
Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations


Abstract
:1. Introduction
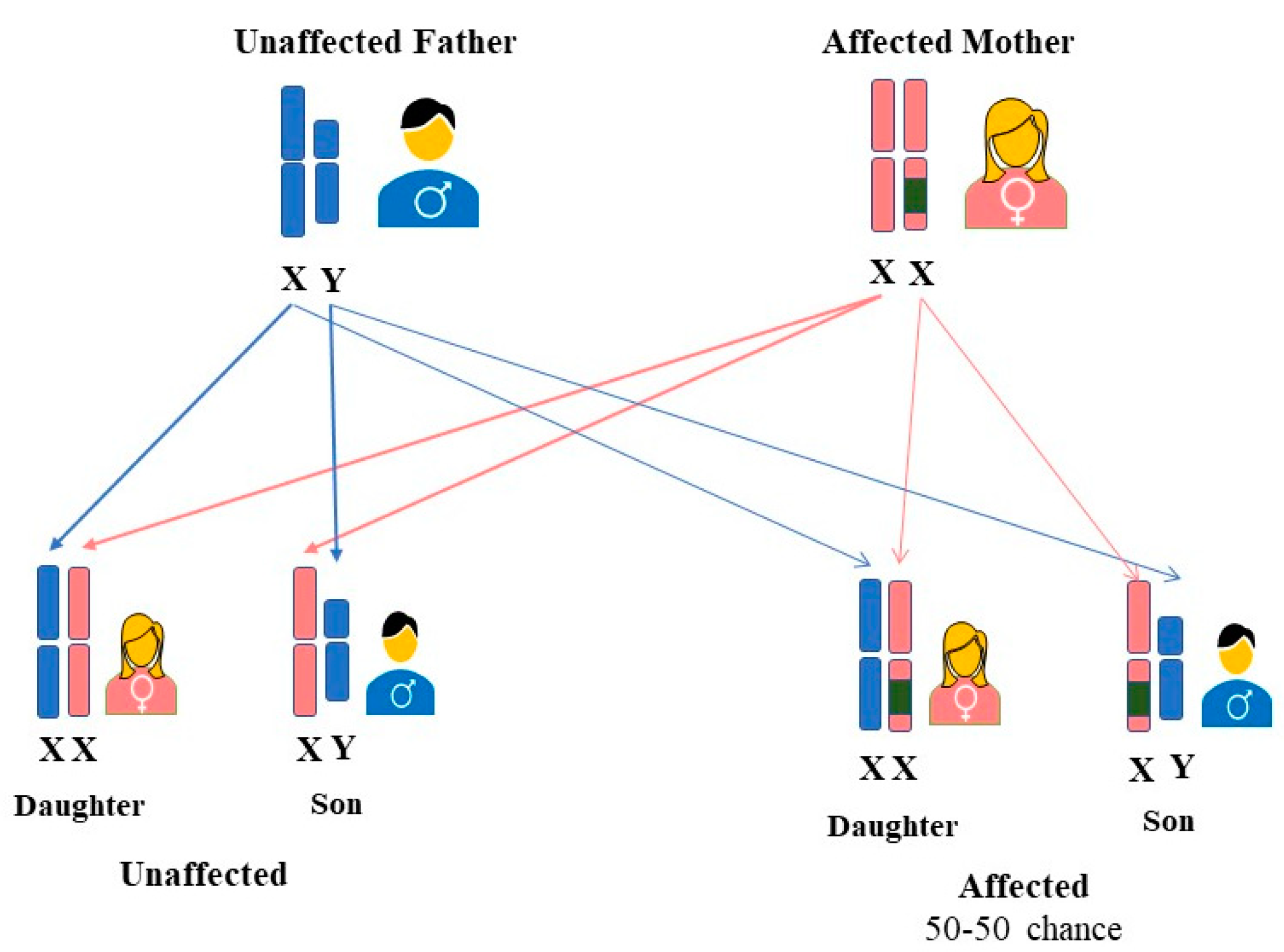
2. FD Inheritance in Females
3. LysoGb3 as a Predictor Biomarker for FD Severity
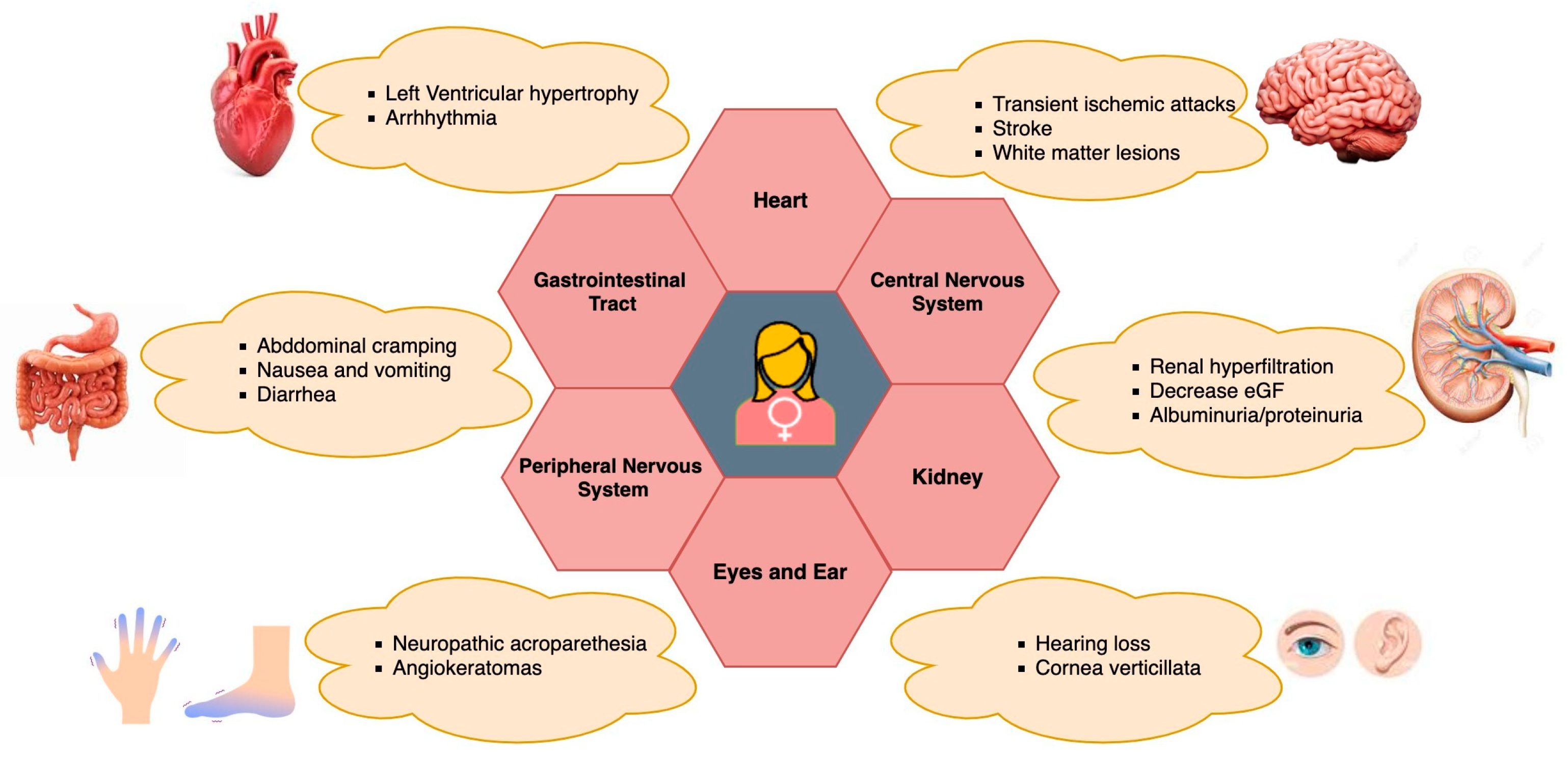
4. FD Symptoms in Women
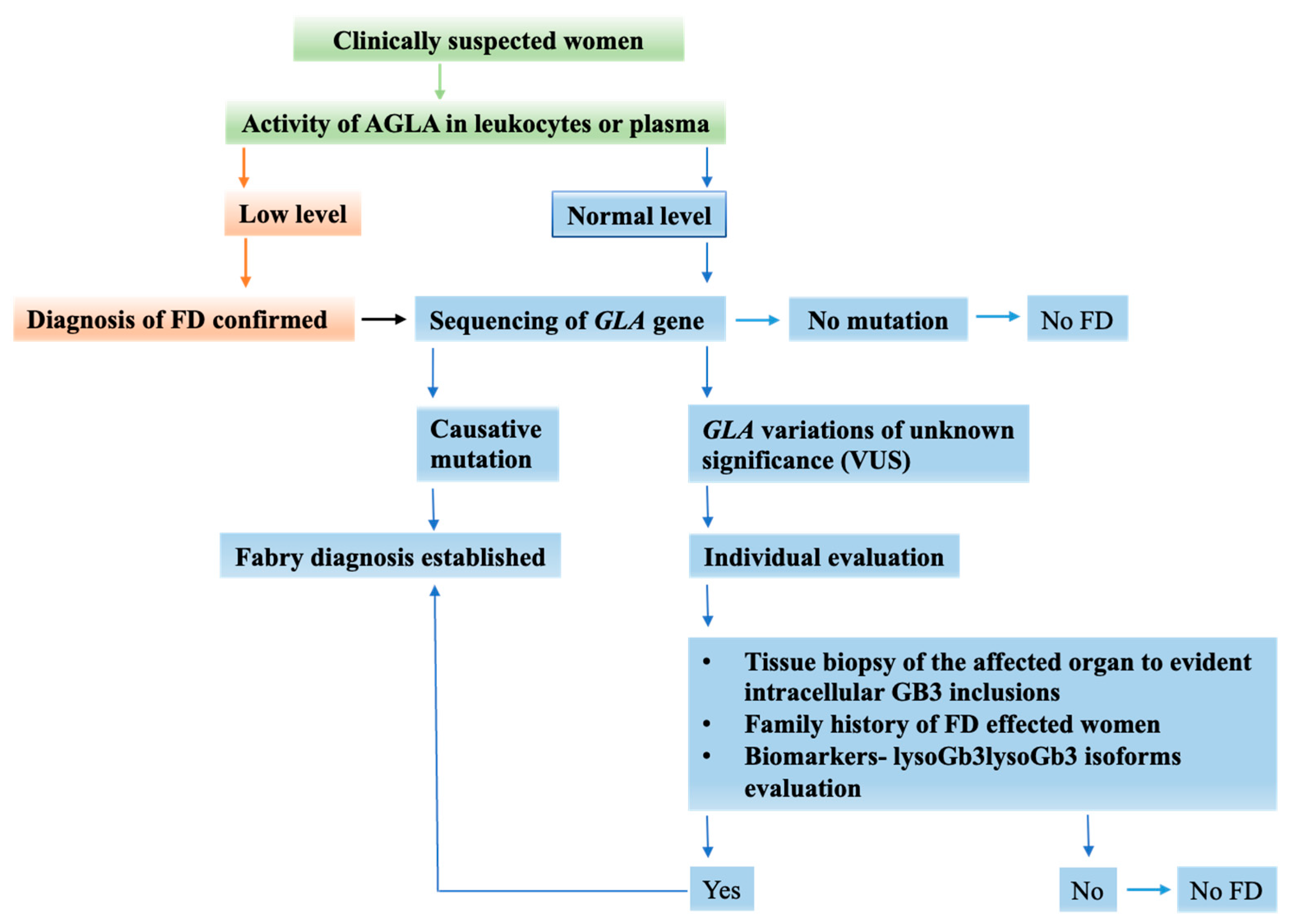
5. FD Diagnosis in Women Is a Challenging Process
6. Disease Management
7. Quality of Life and Psychological Aspects
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Ren, X.; Zhang, Y.; Ding, L.; Huo, M.; Li, Q. Fabry Disease: Mechanism and Therapeutics Strategies. Front. Pharmacol. 2022, 13, 1025740. [Google Scholar] [CrossRef] [PubMed]
- Stamerra, C.A.; Del Pinto, R.; di Giosia, P.; Ferri, C.; Sahebkar, A. Anderson–Fabry Disease: From Endothelial Dysfunction to Emerging Therapies. Adv. Pharmacol. Pharm. Sci. 2021, 2021, 5548445. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-Chromosome Inactivation in Female Patients with Fabry Disease. Clin. Genet. 2015, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Viggiano, E.; Politano, L. X Chromosome Inactivation in Carriers of Fabry Disease: Review and Meta-Analysis. Int. J. Mol. Sci. 2021, 22, 7663. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W. A case of “angeio-keratoma”. Br. J. Dermatol. 1898, 10, 113–117. [Google Scholar] [CrossRef]
- Fabry, J. losa haemorrhagica Hebrae. Arch. Für Dermatol. Und Syph. 1898, 43, 187. [Google Scholar] [CrossRef]
- Desnick, R.J.; Ioannou, Y.A. α-Galactosidase a Deficiency: Fabry Disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Whybra, C.; Kampmann, C.; Willers, I.; Davies, J.; Winchester, B.; Kriegsmann, J.; Brühl, K.; Gal, A.; Bunge, S.; Beck, M. Anderson–Fabry disease: Clinical manifestations of disease in female heterozygotes. J. Inherit. Metab. Dis. 2001, 24, 715–724. [Google Scholar] [CrossRef] [PubMed]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry Disease: Clinical Manifestations and Impact of Disease in a Cohort of 60 Obligate Carrier Females. J. Med. Genet. 2001, 38, 769–775. [Google Scholar] [CrossRef]
- Fuller, M.; Meikle, P.J.; Hopwood, J.J. Epidemiology of Lysosomal Storage Diseases: An Overview. In FabryDisease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 190353903X. [Google Scholar]
- Wang, R.Y.; Lelis, A.; Mirocha, J.; Wilcox, W.R. Heterozygous Fabry Women Are Not Just Carriers, but Have a Significant Burden of Disease and Impaired Quality of Life. Genet. Med. 2007, 9, 34–45. [Google Scholar] [CrossRef]
- Abensur, H.; dos Reis, M.A. Renal Involvement in Fabry Disease. J. Bras. De Nefrol. 2016, 38, 245–254. [Google Scholar] [CrossRef]
- Mehta, A.; Beck, M.; Eyskens, F.; Feliciani, C.; Kantola, I.; Ramaswami, U.; Rolfs, A.; Rivera, A.; Waldek, S.; Germain, D.P. Fabry Disease: A Review of Current Management Strategies. QJM 2010, 103, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Jain, G.; Warnock, D.G. Blood Pressure, Proteinuria and Nephropathy in Fabry Disease. Nephron Clin. Pract. 2011, 118, c43–c48. [Google Scholar] [CrossRef] [PubMed]
- Torra, R. Renal manifestations in Fabry disease and therapeutic options. Kidney Int. 2008, 74, S29–S32. [Google Scholar] [CrossRef] [PubMed]
- Barba-Romero, M.-Ã.; Deegan, P.; Giugliani, R.; Hughes, D. Does Geographical Location Influence the Phenotype of Fabry Disease in Women in Europe? Clin. Genet. 2010, 77, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Schiffmann, R.; Sabnis, S.G.; Murray, G.J.; Quirk, J.M.; Altarescu, G.; Goldfarb, L.; Brady, R.O.; Balow, J.E.; Austin, H.A.; et al. Natural History of Fabry Renal Disease. Medicine 2002, 81, 122–138. [Google Scholar] [CrossRef]
- Tsakiris, D.; Simpson, H.K.L.; Jones, E.H.P.; Briggs, J.D.; Elinder, C.-G.; Mendel, S.; Piccoli, G.; dos Santos, J.P.; Tognoni, G.; Vanrenterghem, Y.; et al. Rare Diseases in Renal Replacement Therapy in the ERA-EDTA Registry. Nephrol. Dial. Transplant. 1996, 11 (Suppl. S7), 4–20. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Wolf, M.; West, M.L.; Tonelli, M.; Ruthazer, R.; Pastores, G.M.; Obrador, G.T. Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002, 61, 249–255. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, M.J.; Rombach, S.M.; Dekker, N.; Poorthuis, B.J.; Linthorst, G.E.; Zwinderman, A.H.; Breunig, F.; Wanner, C.; Aerts, J.M.; Hollak, C.E. Reduction of Elevated Plasma Globotriaosylsphingosine in Patients with Classic Fabry Disease Following Enzyme Replacement Therapy. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 70–76. [Google Scholar] [CrossRef]
- von der Lippe, C.; Frich, J.C.; Harris, A.; Solbrække, K.N. Experiences of Being Heterozygous for Fabry Disease: A Qualitative Study. J. Genet. 2016, 25, 1085–1092. [Google Scholar] [CrossRef]
- Juchniewicz, P.; Kloska, A.; Tylki-Szymańska, A.; Jakóbkiewicz-Banecka, J.; Węgrzyn, G.; Moskot, M.; Gabig-Cimińska, M.; Piotrowska, E. Female Fabry Disease Patients and X-Chromosome Inactivation. Gene 2018, 641, 259–264. [Google Scholar] [CrossRef]
- Gibas, A.L.; Klatt, R.; Johnson, J.; Joe, T.R.C.; Katz, J. A Survey of the Pain Experienced by Males and Females with Fabry Disease. Pain Res. Manag. 2006, 11, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Dobyns, W.B.; Filauro, A.; Tomson, B.N.; Chan, A.S.; Ho, A.W.; Ting, N.T.; Oosterwijk, J.C.; Ober, C. Inheritance of Most X-Linked Traits Is Not Dominant or Recessive, Just X-Linked. Am. J. Med. Genet. 2004, 129A, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Beck, M.; Sunder-Plassmann, G. (Eds.) Fabry Disease: Perspectives from 5 Years of FOS; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Yoshimitsu, M.; Higuchi, K.; Miyata, M.; Devine, S.; Mattman, A.; Sirrs, S.; Medin, J.A.; Tei, C.; Takenaka, T. Identification of Novel Mutations in the α-Galactosidase a Gene in Patients with Fabry Disease: Pitfalls of Mutation Analyses in Patients with Low α-Galactosidase a Activity. J. Cardiol. 2011, 57, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Germain, D.P.; Banikazemi, M.; Warnock, D.G.; Wanner, C.; Hopkin, R.J.; Bultas, J.; Lee, P.; Sims, K.; Brodie, S.E.; et al. Fabry Disease: Guidelines for the Evaluation and Management of Multi-Organ System Involvement. Genet. Med. 2006, 8, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia de Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry Disease Defined: Baseline Clinical Manifestations of 366 Patients in the Fabry Outcome Survey. Eur. J. Clin. Investig. 2004, 34, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.; Rodriguez Botta, G.; Rozenfeld, P. New Mutation in Fabry Disease: C.448delG, First Phenotypic Description. Mol. Genet. Metab. Rep. 2021, 27, 100708. [Google Scholar] [CrossRef]
- Altarescu, G.; Goldfarb, L.; Park, K.-Y.; Kaneski, C.; Jeffries, N.; Litvak, S.; Nagle, J.; Schiffmann, R. Identification of Fifteen Novel Mutations and Genotype-Phenotype Relationship in Fabry Disease. Clin. Genet. 2001, 60, 46–51. [Google Scholar] [CrossRef]
- Juchniewicz, P.; Piotrowska, E.; Kloska, A.; Podlacha, M.; Mantej, J.; Węgrzyn, G.; Tukaj, S.; Jakóbkiewicz-Banecka, J. Dosage Compensation in Females with X-Linked Metabolic Disorders. Int. J. Mol. Sci. 2021, 22, 4514. [Google Scholar] [CrossRef]
- Beck, M. Fabry Disease; Springer eBooks: Berlin/Heidelberg, Germany, 2022; pp. 821–830. [Google Scholar] [CrossRef]
- Morrone, A. Fabry Disease: Molecular Studies in Italian Patients and X Inactivation Analysis in Manifesting Carriers. J. Med. Genet. 2003, 40, e103. [Google Scholar] [CrossRef]
- Bouwman, M.; Rombach, S.; Linthorst, G.; Poorthuis, B.; Lekanne Deprez, R.; Aerts, J.; Wijburg, F. Early Cerebral Manifestations in a Young Female with Fabry Disease with Skewed X-Inactivation. Clin. Genet. 2011, 80, 500–502. [Google Scholar] [CrossRef]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-Inactivation and Clinical Involvement in Fabry Heterozygotes. Eleven Novel Mutations in the α-Galactosidase a Gene in the Czech and Slovak Population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.; Osterrieder, S.; Whybra, C.; Ries, M.; Gal, A.; Beck, M.; Roscher, A.; Muntau, A. Disease Manifestations and X Inactivation in Heterozygous Females with Fabry Disease. Acta Paediatr. 2006, 95, 30–38. [Google Scholar] [CrossRef]
- Elstein, D.; Schachamorov, E.; Beeri, R.; Altarescu, G. X-Inactivation in Fabry Disease. Gene 2012, 505, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nature Reviews. Genetics 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Di Risi, T.; Vinciguerra, R.; Cuomo, M.; Della Monica, R.; Riccio, E.; Cocozza, S.; Imbriaco, M.; Duro, G.; Pisani, A.; Chiariotti, L. DNA Methylation Impact on Fabry Disease. Clin. Epigenetics 2021, 13, 24. [Google Scholar] [CrossRef]
- Hossain, M.A.; Yanagisawa, H.; Miyajima, T.; Wu, C.; Takamura, A.; Akiyama, K.; Itagaki, R.; Eto, K.; Iwamoto, T.; Yokoi, T.; et al. The Severe Clinical Phenotype for a Heterozygous Fabry Female Patient Correlates to the Methylation of Non-Mutated Allele Associated with Chromosome 10q26 Deletion Syndrome. Mol. Genet. Metab. 2017, 120, 173–179. [Google Scholar] [CrossRef]
- Hossain, M.A.; Wu, C.; Yanagisawa, H.; Miyajima, T.; Akiyama, K.; Eto, Y. Future Clinical and Biochemical Predictions of Fabry Disease in Females by Methylation Studies of the GLA Gene. Mol. Genet. Metab. Rep. 2019, 20, 100497. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Simonetta, I.; Riolo, R.; Todaro, F.; Di Chiara, T.; Miceli, S.; Pinto, A. Pathogenesis and Molecular Mechanisms of Anderson–Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 10088. [Google Scholar] [CrossRef]
- Amodio, F.; Caiazza, M.; Monda, E.; Rubino, M.; Capodicasa, L.; Chiosi, F.; Simonelli, V.; Dongiglio, F.; Fimiani, F.; Pepe, N.; et al. An Overview of Molecular Mechanisms in Fabry Disease. Biomolecules 2022, 12, 1460. [Google Scholar] [CrossRef]
- Germain, D.P. Fabry Disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of Inflammatory Pathways to Fabry Disease Pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide Induces Oxidative Stress and Up-Regulates Cell Adhesion Molecule Expression in Fabry Disease Endothelial Cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Sanchez-Niño, M.D.; Politei, J.; Oliveira, J.-P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A Key Feature of Fabry Disease with Potential Therapeutic Implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.J.; Kanack, A.J.; Dahms, N.M. Progress in the Understanding and Treatment of Fabry Disease. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 2020, 1864, 129437. [Google Scholar] [CrossRef] [PubMed]
- Hůlková, H.; Ledvinová, J.; Poupĕtová, H.; Bultas, J.; Zeman, J.; Elleder, M. Pitevní diagnóza Fabryho nemoci u heterozygotky, vedoucí k rozpoznání nediagnostikované manifestní nemoci v rodinĕ [Postmortem diagnosis of Fabry disease in a female heterozygote leading to the detection of undiagnosed manifest disease in the family]. Cas Lek Cesk. 1999, 138, 660–664, Erratum in Cas Lek Cesk 2000, 139, 123. [Google Scholar]
- Nowak, A.; Mechtler, T.P.; Desnick, R.J.; Kasper, D.C. Plasma LysoGb3: A Useful Biomarker for the Diagnosis and Treatment of Fabry Disease Heterozygotes. Mol. Genet. Metab. 2017, 120, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry Disease Revisited: Management and Treatment Recommendations for Adult Patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Whitfield, P.D.; Calvin, J.; Hogg, S.; O’Driscoll, E.; Halsall, D.; Burling, K.; Maguire, G.; Wright, N.; Cox, T.M.; Meikle, P.J.; et al. Monitoring Enzyme Replacement Therapy in Fabry Disease—Role of Urine Globotriaosylceramide. J. Inherit. Metab. Dis. 2005, 28, 21–33. [Google Scholar] [CrossRef]
- Schiffmann, R.; Waldek, S.; Benigni, A.; Auray-Blais, C. Biomarkers of Fabry Disease Nephropathy. Clin. J. Am. Soc. Nephrol. 2009, 5, 360–364. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated Globotriaosylsphingosine Is a Hallmark of Fabry Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Rombach, S.M.; Dekker, N.; Bouwman, M.G.; Linthorst, G.E.; Zwinderman, A.H.; Wijburg, F.A.; Kuiper, S.; vd Bergh Weerman, M.A.; Groener, J.E.M.; Poorthuis, B.J.; et al. Plasma Globotriaosylsphingosine: Diagnostic Value and Relation to Clinical Manifestations of Fabry Disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Polo, G.; Burlina, A.P.; Kolamunnage, T.B.; Zampieri, M.; Dionisi-Vici, C.; Strisciuglio, P.; Zaninotto, M.; Plebani, M.; Burlina, A.B. Diagnosis of Sphingolipidoses: A New Simultaneous Measurement of Lysosphingolipids by LC-MS/MS. Clin. Chem. Lab. Med. (CCLM) 2017, 55, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Ntwari, A.; Clarke, J.T.R.; Warnock, D.G.; Oliveira, J.P.; Young, S.P.; Millington, D.S.; Bichet, D.G.; Sirrs, S.; West, M.L.; et al. How Well Does Urinary Lyso-Gb3 Function as a Biomarker in Fabry Disease? Clin. Chim. Acta 2010, 411, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? Int. J. Mol. Sci. 2018, 19, 3726. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Miyata, K.; Mikame, M.; Taguchi, A.; Guili, C.; Shimura, M.; Murayama, K.; Inoue, T.; Yamamoto, S.; Sugimura, K.; et al. Effectiveness of Plasma Lyso-Gb3 as a Biomarker for Selecting High-Risk Patients with Fabry Disease from Multispecialty Clinics for Genetic Analysis. Genet. Med. 2019, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Takata, T.; Tsubata, Y.; Tazawa, R.; Goto, K.; Tohyama, J.; Narita, I.; Yoshioka, H.; Ishii, S. Screening of Male Dialysis Patients for Fabry Disease by Plasma Globotriaosylsphingosine. Clin. J. Am. Soc. Nephrol. 2013, 8, 629–636. [Google Scholar] [CrossRef]
- Levstek, T.; Vujkovac, B.; Podkrajsek, K.T. Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes 2020, 11, 1091. [Google Scholar] [CrossRef]
- Togawa, T.; Kodama, T.; Suzuki, T.; Sugawara, K.; Tsukimura, T.; Ohashi, T.; Ishige, N.; Suzuki, K.; Kitagawa, T.; Sakuraba, H. Plasma Globotriaosylsphingosine as a Biomarker of Fabry Disease. Mol. Genet. Metab. 2010, 100, 257–261. [Google Scholar] [CrossRef]
- Smid, B.E.; van der Tol, L.; Biegstraaten, M.; Linthorst, G.E.; Hollak, C.E.M.; Poorthuis, B.J.H.M. Plasma Globotriaosylsphingosine in Relation to Phenotypes of Fabry Disease. J. Med. Genet. 2015, 52, 262–268. [Google Scholar] [CrossRef]
- Pettazzoni, M.; Froissart, R.; Pagan, C.; Vanier, M.T.; Ruet, S.; Latour, P.; Guffon, N.; Fouilhoux, A.; Germain, D.P.; Levade, T.; et al. LC-MS/MS Multiplex Analysis of Lysosphingolipids in Plasma and Amniotic Fluid: A Novel Tool for the Screening of Sphingolipidoses and Niemann-Pick Type c Disease. PLoS ONE 2017, 12, e0181700. [Google Scholar] [CrossRef]
- Simonetta, I.; Tuttolomondo, A.; Daidone, M.; Pinto, A. Biomarkers in Anderson–Fabry Disease. Int. J. Mol. Sci. 2020, 21, 8080. [Google Scholar] [CrossRef] [PubMed]
- Baydakova, G.; Ilyushkina, A.; Moiseev, S.; Bychkov, I.; Nikitina, N.V.; Buruleva, T.A.; Zakharova, E. α-Galactosidase A/LysoGb3 Ratio as a Potential Marker for Fabry Disease in Females. Clin. Chim. Acta 2020, 501, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Young, E.; Mills, K.; Morris, P.; Vellodi, A.; Lee, P.; Waldek, S.; Winchester, B. Is Globotriaosylceramide a Useful Biomarker in Fabry Disease? Acta Paediatr. 2007, 94, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients with Fabry Disease: A Critical Link to Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Togawa, T.; Tsukimura, T.; Kato, H. Plasma Lyso-Gb3: A Biomarker for Monitoring Fabry Patients during Enzyme Replacement Therapy. Clin. Exp. Nephrol. 2017, 22, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Nino, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine Actions on Human Glomerular Podocytes: Implications for Fabry Nephropathy. Nephrol. Dial. Transplant. 2010, 26, 1797–1802. [Google Scholar] [CrossRef]
- Niemann, M.; Rolfs, A.; Störk, S.; Bijnens, B.; Breunig, F.; Beer, M.; Ertl, G.; Wanner, C.; Weidemann, F. Gene Mutations versus Clinically Relevant Phenotypes. Circ. Cardiovasc. Genet. 2014, 7, 8–16. [Google Scholar] [CrossRef]
- Gold, H.; Mirzaian, M.; Dekker, N.; Ferraz, M.J.; Lugtenburg, J.; Codée, J.D.; van der Marel, G.A.; Overkleeft, H.S.; Linthorst, G.E.; Groener, J.E.; et al. Quantification of Globotriaosylsphingosine in Plasma and Urine of Fabry Patients by Stable Isotope Ultraperformance Liquid Chromatography–Tandem Mass Spectrometry. Clin. Chem. 2013, 59, 547–556. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Blais, C.; Ramaswami, U.; Boutin, M.; Germain, D.P.; Dyack, S.; Bodamer, O.A.; Pintos-Morell, G.; Joe, T.R.C.; Bichet, D.G.; et al. Urinary Biomarker Investigation in Children with Fabry Disease Using Tandem Mass Spectrometry. Clin. Chim. Acta 2015, 438, 195–204. [Google Scholar] [CrossRef]
- Heywood, W.E.; Doykov, I.; Spiewak, J.; Hallqvist, J.; Mills, K.; Nowak, A. Global Glycosphingolipid Analysis in Urine and Plasma of Female Fabry Disease Patients. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2726–2735. [Google Scholar] [CrossRef]
- Winchester, B.; Young, E. Biochemical and Genetic Diagnosis of Fabry Disease; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 190353903X. [Google Scholar]
- Desnick, R.J.; Allen, K.Y.; Desnick, S.J.; Raman, M.K.; Bernlohr, R.W.; Krivit, W. Fabry’s Disease: Enzymatic Diagnosis of Hemizygotes and Heterozygotes. α-Galactosidase Activities in Plasma, Serum, Urine, and Leukocytes. J. Lab. Clin. Med. 1973, 81, 157–171. [Google Scholar] [PubMed]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry Disease: Enzymatic Diagnosis in Dried Blood Spots on Filter Paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef]
- Vedder, A.C.; Linthorst, G.E.; van Breemen, M.J.; Groener, J.E.M.; Bemelman, F.J.; Strijland, A.; Mannens, M.M.A.M.; Aerts, J.M.F.G.; Hollak, C.E.M. The Dutch Fabry Cohort: Diversity of Clinical Manifestations and Gb3 Levels. J. Inherit. Metab. Dis. 2007, 30, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.; Zwiers, K.C.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.M.F.G.; Artola, M. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.T.; Spadaccino, F.; Catalano, V.; Zaza, G.; Stallone, G.; Fiocco, D.; Netti, G.S.; Ranieri, E. Metabolic Fingerprinting of Fabry Disease: Diagnostic and Prognostic Aspects. Metabolites 2022, 12, 703. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Hughes, D.A.; Winchester, B. Toward a Consensus in the Laboratory Diagnostics of Fabry Disease—Recommendations of a European Expert Group. J. Inherit. Metab. Dis. 2011, 34, 509–514. [Google Scholar] [CrossRef]
- Yazd, H.S.; Bazargani, S.F.; Vanbeek, C.A.; King-Morris, K.; Heldermon, C.; Segal, M.S.; Yost, R.; Clapp, W.L.; Garrett, T.J. LC-MS Lipidomics of Renal Biopsies for the Diagnosis of Fabry Disease. J. Mass Spectrom. Adv. Clin. Lab 2021, 22, 71–78. [Google Scholar] [CrossRef]
- Riccio, E.; Sabbatini, M.; Capuano, I.; Pisani, A. Early Biomarkers of Fabry Nephropathy: A Review of the Literature. Nephron 2019, 143, 274–281. [Google Scholar] [CrossRef]
- Beirão, I.; Cabrita, A.; Simas, R.; Silva, F.; Aguiar, P.; Laranjeira, F.; Gomes, A.M. Biomarkers and Imaging Findings of Anderson–Fabry Disease—What We Know Now. Diseases 2017, 5, 15. [Google Scholar] [CrossRef]
- Weidemann, F.; Niemann, M.; Störk, S.; Breunig, F.; Beer, M.; Sommer, C.; Herrmann, S.; Ertl, G.; Wanner, C. Long-Term Outcome of Enzyme-Replacement Therapy in Advanced F Abry Disease: Evidence for Disease Progression towards Serious Complications. J. Intern. Med. 2013, 274, 331–341. [Google Scholar] [CrossRef]
- Niemann, M.; Herrmann, S.; Hu, K.; Breunig, F.; Strotmann, J.; Beer, M.; Machann, W.; Voelker, W.; Ertl, G.; Wanner, C.; et al. Differences in Fabry Cardiomyopathy between Female and Male Patients. JACC Cardiovasc. Imaging 2011, 4, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Lu, D.; Hoepfner, J.; Santer, L.; Gupta, S.; Pfanne, A.; Thum, S.; Lenders, M.; Brand, E.; Nordbeck, P.; et al. Circulating MicroRNAs in Fabry Disease. Sci. Rep. 2019, 9, 15277. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, F.J.; Baig, S.; Rambhatla, S.B.; Vijapurapu, R.; Auray-Blais, C.; Boutin, M.; Steeds, R.; Wheeldon, N.; Dawson, C.; Geberhiwot, T. The Clinical Utility of Total Concentration of Urinary Globotriaosylsphingosine plus Its Analogues in the Diagnosis of Fabry Disease. Clin. Chim. Acta 2020, 500, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Svarstad, E.; Marti, H.P. The Changing Landscape of Fabry Disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Mitobe, S.; Togawa, T.; Tsukimura, T.; Kodama, T.; Tanaka, T.; Doi, K.; Noiri, E.; Akai, Y.; Saito, Y.; Yoshino, M.; et al. Mutant α-Galactosidase a with M296I Does Not Cause Elevation of the Plasma Globotriaosylsphingosine Level. Mol. Genet. Metab. 2012, 107, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Boutin, M.; Gagnon, R.; Dupont, F.O.; Lavoie, P.; Clarke, J.T.R. Urinary Globotriaosylsphingosine-Related Biomarkers for Fabry Disease Targeted by Metabolomics. Anal. Chem. 2012, 84, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Dupont, F.O.; Gagnon, R.; Boutin, M.; Auray-Blais, C. A Metabolomic Study Reveals Novel Plasma Lyso-Gb3 Analogs as Fabry Disease Biomarkers. Curr. Med. Chem. 2013, 20, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Manwaring, V.; Boutin, M.; Auray-Blais, C. A Metabolomic Study to Identify New Globotriaosylceramide-Related Biomarkers in the Plasma of Fabry Disease Patients. Anal. Chem. 2013, 85, 9039–9048. [Google Scholar] [CrossRef]
- Boutin, M.; Auray-Blais, C. Metabolomic Discovery of Novel Urinary Galabiosylceramide Analogs as Fabry Disease Biomarkers. J. Am. Soc. For Mass Spectrom. 2015, 26, 499–510. [Google Scholar] [CrossRef]
- Boutin, M.; Auray-Blais, C. Multiplex Tandem Mass Spectrometry Analysis of Novel Plasma Lyso-Gb3-Related Analogues in Fabry Disease. Anal. Chem. 2014, 86, 3476–3483. [Google Scholar] [CrossRef]
- Lavoie, P.; Boutin, M.; Auray-Blais, C. Multiplex Analysis of Novel Urinary Lyso-Gb3-Related Biomarkers for Fabry Disease by Tandem Mass Spectrometry. Anal. Chem. 2013, 85, 1743–1752. [Google Scholar] [CrossRef]
- Ferreira, S.; Auray-Blais, C.; Boutin, M.; Lavoie, P.; Nunes, P.L.; Martins, E.; Garman, S.C.; Oliveira, J.P. Variations in the GLA Gene Correlate with Globotriaosylceramide and Globotriaosylsphingosine Analog Levels in Urine and Plasma. Clin. Chim. Acta 2015, 447, 96–104. [Google Scholar] [CrossRef]
- Boutin, M.; Gagnon, R.; Lavoie, P.; Auray-Blais, C. LC–MS/MS Analysis of Plasma Lyso-Gb3 in Fabry Disease. Clin. Chim. Acta 2012, 414, 273–280. [Google Scholar] [CrossRef]
- Sueoka, H.; Ichihara, J.; Tsukimura, T.; Togawa, T.; Sakuraba, H. Nano-LC-MS/MS for Quantification of Lyso-Gb3 and Its Analogues Reveals a Useful Biomarker for Fabry Disease. PLoS ONE 2015, 10, e0127048. [Google Scholar] [CrossRef]
- Bertoldi, G.; Caputo, I.; Driussi, G.; Stefanelli, L.F.; Di Vico, V.; Carraro, G.; Nalesso, F.; Calò, L.A. Biochemical Mechanisms beyond Glycosphingolipid Accumulation in Fabry Disease: Might They Provide Additional Therapeutic Treatments? J. Clin. Med. 2023, 12, 2063. [Google Scholar] [CrossRef]
- Brady, R.O. Clinical Features of and Recent Advances in Therapy for Fabry Disease. JAMA 2000, 284, 2771. [Google Scholar] [CrossRef]
- Lai, S.; Perrotta, A.M.; Bagordo, D.; Mazzaferro, S.; Menè, P.; Errigo, F.; Tinti, F.; Rotondi, S.; Molfino, A.; Simeoni, M.; et al. Literature Review on the Cross-Link between Ocular and Renal Disease: Renin Angiotensin Aldosterone System Is a Main Actor. PubMed 2022, 26, 4774–4788. [Google Scholar] [CrossRef]
- Deegan, P.B. Natural History of Fabry Disease in Females in the Fabry Outcome Survey. J. Med. Genet. 2005, 43, 347–352. [Google Scholar] [CrossRef]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Females with Fabry Disease Frequently Have Major Organ Involvement: Lessons from the Fabry Registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar] [CrossRef]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Natural history of Fabry disease in affected males and obligate carrier females. J. Inherit. Metab. Dis. 2001, 24 (Suppl. S2), 13–14. [Google Scholar] [CrossRef]
- Ortiz, A.; Oliveira, J.P.; Waldek, S.; Warnock, D.G.; Cianciaruso, B.; Wanner, C. Nephropathy in Males and Females with Fabry Disease: Cross-Sectional Description of Patients before Treatment with Enzyme Replacement Therapy. Nephrol. Dial. Transplant. 2008, 23, 1600–1607. [Google Scholar] [CrossRef]
- Zar-Kessler, C.; Karaa, A.; Sims, K.B.; Clarke, V.; Kuo, B. Understanding the Gastrointestinal Manifestations of Fabry Disease: Promoting Prompt Diagnosis. Ther. Adv. Gastroenterol. 2016, 9, 626–634. [Google Scholar] [CrossRef]
- Simeoni, M.; Damiano, S.; Capolongo, G.; Trepiccione, F.; Zacchia, M.; Fuiano, G.; Capasso, G. Rare Renal Diseases Can Be Used as Tools to Investigate Common Kidney Disorders. Kidney Dis. 2017, 3, 43–49. [Google Scholar] [CrossRef]
- Coppola, A.; Lombari, P.; Mazzella, E.; Capolongo, G.; Simeoni, M.; Perna, A.F.; Ingrosso, D.; Borriello, M. Zebrafish as a Model of Cardiac Pathology and Toxicity: Spotlight on Uremic Toxins. Int. J. Mol. Sci. 2023, 24, 5656. [Google Scholar] [CrossRef]
- Holmes, A.; Laney, D. A Retrospective Survey Studying the Impact of Fabry Disease on Pregnancy. JIMD Rep. 2014, 21, 57–63. [Google Scholar] [CrossRef]
- Vedder, A.C.; Strijland, A.; Weerman, M.A.V.B.; Florquin, S.; Aerts, J.M.F.G.; Hollak, C.E.M. Manifestations of Fabry Disease in Placental Tissue. J. Inherit. Metab. Dis. 2006, 29, 106–111. [Google Scholar] [CrossRef]
- Bouwman, M.; Hollak, C.; Weerman, M.v.D.B.; Wijburg, F.; Linthorst, G. Analysis of Placental Tissue in Fabry Disease with and without Enzyme Replacement Therapy. Placenta 2010, 31, 344–346. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Politei, J.M. Histologic Abnormalities of Placental Tissues in Fabry Disease: A Case Report and Review of the Literature. Hum. Pathol. 2012, 43, 610–614. [Google Scholar] [CrossRef]
- Macdonald-Wallis, C.; Lawlor, D.A.; Heron, J.; Fraser, A.; Nelson, S.M.; Tilling, K. Relationships of Risk Factors for Pre-Eclampsia with Patterns of Occurrence of Isolated Gestational Proteinuria during Normal Term Pregnancy. PLoS ONE 2011, 6, e22115. [Google Scholar] [CrossRef]
- Laney, D.A.; Clarke, V.; Foley, A.; Hall, E.W.; Gillespie, S.E.; Holida, M.; Simmons, M.; Wadley, A. The Impact of Fabry Disease on Reproductive Fitness. JIMD Rep. 2017, 37, 85–97. [Google Scholar] [CrossRef]
- Chandra, A.; Copen, C.E.; Stephen, E.H. Infertility and impaired fecundity in the United States, 1982–2010: Data from the National Survey of Family Growth. Natl. Health Stat. Rep. 2013, 67, 1–19. [Google Scholar]
- Chandra, A.; Copen, C.E.; Stephen, E.H. Infertility service use in the United States: Data from the National Survey of Family Growth, 1982–2010. Natl. Health Stat. Rep. 2014, 73, 1–21. [Google Scholar]
- Kessler, L.M.; Craig, B.M.; Plosker, S.M.; Reed, D.R.; Quinn, G.P. Infertility Evaluation and Treatment among Women in the United States. Fertil. Steril. 2013, 100, 1025–1032.e2. [Google Scholar] [CrossRef]
- Bouwman, M.G.; Rombach, S.M.; Schenk, E.; Sweeb, A.; Wijburg, F.A.; Hollak, C.E.M.; Linthorst, G.E. Prevalence of Symptoms in Female Fabry Disease Patients: A Case-Control Survey. J. Inherit. Metab. Dis. 2012, 35, 891–898. [Google Scholar] [CrossRef]
- Simon, J.A. Low Sexual Desire—Is It All in Her Head? Pathophysiology, Diagnosis, and Treatment of Hypoactive Sexual Desire Disorder. Postgrad. Med. 2010, 122, 128–136. [Google Scholar] [CrossRef]
- SSessa, A.; Meroni, M.; Battini, G.; Maglio, A.; Brambilla, P.L.; Bertella, M.; Nebuloni, M.; Pallotti, F.; Giordano, F.; Bertagnolio, B.; et al. Renal Pathological Changes in Fabry Disease. J. Inherit. Metab. Dis. 2001, 24, 66–70. [Google Scholar] [CrossRef]
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am. J. Kidney Dis. 2002, 39 (Suppl. S1), S1–S266. [Google Scholar]
- Kriegsmann, J.; Otto, M.; Wandel, E.; Schwarting, A.; Faust, J.; Hansen, T.; Beck, J.; Whybra, C.; Beck, M. Morbus Fabry, Glomerulonephritis Mit Halbmondbildung Und Granulomatöse Interstitielle Nephritis. Der Pathol. 2003, 24, 439–443. [Google Scholar] [CrossRef]
- Martins, A.M.; D’Almeida, V.; Kyosen, S.O.; Takata, E.T.; Delgado, A.G.; Gonçalves, M.B.F.; Filho, C.C.B.; Filho, D.M.; Biagini, G.; Pimentel, H.; et al. Guidelines to Diagnosis and Monitoring of Fabry Disease and Review of Treatment Experiences. J. Pediatr. 2009, 155, S19–S31. [Google Scholar] [CrossRef]
- Eng, C.M.; Fletcher, J.; Wilcox, W.R.; Waldek, S.; Scott, C.R.; Sillence, D.O.; Breunig, F.; Charrow, J.; Germain, D.P.; Nicholls, K.; et al. Fabry Disease: Baseline Medical Characteristics of a Cohort of 1765 Males and Females in the Fabry Registry. J. Inherit. Metab. Dis. 2007, 30, 184–192. [Google Scholar] [CrossRef]
- Whybra, C.; Bähner, F.; Baron, K. Measurement of disease severity and progression in Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Kampmann, C.; Baehner, F.; Whybra, C.; Martin, C.; Wiethoff, C.M.; Ries, M.; Gal, A.; Beck, M. Cardiac Manifestations of Anderson–Fabry Disease in Heterozygous Females. J. Am. Coll. Cardiol. 2002, 40, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Galanos, J.; Nicholls, K.; Grigg, L.; Kiers, L.; Crawford, A.; Becker, G. Clinical Features of Fabry’s Disease in Australian Patients. Intern. Med. J. 2002, 32, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry Disease: Genetics, Pathology, and Treatment. Rev. Da Assoc. Médica Bras. 2020, 66 (Suppl. S1), s10–s16. [Google Scholar] [CrossRef] [PubMed]
- Curiati, M.A.; Aranda, C.S.; Kyosen, S.O.; Varela, P.; Pereira, V.G.; D’Almeida, V.; Pesquero, J.B.; Martins, A.M. The Challenge of Diagnosis and Indication for Treatment in Fabry Disease. J. Inborn Errors Metab. Screen. 2017, 5, 232640981668573. [Google Scholar] [CrossRef]
- Varela-Calais, P.; Nicolicht, P.; Martin, R.P.; Yamamoto, J.; D’Almeida, V.; Martins, A.M.; Pesquero, J.B. Functional Characterization of Novel Variants Found in Patients with Suspected Fabry Disease. Clin. Chim. Acta 2022, 534, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Jehn, U.; Bayraktar, S.; Pollmann, S.; Van Marck, V.; Weide, T.; Pavenstädt, H.; Brand, E.; Lenders, M. α-Galactosidase a Deficiency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes. Int. J. Mol. Sci. 2021, 22, 11339. [Google Scholar] [CrossRef]
- Froissart, R. Fabry Disease: D313Y Is an α-Galactosidase a Sequence Variant That Causes Pseudodeficient Activity in Plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef]
- Daitx, V.V.; Mezzalira, J.; Goldim, M.P.d.S.; Coelho, J.C. Comparison between α-Galactosidase a Activity in Blood Samples Collected on Filter Paper, Leukocytes and Plasma. Clin. Biochem. 2012, 45, 1233–1238. [Google Scholar] [CrossRef]
- Linthorst, G.E.; Poorthuis, B.J.H.M.; Hollak, C.E.M. Enzyme Activity for Determination of Presence of Fabry Disease in Women Results in 40% False-Negative Results. J. Am. Coll. Cardiol. 2008, 51, 2082. [Google Scholar] [CrossRef]
- Germain, D.P.; Benistan, K.; Angelova, L. X-Linked Inheritance and Its Implication in the Diagnosis and Management of Female Patients in Fabry Disease. La Rev. De Médecine Interne 2010, 31, S209–S213. [Google Scholar] [CrossRef]
- Pastores, G.M.; Lien, Y.H. Biochemical and molecular genetic basis of Fabry disease. J. Am. Soc. Nephrol. 2002, 13 (Suppl. S2), S130–S133. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Beck, M.; Höppner, W.; Germain, D.P. Clinical Utility Gene Card For: Fabry Disease—Update 2016. Eur. J. Hum. Genet. 2017, 25, e1. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Resnick-Silverman, L.A.; Niehaus, D.J.; Astrin, K.H.; Desnick, R.J. Nature and frequency of mutations in the α-galactosidase A gene that cause Fabry disease. Am. J. Hum. Genet. 1993, 53, 1186–1197. [Google Scholar] [PubMed]
- Mogensen, J.; van Tintelen, J.P.; Fokstuen, S.; Elliott, P.; van Langen, I.M.; Meder, B.; Richard, P.; Syrris, P.; Caforio, A.L.; Adler, Y.; et al. The Current Role of Next-Generation DNA Sequencing in Routine Care of Patients with Hereditary Cardiovascular Conditions: A Viewpoint Paper of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases and Members of the European Society of Human Genetics. Eur. Hear. J. 2015, 36, 1367–1370. [Google Scholar] [CrossRef]
- Germain, D.P.; Fouilhoux, A.; Decramer, S.; Tardieu, M.; Pillet, P.; Fila, M.; Rivera, S.; Deschênes, G.; Lacombe, D. Consensus Recommendations for Diagnosis, Management and Treatment of Fabry Disease in Paediatric Patients. Clin. Genet. 2019, 96, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, U.; Whybra, C.; Parini, R.; Pintos-Morell, G.; Mehta, A.; Sunder-Plassmann, G.; Widmer, U.; Beck, M. Clinical Manifestations of Fabry Disease in Children: Data from the Fabry Outcome Survey. Acta Paediatr. 2006, 95, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.L.; Costa, O.; do Sameiro Faria, M.; Almeida, P.; Lacerda, L. Cardiac Fabry’s Disease: An Unusual Cause of Left Ventricular Hypertrophy. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 630–633. [Google Scholar] [CrossRef]
- Ishii, S.; Nakao, S.; Minamikawa-Tachino, R.; Desnick, R.J.; Fan, J.-Q. Alternative Splicing in the α-Galactosidase a Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am. J. Hum. Genet. 2002, 70, 994–1002. [Google Scholar] [CrossRef]
- Kornreich, R.; Bishop, D.F.; Desnick, R.J. α-galactosidase A gene rearrangements causing Fabry disease. Identification of short direct repeats at breakpoints in an Alu-rich gene. J. Biol. Chem. 1990, 265, 9319–9326. [Google Scholar] [CrossRef]
- Bernstein, H.S.; Bishop, D.F.; Astrin, K.H.; Kornreich, R.; Eng, C.M.; Sakuraba, H.; Desnick, R.J. Fabry Disease: Six Gene Rearrangements and an Exonic Point Mutation in the α-Galactosidase Gene. J. Clin. Investig. 1989, 83, 1390–1399. [Google Scholar] [CrossRef]
- Ashton-Prolla, P.; Tong, B.; Shabbeer, J.; Astrin, K.H.; Eng, C.M.; Desnick, R.J. Fabry disease: Twenty-two novel mutations in the α-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes. J. Investig. Med. 2000, 48, 227–235. [Google Scholar] [PubMed]
- Laney, D.A.; Bennett, R.L.; Clarke, V.; Fox, A.; Hopkin, R.J.; Johnson, J.; O’Rourke, E.; Sims, K.; Walter, G. Fabry Disease Practice Guidelines: Recommendations of the National Society of Genetic Counselors. J. Genet. Couns. 2013, 22, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Bernstein, H.S.; Astrin, K.H.; Bishop, D.F. Fabry Disease: Molecular Diagnosis of Hemizygotes and Heterozygotes. Enzyme 1987, 38, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Schelleckes, M.; Lenders, M.; Guske, K.; Schmitz, B.; Tanislav, C.; Ständer, S.; Metze, D.; Katona, I.; Weis, J.; Brand, S.-M.; et al. Cryptogenic Stroke and Small Fiber Neuropathy of Unknown Etiology in Patients with α-Galactosidase A-10T Genotype. Orphanet J. Rare Dis. 2014, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Duro, G.; Pecoraro, R.; Simonetta, I.; Miceli, S.; Colomba, P.; Zizzo, C.; Di Chiara, T.; Scaglione, R.; Della Corte, V.; et al. A Family with Various Symptomatology Suggestive of Anderson–Fabry Disease and a Genetic Polymorphism of α Galactosidase a Gene. Clin. Biochem. 2015, 48, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Oder, D.; Liu, D.; Üçeyler, N.; Sommer, C.; Hu, K.; Salinger, T.; Müntze, J.; Petritsch, B.; Ertl, G.; Wanner, C.; et al. Clinical Impact of the α-Galactosidase a Gene Single Nucleotide Polymorphism -10C>T. Medicine 2018, 97, e10669. [Google Scholar] [CrossRef]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. α-Galactosidase a Deficient Mice: A Model of Fabry Disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef]
- Houge, G.; Tøndel, C.; Kaarbøe, Ø.; Hirth, A.; Bostad, L.; Svarstad, E. Fabry or Not Fabry—A Question of Ascertainment. Eur. J. Hum. Genet. 2011, 19, 1111. [Google Scholar] [CrossRef]
- Jaurretche, S.; Perez, G.R.; Venera, G. High Lyso-Gb3 Plasma Levels Associated with Decreased MiR-29 and MiR-200 Urinary Excretion in Young Non-Albuminuric Male Patient with Classic Fabry Disease. Case Rep. Nephrol. 2019, 2019, 4980942. [Google Scholar] [CrossRef]
- Jaurretche, S.; Venera, G.; Antongiovanni, N.; Perretta, F.; Perez, G.R. Urinary excretion of microRNAs in young Fabry disease patients with mild or absent nephropathy. Open J. Nephrol. 2018, 8, 71–83. [Google Scholar] [CrossRef]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A.I.; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme Replacement Therapy in Fabry Disease. JAMA 2001, 285, 2743. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, P.; Neumann, P.M. Treatment of Fabry Disease: Current and Emerging Strategies. Curr. Pharm. Biotechnol. 2011, 12, 916–922. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the Treatment of Fabry Disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Cianfrone, P.; Simeoni, M.; Comi, N.; Piraina, V.; Talarico, R.; Cerantonio, A.; Gentile, I.; Fabiano, F.F.; Lucisano, G.; Foti, D.; et al. How to Improve Duration and Efficiency of the Antiproteinuric Response to Ramipril: RamiPROT—A Prospective Cohort Study. J. Nephrol. 2015, 30, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, M.M.; Cianfrone, P.; Comi, N.; Gentile, I.; Fabiano, F.F.; Piraina, V.; Talarico, R.; Lucisano, G.; Rivoli, L.; Andreucci, M. È possibile migliorare la durata e l’efficacia della risposta anti-proteinurica di Ramipril? [Is it feasible to improve the duration and the efficiency of Ramipril anti-proteinuric response?]. G Ital. Nefrol. 2015, 32, 1724–5590. [Google Scholar]
- Simeoni, M.; Nicotera, R.; Colao, M.; Citraro, M.L.; Pelagi, E.; Cerantonio, A.; Comi, N.; Coppolino, G.; Fuiano, G. Direct Inhibition of Plasmatic Renin Activity with Aliskiren: A Promising but Under-Investigated Therapeutic Option for Non-Diabetic Glomerulonephritis. Int. Urol. Nephrol. 2015, 48, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, P.A. Fabry Disease: Treatment and Diagnosis. IUBMB Life 2009, 61, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and Efficacy of Recombinant Human α-Galactosidase a Replacement Therapy in Fabry’s Disease. New Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef]
- Weidemann, F.; Niemann, M.; Sommer, C.; Beer, M.; Breunig, F.; Wanner, C. Interdisciplinary Approach towards Female Patients with Fabry Disease. Eur. J. Clin. Investig. 2011, 42, 455–462. [Google Scholar] [CrossRef]
- Whybra, C.; Miebach, E.; Mengel, E.; Gal, A.; Baron, K.; Beck, M.; Kampmann, C. A 4-Year Study of the Efficacy and Tolerability of Enzyme Replacement Therapy with Agalsidase Alfa in 36 Women with Fabry Disease. Genet. Med. 2009, 11, 441–449. [Google Scholar] [CrossRef]
- Baehner, F.; Kampmann, C.; Whybra, C.; Miebach, E.; Wiethoff, C.M.; Beck, M.; Beck, M. Enzyme Replacement Therapy in Heterozygous Females with Fabry Disease: Results of a Phase IIIB Study. J. Inherit. Metab. Dis. 2003, 26, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Wendt, S.; Whybra, C.; Kampmann, C.; Teichmann, E.; Beck, M. Successful Pregnancy Outcome in a Patient with Fabry Disease Receiving Enzyme Replacement Therapy with Agalsidase Alfa. J. Inherit. Metab. Dis. 2005, 28, 787–788. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B.; Bähner, F.; Barba, M.; Hughes, D.A.; Beck, M. Fabry disease in females: Clinical characteristics and effects of enzyme replacement therapy. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Goláň, L.; Goker-Alpan, O.; Holida, M.; Kantola, I.; Klopotowski, M.; Kuusisto, J.; Linhart, A.; Musial, J.; Nicholls, K.; Gonzalez-Rodriguez, D.; et al. Evaluation of the Efficacy and Safety of Three Dosing Regimens of Agalsidase Alfa Enzyme Replacement Therapy in Adults with Fabry Disease. Drug Des. Dev. Ther. 2015, 9, 3435–3444. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Banypersad, S.; Woolfson, P.; Waldek, S. Enzyme Replacement Therapy Improves Cardiac Features and Severity of Fabry Disease. Mol. Genet. Metab. 2012, 107, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Feriozzi, S.; Schwarting, A.; Sunder-Plassmann, G.; West, M.; Cybulla, M. Agalsidase Alfa Slows the Decline in Renal Function in Patients with Fabry Disease. Am. J. Nephrol. 2008, 29, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Feriozzi, S.; Torras, J.; Cybulla, M.; Nicholls, K.; Sunder-Plassmann, G.; West, M. The Effectiveness of Long-Term Agalsidase Alfa Therapy in the Treatment of Fabry Nephropathy. Clin. J. Am. Soc. Nephrol. 2012, 7, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Sergi, B.; Conti, G.; Paludetti, G.; Interdisciplinary Study Group On Fabry Disease. Inner Ear Involvement in Anderson-Fabry Disease: Long-Term Follow-up during Enzyme Replacement Therapy. Acta Otorhinolaryngol. Ital. 2010, 30, 87–93. [Google Scholar] [PubMed]
- Hughes, D.A.; Barba Romero, M.-Á.; Hollak, C.E.M.; Giugliani, R.; Deegan, P.B. Response of Women with Fabry Disease to Enzyme Replacement Therapy: Comparison with Men, Using Data from FOS—The Fabry Outcome Survey. Mol. Genet. Metab. 2011, 103, 207–214. [Google Scholar] [CrossRef]
- Warnock, D.G.; Ortiz, A.; Mauer, M.; Linthorst, G.E.; Oliveira, J.P.; Serra, A.L.; Marodi, L.; Mignani, R.; Vujkovac, B.; Beitner-Johnson, D.; et al. Renal Outcomes of Agalsidase β Treatment for Fabry Disease: Role of Proteinuria and Timing of Treatment Initiation. Nephrol. Dial. Transplant. 2011, 27, 1042–1049. [Google Scholar] [CrossRef]
- Hoffmann, B.; Schwarz, M.; Mehta, A.; Keshav, S. Gastrointestinal Symptoms in 342 Patients with Fabry Disease: Prevalence and Response to Enzyme Replacement Therapy. Clin. Gastroenterol. Hepatol. 2007, 5, 1447–1453. [Google Scholar] [CrossRef]
- Hoffmann, B. Fabry Disease: Recent Advances in Pathology, Diagnosis, Treatment and Monitoring. Orphanet J. Rare Dis. 2009, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, G.; Simeoni, M.; Rizza, P.; Caroleo, M.; Capria, M.; Mazzitello, G.; Sacco, T.; Mazzuca, E.; Panzino, M.T.; Cerantonio, A.; et al. Quality of Life, Clinical Outcome, Personality and Coping in Chronic Hemodialysis Patients. Ren. Fail. 2016, 39, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Morand, O.; Johnson, J.; Walter, J.; Atkinson, L.; Kline, G.; Frey, A.; Politei, J.; Schiffmann, R. Symptoms and Quality of Life in Patients with Fabry Disease: Results from an International Patient Survey. Adv. Ther. 2019, 36, 2866. [Google Scholar] [CrossRef] [PubMed]
- Street, N.J.; Yi, M.S.; Bailey, L.A.; Hopkin, R.J. Comparison of Health-Related Quality of Life between Heterozygous Women with Fabry Disease, a Healthy Control Population, and Patients with Other Chronic Disease. Genet. Med. 2006, 8, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Silva-Gburek, J.; Rochford, L.; Hopkin, R.J.; Jefferies, J.L. Ventricular Tachycardia in Fabry Disease Detected in a 50-Year-Old Woman during 14-Day Continuous Cardiac Monitoring. Tex. Heart Inst. J. 2016, 43, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.J. Neuropsychiatric and Psychosocial Aspects of Fabry Disease. PubMed. Available online: https://www.ncbi.nlm.nih.gov/books/NBK11618/ (accessed on 18 December 2023).
- Sadek, J.; Shellhaas, R.; Camfield, C.S.; Camfield, P.R.; Burley, J. Psychiatric Findings in Four Female Carriers of Fabry Disease. Psychiatr. Genet. 2004, 14, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Borsini, W.; Giuliacci, G.; Torricelli, F.; Pelo, E.; Martinelli, F.; Scordo, M.R. Anderson-Fabry Disease with Cerebrovascular Complications in Two Italian Families. Neurol. Sci. 2002, 23, 49–53. [Google Scholar] [CrossRef]
- Mendez, M.F.; Stanley, T.M.; Medel, N.M.; Li, Z.; Tedesco, D.T. The Vascular Dementia of Fabry’s Disease. Dement. Geriatr. Cogn. Disord. 1997, 8, 252–257. [Google Scholar] [CrossRef]
- Hoffmann, B. Effects of Enzyme Replacement Therapy on Pain and Health Related Quality of Life in Patients with Fabry Disease: Data from FOS (Fabry Outcome Survey). J. Med. Genet. 2005, 42, 247–252. [Google Scholar] [CrossRef]
- Fellgiebel, A.; Muller, M.J.; Mazanek, M.; Baron, K.; Beck, M.; Stoeter, P. White Matter Lesion Severity in Male and Female Patients with Fabry Disease. Neurology 2005, 65, 600–602. [Google Scholar] [CrossRef]
- Crutchfield, K.E.; Patronas, N.J.; Dambrosia, J.M.; Frei, K.P.; Banerjee, T.K.; Barton, N.W.; Schiffmann, R. Quantitative Analysis of Cerebral Vasculopathy in Patients with Fabry Disease. Neurology 1998, 50, 1746–1749. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, B.L.; Joiner, T. Positive Emotions Trigger Upward Spirals toward Emotional Well-Being. Psychol. Sci. 2002, 13, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Ramaswami, U.; Parini, R.; Lindblad, B.; Whybra, C.; Willers, I.; Gal, A.; Beck, M. The Early Clinical Phenotype of Fabry Disease: A Study on 35 European Children and Adolescents. Eur. J. Pediatr. 2003, 162, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Ries, M. Pediatric Fabry Disease. Pediatrics 2005, 115, e344–e355. [Google Scholar] [CrossRef]
- Hopkin, R.J.; Bissler, J.; Banikazemi, M.; Clarke, L.; Eng, C.M.; Germain, D.P.; Lemay, R.; Tylki-Szymanska, A.; Wilcox, W.R. Characterization of Fabry Disease in 352 Pediatric Patients in the Fabry Registry. Pediatr. Res. 2008, 64, 550–555. [Google Scholar] [CrossRef]
- Tøndel, C.; Bostad, L.; Hirth, A.; Svarstad, E. Renal Biopsy Findings in Children and Adolescents with Fabry Disease and Minimal Albuminuria. Am. J. Kidney Dis. 2008, 51, 767–776. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Biomarker Category | Biomarkers in Women | References |
|---|---|---|
| Plasma | Globotriaosylceramide (Gb3) | [67] |
| Lyso-Gb3 | [62,72] | |
| Urinary | Urinary globotriaosylceramide (Gb3) | [67,73] |
| Urinary lyso-Gb3 | [62,73] | |
| Urinary CDH of Ga2 (long chains isoforms) | [74] | |
| Enzyme activity | α-Galactosidase A/LysoGb3 ratio | [66] |
| α-GalactosidaseA enzyme activity | [75,76,77,78] | |
| Genetic | Genetic mutations in the GLA gene | [79,80,81] |
| Kidney | eGFR, Creatinine, albuminuria, and proteinuria | [82,83] |
| Cardiac related | Troponins | [65] |
| TNF, TNFR1, TNFR2 and IL6 | [65,84] | |
| galectin-3,NT-proBNP, BNP, MRproANP, MMP2 and MMP9 | [65,85] | |
| Detection of Fabry cardiomyopathy by CMR with LE imaging to assess LV hypertrophy and replacement fibrosis | [86] | |
| MicroRNA profiles | Dysregulation of miRNAs associated with Fabry disease, for example, miR21-5p and miR19a-3p in the TGF-β signaling pathways | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izhar, R.; Borriello, M.; La Russa, A.; Di Paola, R.; De, A.; Capasso, G.; Ingrosso, D.; Perna, A.F.; Simeoni, M. Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations. Genes 2024, 15, 37. https://doi.org/10.3390/genes15010037
Izhar R, Borriello M, La Russa A, Di Paola R, De A, Capasso G, Ingrosso D, Perna AF, Simeoni M. Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations. Genes. 2024; 15(1):37. https://doi.org/10.3390/genes15010037
Chicago/Turabian StyleIzhar, Raafiah, Margherita Borriello, Antonella La Russa, Rossella Di Paola, Ananya De, Giovambattista Capasso, Diego Ingrosso, Alessandra F. Perna, and Mariadelina Simeoni. 2024. "Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations" Genes 15, no. 1: 37. https://doi.org/10.3390/genes15010037