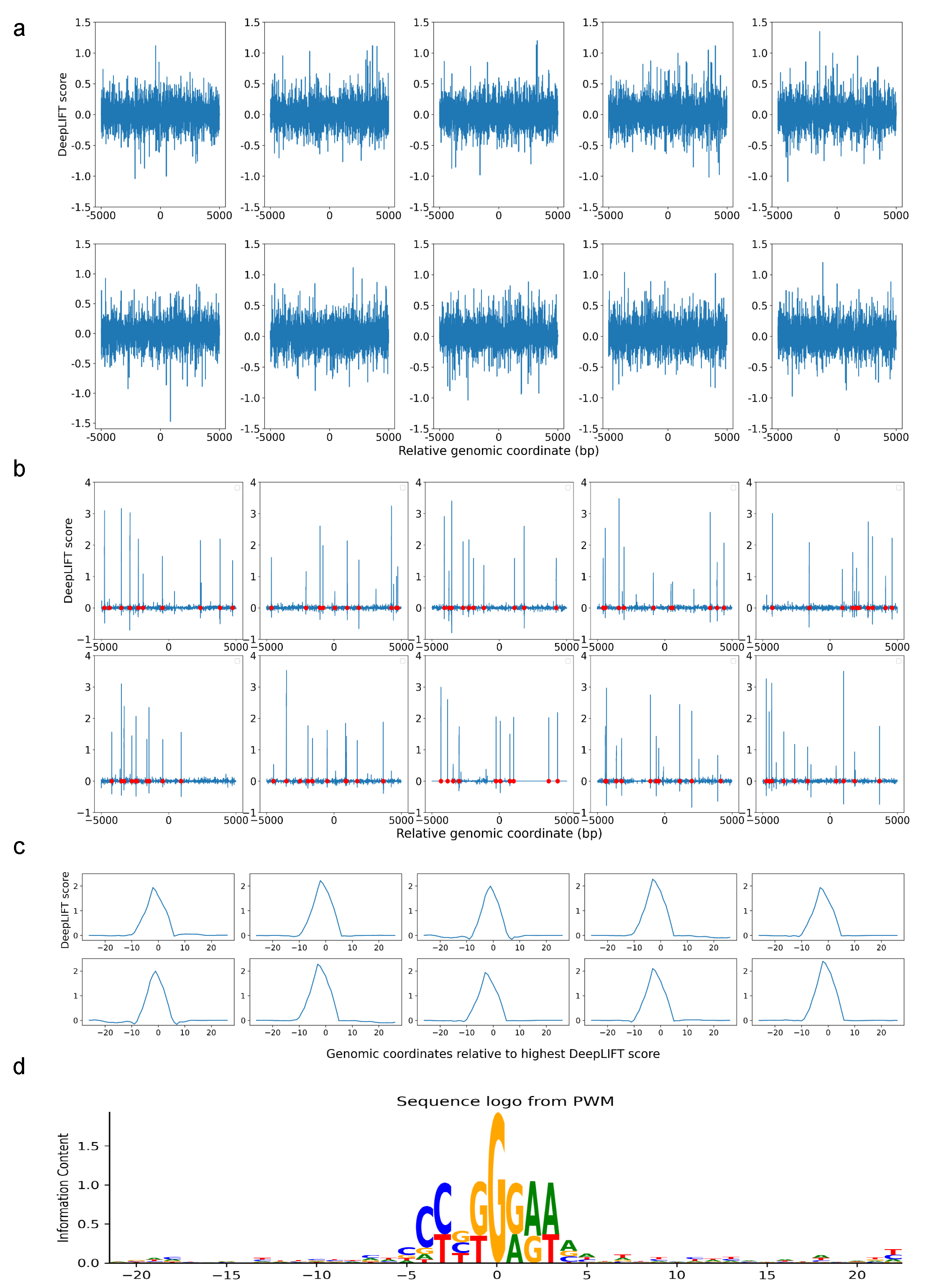
We first demonstrated the assignment of DeepLIFT scores to sequences that were trained using DARDN without any RBPJ sites inserted. Since these sites were randomly selected from actual DNA sequences without any specific criteria, the scores do not exhibit any discernible pattern (
Figure 2a). On the other hand, after DARDN was trained to classify RBPJ-inserted sequences, it was evident that the DeepLIFT scores at those particular locations with RBPJ insertions (indicated with red dots) were significantly greater than those at other locations (
Figure 2b). This provides a clear evidence that DARDN and our score assignment work as expected.
Once the DeepLIFT score at each individual index was computed, we used a sliding window to compute the average scores of subsequences in the input sequences. Specifically, the average score at index was computed using formula , where w indicates the window size. Because the RBPJ consensus sequence we inserted contains 8 bps (CCTGGGAA), for our simulation, we used . In our primary experiments, we tested various values of w to optimize the window size for motif enrichment identification.
Figure 2c shows the peak with the highest average DeepLIFT score for each plot in
Figure 2b, after re-indexing to center at zero. Lastly, in
Figure 2d, we illustrate the sequence logo generated by computing the Position Weight Matrix (PWM) for the sequences that center at the highest peak at each RBPJ-inserted site. Evidently, the inserted sequence of CCTGGGAA is displayed with the highest frequency in the center, which further validates our pipeline.
3.2. Robustness Evaluation
To comprehensively evaluate the robustness of DARDN, we subjected it to four distinct test conditions and observed the enrichment of RBPJ, which we noted in our previous research as the most enriched motif for T-ALL [
4]. The test conditions we considered were (1) modifying subsequence lengths for HOMER input: this scenario involves examining how changes in subsequence lengths influence motif rankings. This is equivalent to the window size with which we compute the running average DeepLIFT scores; (2) altering input sequence lengths: we explore how motif enrichment changes with input sequences of various lengths, specifically 5000, 10,000, and 20,000 base pairs (bps); (3) sampling background control sequences from constitutive CTCF sites: this entails studying the effect of sampling constitutive sites on motif rankings; (4) sampling foreground sequences from cancer-specific CTCF sites: we investigate the impact on motif rankings when gained sites are sampled. In our experiments, we selected 150 most statistically significant T-ALL-specific CTCF gained sites and 22,097 constitutive CTCF sites, which were subsequently centered within the sequences.
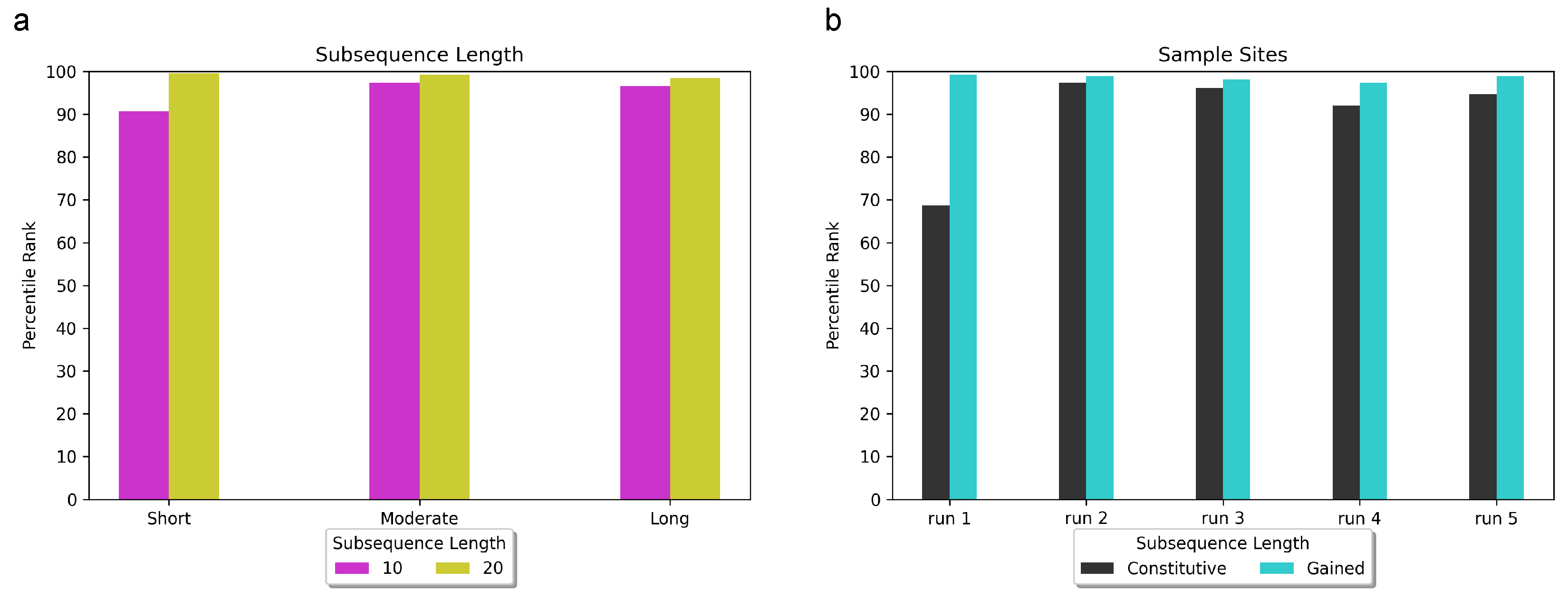
In our investigation, we explored subsequence lengths ranging from 10 to 20 base pairs (bps) and discovered that adopting a subsequence length of 20 bps consistently yielded superior rankings for RBPJ, irrespective of input sequence length (5 kbps, 10 kbps, or 20 kbps). In
Figure 3a, we present the percentile rank of RBPJ across various combinations of input sequence length and subsequence length. The x-axis represents the sequence lengths of 5 kbps, 10 kbps, and 20 kbps, denoted as “short”, “moderate”, and “long”, respectively.
Using 10 bp subsequences, RBPJ achieved the 91st percentile (3 out of 32 enriched motifs) for the short input sequence length, the 97.3th percentile (7 out of 264 enriched motifs) for the moderate input sequence length, and the 96.4th percentile (9 out of 264 enriched motifs) for the long input sequence length. On the other hand, when utilizing 20 bp subsequences, RBPJ achieved the 99.7th percentile (1st out of 264 enriched motifs) for the short input sequence length, 99.2th percentile (2nd rank among 264 enriched motifs) for the moderate input sequence length, and 98.5th percentile (4th rank among 264 enriched motifs) for long input sequence length. Regarding classification accuracy, DARDN demonstrated a Matthews correlation coefficient (MCC) of 0.91 for short input sequence length, as well as 0.87 for both moderate and long input sequence lengths.
To further evaluate the robustness of DARDN, we conducted five samplings of the background constitutive sites and five separate samplings of T-ALL-specific gained sites. In each trial, we randomly selected 15,000 out of 22,097 (approximately 68%) background constitutive sites and 72 out of 150 T-ALL-specific gained sites. The performance of DARDN was individually evaluated on each set of sampled sites, and the respective results are presented in
Figure 3b.
During one of the tests using sampled background constitutive sites (Run 1 in
Figure 3b), we observed a decline in the average rank of RBPJ compared to our previous trials that involved the complete set of 22,097 constitutive sites. RBPJ achieved the following percentiles and rankings in the five trials: 69 percentile (10th out of 32 enriched motifs), 97.3 percentile (7th out of 264 enriched motifs), 96.2 percentile (10th out of 264 enriched motifs), 92.1 percentile (21st out of 264 enriched motifs), and 94.7 percentile (14th out of 264 enriched motifs). The corresponding MCC values for classification were 0.88, 0.92, 0.81, 0.80, and 0.84, respectively. This outcome was expected as reducing the number of background constitutive sites not only diminishes the pool of negative samples, but also can weaken the robustness of DeepLIFT reference values.
For the first sampling of foreground-specific sites, we specifically sampled 72 most significant T-ALL-specific CTCF sites, measured by the specificity and the enrichment of the occurrences. Samplings 2 through 5 involved random samplings of 72 sites from the top 150 gained sites. The MCC scores for classification and the rankings of RBPJ for these trials were notably higher than those observed in the classification involving sampled constitutive sites: 99.2nd percentile (2nd out of 264 enriched motifs), 98.9th percentile (3rd out of 264 enriched motifs), 98.1st percentile (5th out of 264 enriched motifs), 96.4th percentile (7th out of 264 enriched motifs), and 98.1st percentile (3rd out of 264 enriched motifs). Both the classification accuracy and the ranking of RBPJ reached the most significant values among the five sampling experiments of the gained sites.
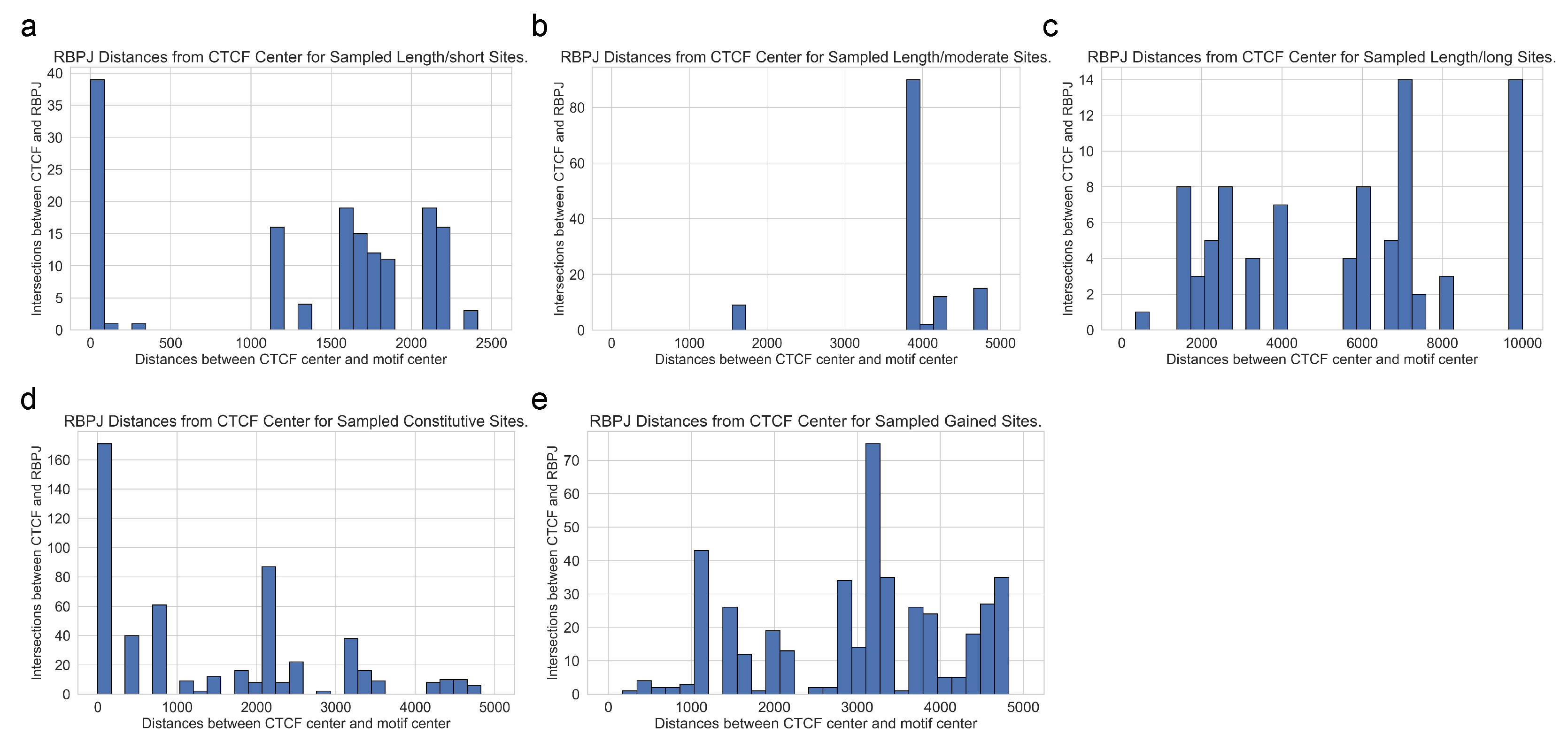
In
Figure 4, we present the distributions of distances between each specific CTCF site and enriched RBPJ site under the five test criteria.
Figure 4a–c showcase the distance variations for different input sequence lengths of 5 k, 10 k, and 20 kbps, respectively.
Figure 4d,e visualize the distances obtained by independently sampling constitutive and gained sites five times. For any input sequence length, the identified RBPJ sites may occur at any distance from foreground-specific CTCF sites, suggesting that long-range interactions exist between cooperating transcription factors and specific CTCF.
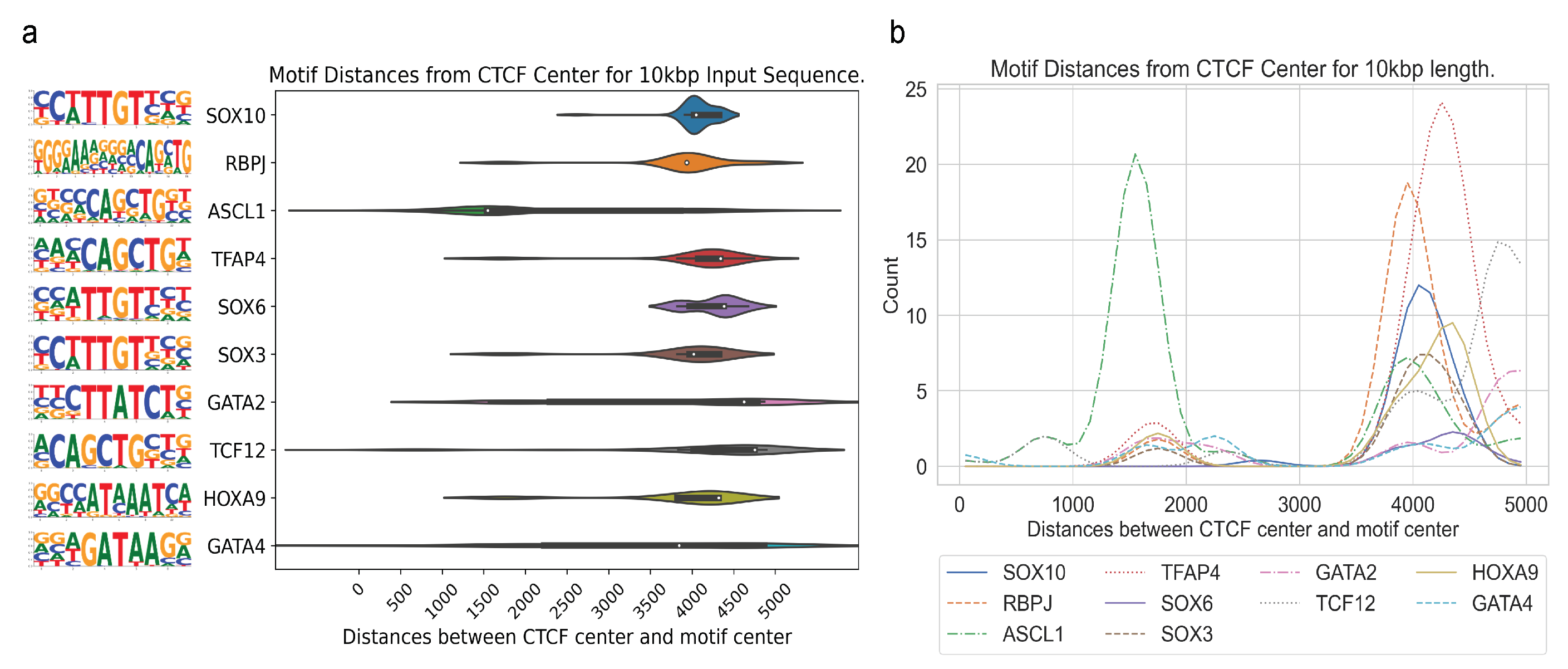
In
Figure 5, we show the CTCF center to motif center distance distributions for the most enriched motifs for T-ALL, including RBPJ. As shown in
Figure 5a, individual motif’s distances vary widely, while the median remains in the range from 3500 to 5000 bps away from the CTCF center. In
Figure 5b, the distance distributions for the motifs in
Figure 5a are plotted using 1D Gaussian smoothing. We do not observe a trend of close genomic distance between the specific CTCF binding and identified motif sites for transcription factor binding, further indicating that long-range interactions can occur at a long distance through DNA looping.
Although we also have Hi-C data pertaining to T-ALL, its resolution is inadequate for deriving meaningful insights for this project. This is because the existing resolution of Hi-C data is 10 kb, and we only search for motifs within 10 kb.
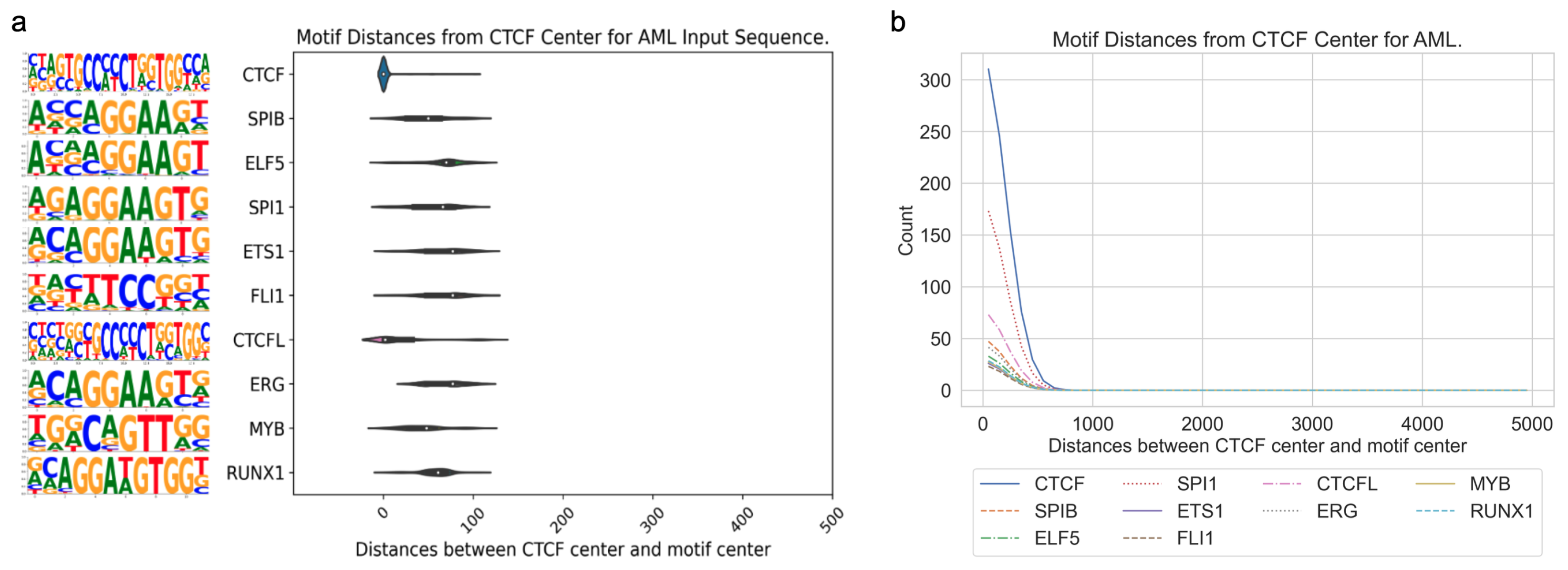
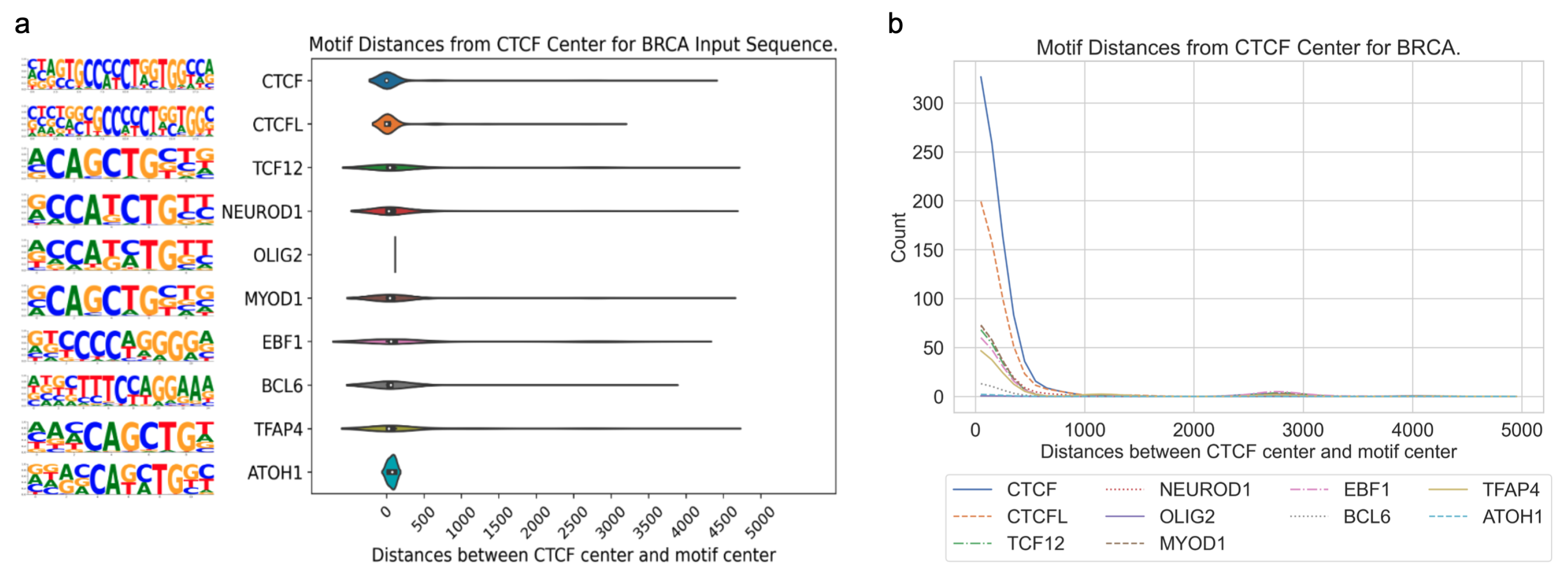
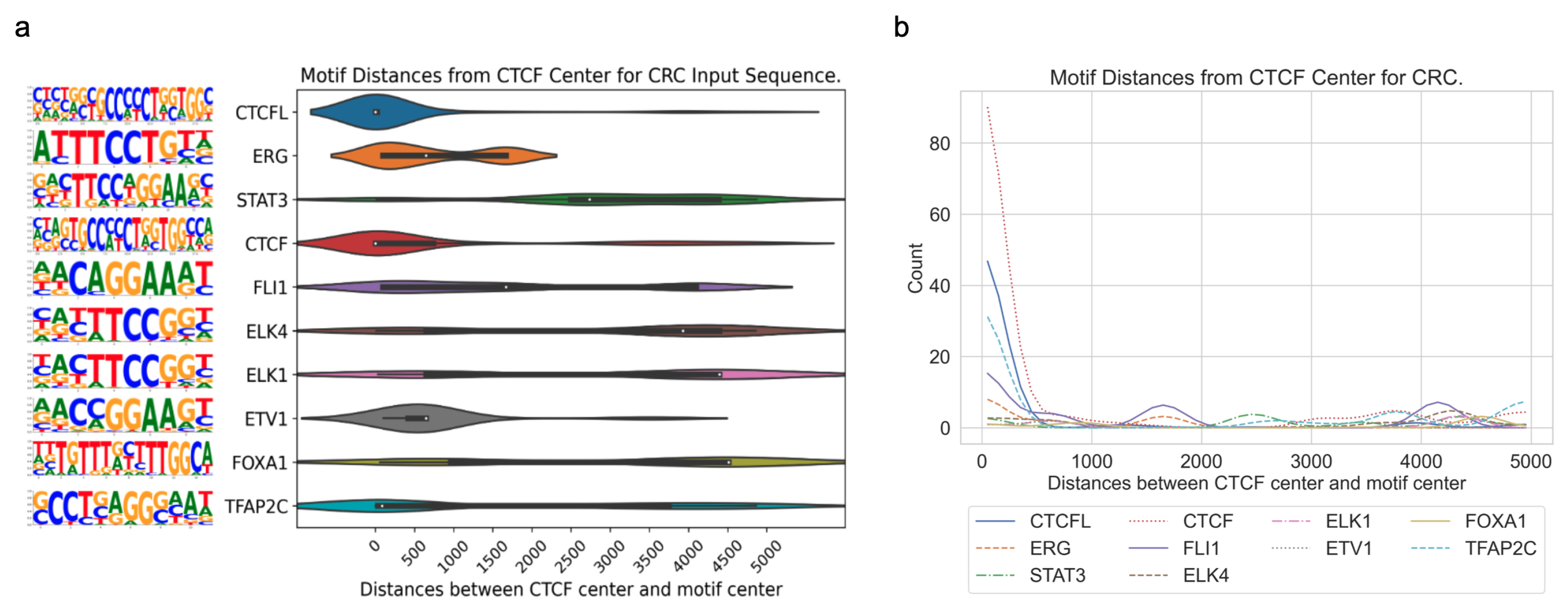
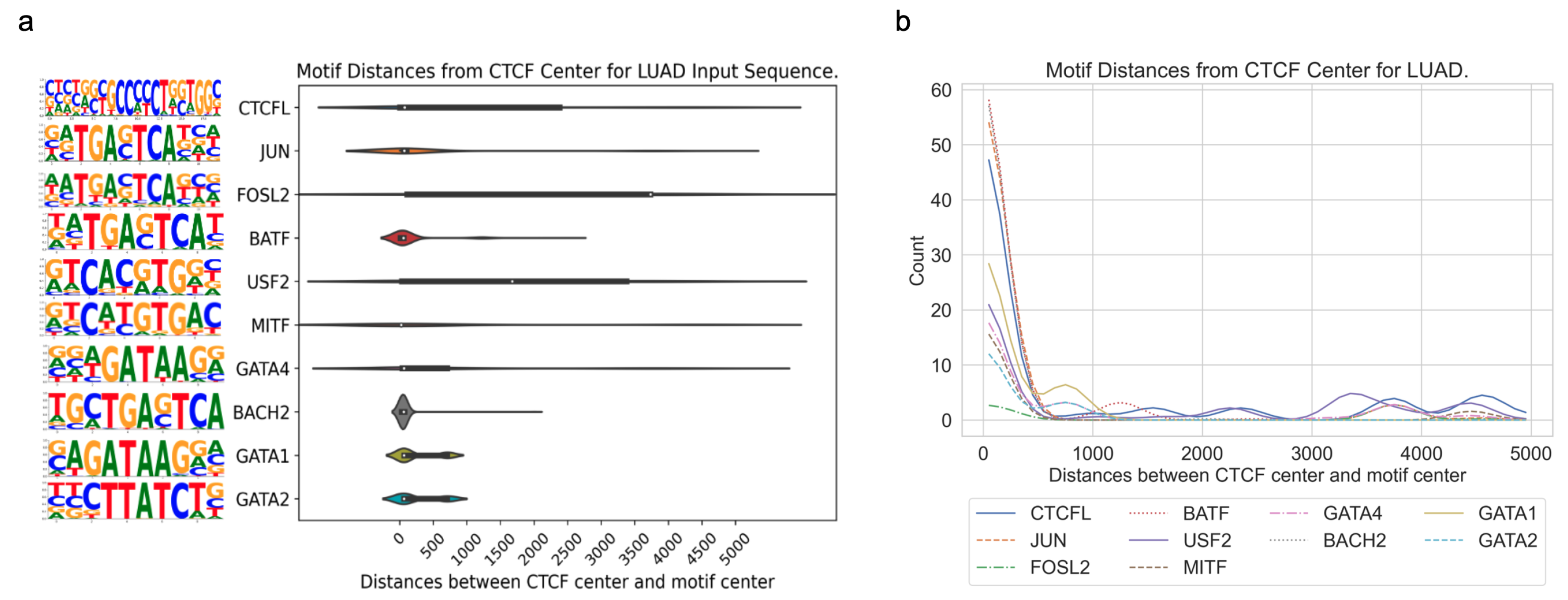
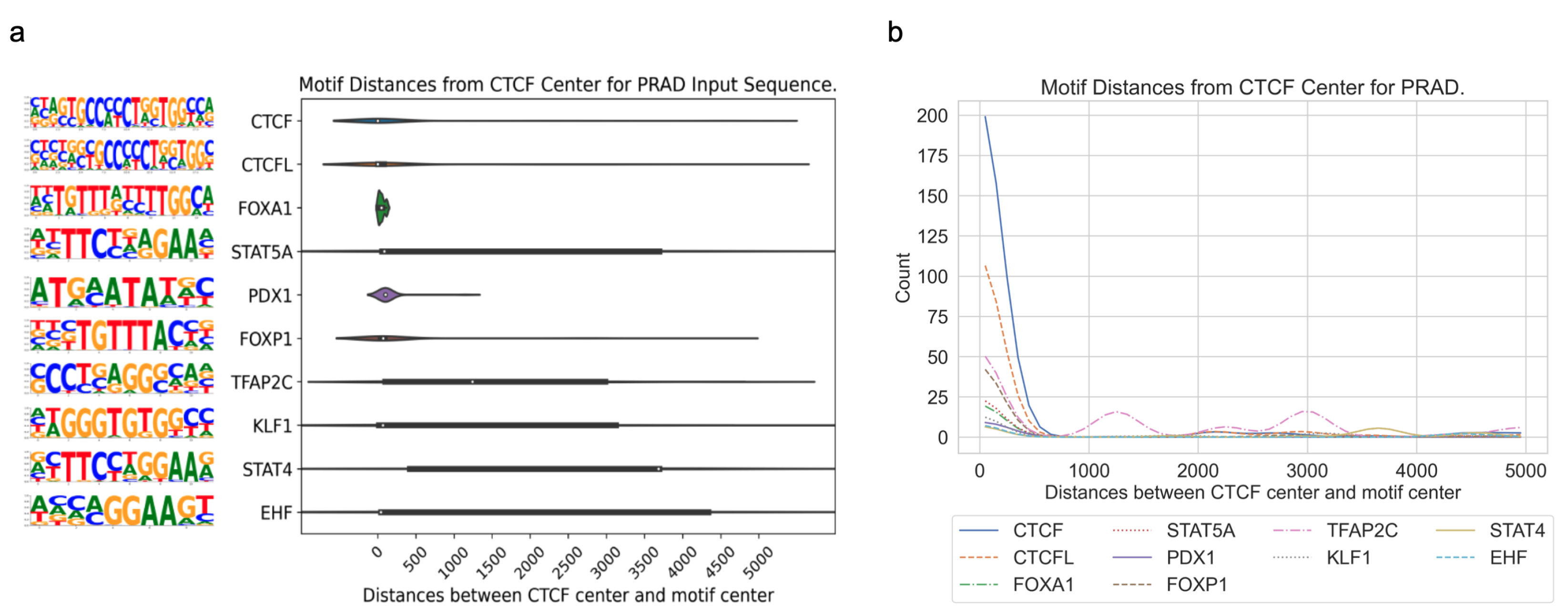
3.3. Application of DARDN to Diverse Cancer Types
To evaluate the adaptability of the DARDN sequence feature identification method, we applied it on five other cancer types where cancer-specific CTCF sites were identified in our previous work [
4], acute myeloid leukemia (AML), breast invasive carcinoma (BRCA), colorectal cancer (CRC), lung adenocarcinoma (LUAD), and prostate adenocarcinoma (PRAD), using the moderate input sequence length of 10 kbps.
In all five cancer types, the motifs for CTCF or CTCFL (a.k.a. BORIS, a paralog of CTCF) are highly enriched near the CTCF center. As both the foreground and background sequences are centered at specific or constitutive CTCF binding sites, respectively, the enrichment of the CTCF motif indicates additional CTCF occupancy near these specific sites. This is consistent with the fact that CTCF binding exhibits a clustered pattern in the genome to maintain the higher-order chromatin structure [
23].
Overall, this pattern of enrichment and distribution of different sequence motifs surrounding cancer-specific CTCF sites suggests that the regulatory mechanisms governing gene expression are specific to each cancer type and potentially involved in the specific CTCF binding events to facilitate enhancer–promoter interactions for oncogenic transcription factors to regulate their target genes.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









